Characterization of the Escherichia coli Virulent Myophage ST32

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Strain
2.2. Phage Isolation and Purification
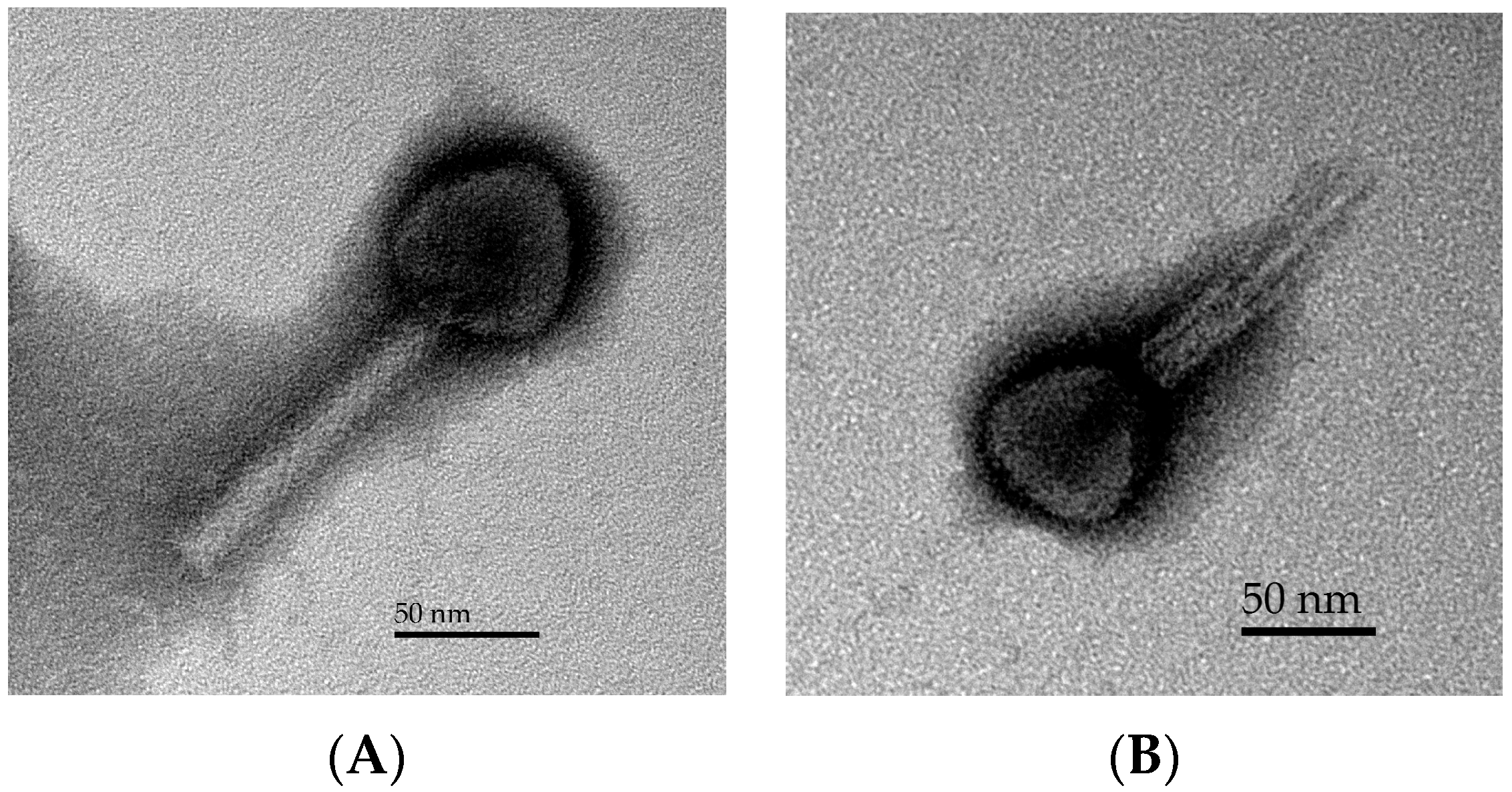
2.3. Phage Morphology
2.4. Host Range
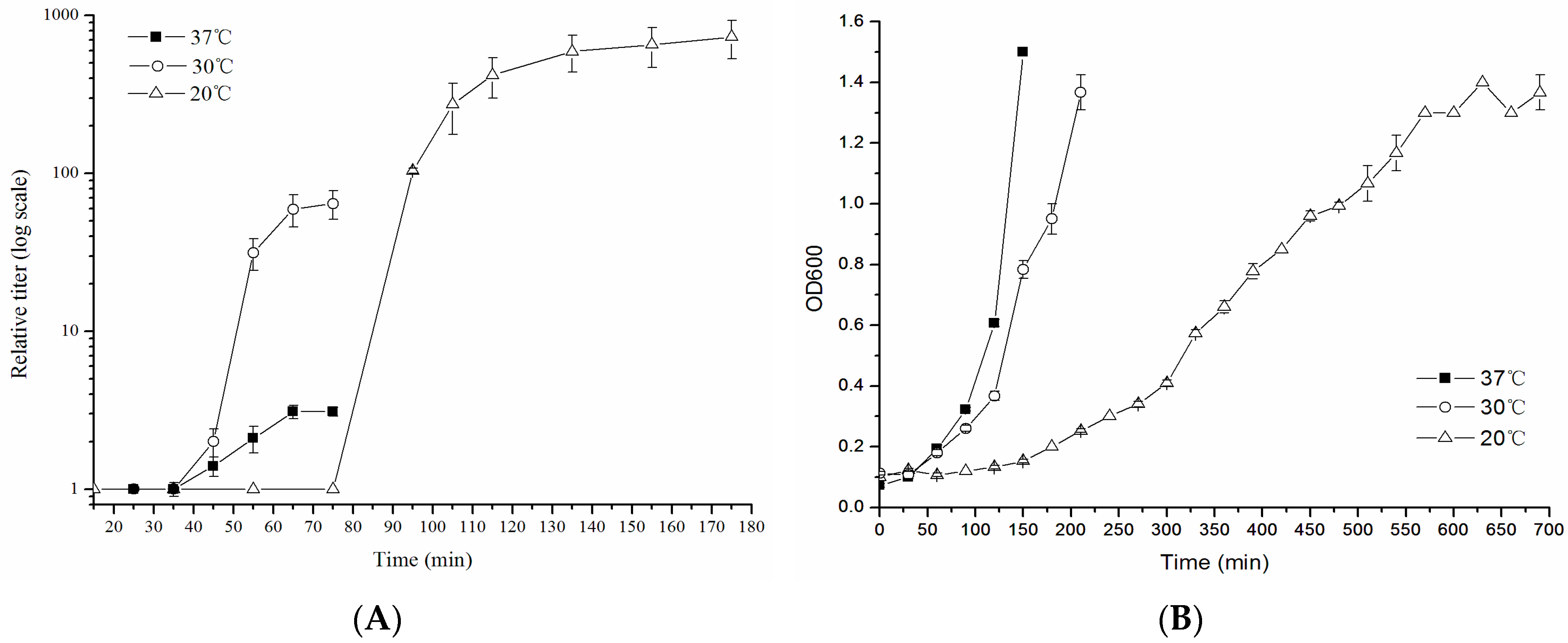
2.5. One-Step Growth Curve Assay
2.6. E. coli ST130 Growth
2.7. Sequencing and Analysis
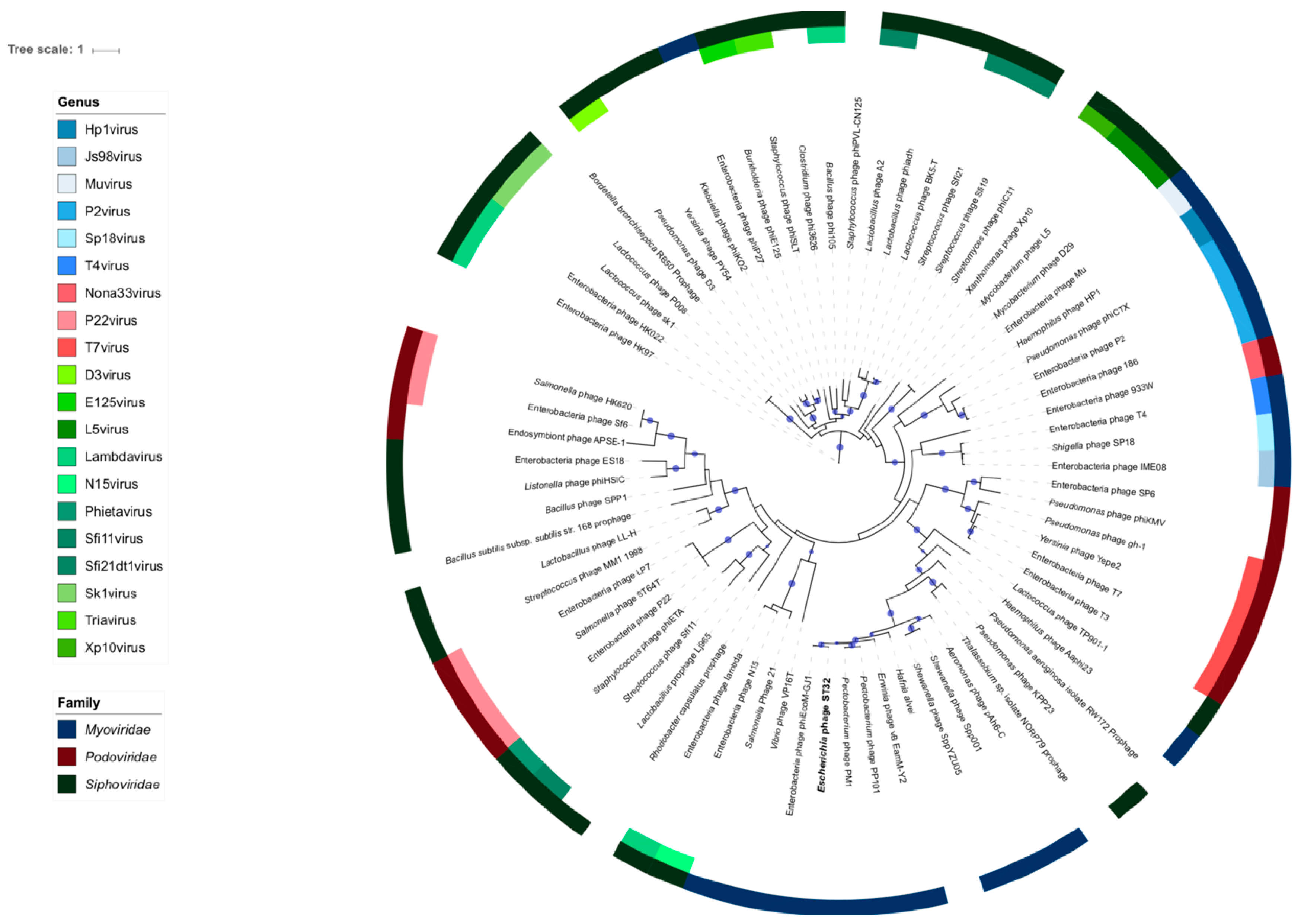
2.8. Terminase Tree
2.9. Nucleotide Sequence Accession Number
3. Results and Discussion
3.1. Phage Morphology
3.2. Host Range
3.3. One-Step Growth Curve
3.4. Genomic Features of Phage ST32
3.5. Phylogeny of Phage ST32
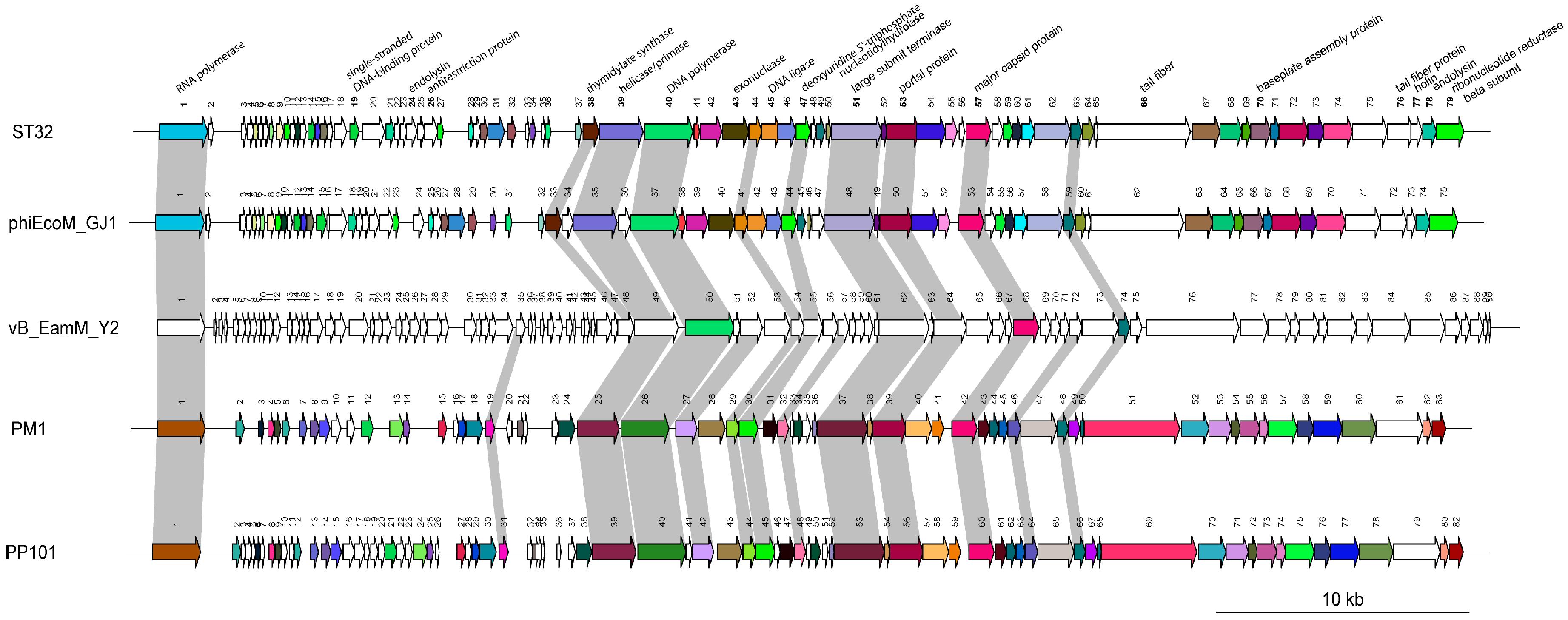
3.6. Comparative Genomic Analysis
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Caprioli, A.; Scavia, G.; Morabito, S. Public health microbiology of Shiga toxin-producing Escherichia coli. Microbiol. Spectr. 2014, 2. [Google Scholar] [CrossRef] [PubMed]
- Melton-Celsa, A.; Mohawk, K.; Teel, L.; O’Brien, A. Pathogenesis of Shiga-toxin producing Escherichia coli. In Ricin and Shiga Toxins; Current Topics in Microbiology and Immunology; Springer: Berlin/Heidelberg, Germany, 2011; Volume 357, pp. 67–103. [Google Scholar]
- Kaper, J.B.; Nataro, J.P.; Mobley, H.L.T. Pathogenic Escherichia coli. Nat. Rev. Microbiol. 2004, 2, 123. [Google Scholar] [CrossRef] [PubMed]
- Suojala, L.; Kaartinen, L.; Pyörälä, S. Treatment for bovine Echerichia coli mastitis—An evidence-based approach. J. Vet. Pharmacol. Ther. 2013, 36, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Paton, J.C.; Paton, A.W. Pathogenesis and diagnosis of Shiga toxin-producing Escherichia coli infections. Clin. Microbiol. Rev. 1998, 11, 450–479. [Google Scholar] [CrossRef] [PubMed]
- Blaser, M. Antibiotic overuse: Stop the killing of beneficial bacteria. Nature 2011, 476, 393. [Google Scholar] [CrossRef] [PubMed]
- Safwat Mohamed, D.; Farouk Ahmed, E.; Mohamed Mahmoud, A.; Abd El-Baky, R.M.; John, J. Isolation and evaluation of cocktail phages for the control of multidrug-resistant Escherichia coli serotype O104: H4 and E. coli O157: H7 isolates causing diarrhea. FEMS Microbiol. Lett. 2017, 365, fnx275. [Google Scholar] [CrossRef] [PubMed]
- Cieplak, T.; Soffer, N.; Sulakvelidze, A.; Nielsen, D.S. A bacteriophage cocktail targeting Escherichia coli reduces E. coli in simulated gut conditions, while preserving a non-targeted representative commensal normal microbiota. Gut Microbes 2018, 9, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Poirel, L.; Madec, J.-Y.; Lupo, A.; Schink, A.-K.; Kieffer, N.; Nordmann, P.; Schwarz, S. Antimicrobial resistance in Escherichia coli. Microbiol. Spectr. 2018, 6. [Google Scholar] [CrossRef]
- Cabello, F.C. Heavy use of prophylactic antibiotics in aquaculture: A growing problem for human and animal health and for the environment. Environ. Microbiol. 2006, 8, 1137–1144. [Google Scholar] [CrossRef] [PubMed]
- Tajbakhsh, E.; Khamesipour, F.; Ranjbar, R.; Ugwu, I.C. Prevalence of class 1 and 2 integrons in multi-drug resistant Escherichia coli isolated from aquaculture water in Chaharmahal Va Bakhtiari province, Iran. Ann. Clin. Microbiol. Antimicrob. 2015, 14, 37. [Google Scholar] [CrossRef] [PubMed]
- Silva, Y.J.; Costa, L.; Pereira, C.; Cunha, Â.; Calado, R.; Gomes, N.C.M.; Almeida, A. Influence of environmental variables in the efficiency of phage therapy in aquaculture. Microb. Biotechnol. 2014, 7, 401–413. [Google Scholar] [CrossRef] [PubMed]
- Mohan Raj, J.R.; Vittal, R.; Huilgol, P.; Bhat, U.; Karunasagar, I. T4-like Escherichia coli phages from the environment carry blaCTX-M. Lett. Appl. Microbiol. 2018, 67, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Heuer, O.E.; Kruse, H.; Grave, K.; Collignon, P.; Karunasagar, I.; Angulo, F.J. Human health consequences of use of antimicrobial agents in aquaculture. Clin. Infect. Dis. 2009, 49, 1248–1253. [Google Scholar] [CrossRef] [PubMed]
- Van den Bogaard, A.E.; London, N.; Driessen, C.; Stobberingh, E.E. Antibiotic resistance of faecal Escherichia coli in poultry, poultry farmers and poultry slaughterers. J. Antimicrob. Chemother. 2001, 47, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Hammerum, A.M.; Heuer, O.E. Human health hazards from antimicrobial-resistant Escherichia coli of animal origin. Clin. Infect. Dis. 2009, 48, 916–921. [Google Scholar] [CrossRef] [PubMed]
- Reinthaler, F.F.; Posch, J.; Feierl, G.; Wüst, G.; Haas, D.; Ruckenbauer, G.; Mascher, F.; Marth, E. Antibiotic resistance of E. coli in sewage and sludge. Water Res. 2003, 37, 1685–1690. [Google Scholar] [CrossRef]
- Mesa, R.J.; Blanc, V.; Blanch, A.R.; Cortés, P.; Gonzalez, J.J.; Lavilla, S.; Miro, E.; Muniesa, M.; Saco, M.; Tórtola, M.T. Extended-spectrum β-lactamase-producing Enterobacteriaceae in different environments (humans, food, animal farms and sewage). J. Antimicrob. Chemother. 2006, 58, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Watkinson, A.J.; Micalizzi, G.B.; Graham, G.M.; Bates, J.B.; Costanzo, S.D. Antibiotic-resistant Escherichia coli in wastewaters, surface waters, and oysters from an urban riverine system. Appl. Environ. Microbiol. 2007, 73, 5667–5670. [Google Scholar] [CrossRef] [PubMed]
- Korzeniewska, E.; Korzeniewska, A.; Harnisz, M. Antibiotic resistant Escherichia coli in hospital and municipal sewage and their emission to the environment. Ecotoxicol. Environ. Saf. 2013, 91, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Nakai, T.; Park, S.C. Bacteriophage therapy of infectious diseases in aquaculture. Res. Microbiol. 2002, 153, 13–18. [Google Scholar] [CrossRef]
- Wagenaar, J.A.; Van Bergen, M.A.P.; Mueller, M.A.; Wassenaar, T.M.; Carlton, R.M. Phage therapy reduces Campylobacter jejuni colonization in broilers. Vet. Microbiol. 2005, 109, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Diaz, S.F.; Hipólito-Morales, A. Efficacy of phage therapy to prevent mortality during the vibriosis of brine shrimp. Aquaculture 2013, 400, 120–124. [Google Scholar] [CrossRef]
- Litt, P.K.; Saha, J.; Jaroni, D. Characterization of bacteriophages targeting Non-O157 Shiga toxigenic Escherichia coli. J. Food Prot. 2018, 81, 785–794. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, K.; Cazarez-Montoya, C.; Lopez-Moreno, H.S.; Castro-del Campo, N. Bacteriophage cocktail for biocontrol of Escherichia coli O157: H7: Stability and potential allergenicity study. PLoS ONE 2018, 13, e0195023. [Google Scholar] [CrossRef] [PubMed]
- Duarte, V.S.; Dias, R.S.; Kropinski, A.M.; Campanaro, S.; Treu, L.; Siqueira, C.; Vieira, M.S.; Paes, I.S.; Santana, G.R.; Martins, F. Genomic analysis and immune response in a murine mastitis model of vB_EcoM-UFV13, a potential biocontrol agent for use in dairy cows. Sci. Rep. 2018, 8, 6845. [Google Scholar] [CrossRef] [PubMed]
- Cisek, A.A.; Dąbrowska, I.; Gregorczyk, K.P.; Wyżewski, Z. Phage therapy in bacterial infections treatment: One hundred years after the discovery of bacteriophages. Curr. Microbiol. 2017, 74, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Bhensdadia, D.V.; Bhimani, H.D.; Rawal, C.M.; Kothari, V.V.; Raval, V.H.; Kothari, C.R.; Patel, A.B.; Bhatt, V.D.; Parmar, N.R.; Sajnani, M.R. Complete genome sequence of Escherichia phage ADB-2 isolated from a fecal sample of poultry. Genome Announc. 2013, 1, e00043-13. [Google Scholar] [CrossRef] [PubMed]
- Von lytischen Bakteriophagen, P.A. Post-harvest application of lytic bacteriophages for biocontrol of food-borne pathogens and spoilage bacteria. Berl. Munch. Tierarztl. Wochenschr. 2013, 126, 9. [Google Scholar]
- Lynch, M.F.; Tauxe, R.V.; Hedberg, C.W. The growing burden of foodborne outbreaks due to contaminated fresh produce: Risks and opportunities. Epidemiol. Infect. 2009, 137, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M. Lytic bacteriophages: Potential interventions against enteric bacterial pathogens on produce. Bacteriophage 2013, 3, e25518. [Google Scholar] [CrossRef] [PubMed]
- Jensen, E.C.; Schrader, H.S.; Rieland, B.; Thompson, T.L.; Lee, K.W.; Nickerson, K.W.; Kokjohn, T.A. Prevalence of broad-host-range lytic bacteriophages of Sphaerotilus natans, Escherichia coli, and Pseudomonas aeruginosa. Appl. Environ. Microbiol. 1998, 64, 575–580. [Google Scholar] [PubMed]
- Koskella, B.; Meaden, S. Understanding bacteriophage specificity in natural microbial communities. Viruses 2013, 5, 806–823. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.H. Bacteriophages; Interscience Publishers: New York, NY, USA, 1959; p. 592. [Google Scholar]
- Azaïez, S.R.C.; Fliss, I.; Simard, R.E.; Moineau, S. Monoclonal antibodies raised against native major capsid proteins of lactococcal c2-like bacteriophages. Appl. Environ. Microbiol. 1998, 64, 4255–4259. [Google Scholar]
- Fortier, L.-C.; Moineau, S. Morphological and genetic diversity of temperate phages in Clostridium difficile. Appl. Environ. Microbiol. 2007, 73, 7358–7366. [Google Scholar] [CrossRef] [PubMed]
- Duplessis, M.; Russell, W.M.; Romero, D.A.; Moineau, S. Global gene expression analysis of two Streptococcus thermophilus bacteriophages using DNA microarray. Virology 2005, 340, 192–208. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.; Russell, D.W. Molecular clonning: A laboratory manual. In Molecular Clonning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2001. [Google Scholar]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Mercanti, D.J.; Rousseau, G.M.; Capra, M.L.; Quiberoni, A.; Tremblay, D.M.; Labrie, S.J.; Moineau, S. Genomic diversity of phages infecting probiotic strains of Lactobacillus paracasei. Appl. Environ. Microbiol. 2016, 82, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Lavigne, R.; Darius, P.; Summer, E.J.; Seto, D.; Mahadevan, P.; Nilsson, A.S.; Ackermann, H.W.; Kropinski, A.M. Classification of Myoviridae bacteriophages using protein sequence similarity. BMC Microbiol. 2009, 9, 224. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.D.; Johnson, R.P.; Xu, Y.; McAllister, T.A.; Sharma, R.; Louie, M.; Stanford, K. Host range and lytic capability of four bacteriophages against bovine and clinical human isolates of Shiga toxin-producing Escherichia coli O157: H7. J. Appl. Microbiol. 2009, 107, 646–656. [Google Scholar] [CrossRef] [PubMed]
- Sillankorva, S.; Oliveira, D.; Moura, A.; Henriques, M.; Faustino, A.; Nicolau, A.; Azeredo, J. Efficacy of a broad host range lytic bacteriophage against E. coli adhered to urothelium. Curr. Microbiol. 2011, 62, 1128–1132. [Google Scholar] [CrossRef] [PubMed]
- Hoang, H.A.; Quy, N.T.C.; Chi, N.V.T. Detection of Escherichia coli in ready-to-eat fresh vegetables using broad-host-range recombinant phages. J. Appl. Microbiol. 2018, 124, 1610–1616. [Google Scholar] [CrossRef] [PubMed]
- Ross, A.; Ward, S.; Hyman, P. More is better: Selecting for broad host range bacteriophages. Front. Microbiol. 2016, 7, 1352. [Google Scholar] [CrossRef] [PubMed]
- Keen, E.C.; Adhya, S.L. Phage therapy: Current research and applications. Clin. Infect. Dis. 2015, 61, 141–142. [Google Scholar] [CrossRef]
- Kaliniene, L.; Truncaitė, L.; Šimoliūnas, E.; Zajančkauskaitė, A.; Vilkaitytė, M.; Kaupinis, A.; Skapas, M.; Meškys, R. Molecular analysis of the low-temperature Escherichia coli phage vB_EcoS_NBD2. Arch. Virol. 2018, 163, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Jamalludeen, N.; Kropinski, A.M.; Johnson, R.P.; Lingohr, E.; Harel, J.; Gyles, C.L. Complete genomic sequence of bacteriophage φEcoM-GJ1, a novel phage that has myovirus morphology and a podovirus-like RNA polymerase. Appl. Environ. Microbiol. 2008, 74, 516–525. [Google Scholar] [CrossRef] [PubMed]
- Fouts, D.E. Phage_Finder: Automated identification and classification of prophage regions in complete bacterial genome sequences. Nucleic Acids Res. 2006, 34, 5839–5851. [Google Scholar] [CrossRef] [PubMed]
- Maluf, N.K.; Yang, Q.; Catalano, C.E. Self-association properties of the bacteriophage λ terminase holoenzyme: Implications for the DNA packaging motor. J. Mol. Biol. 2005, 347, 523–542. [Google Scholar] [CrossRef] [PubMed]
- Catalano, C.E. The terminase enzyme from bacteriophage lambda: A DNA-packaging machine. Cell. Mol. Life Sci. C 2000, 57, 128–148. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.-A.; Shin, H.; Lee, D.H.; Han, S.-W.; Lee, J.-H.; Ryu, S.; Heu, S. Complete genome sequence of the Pectobacterium carotovorum subsp. carotovorum virulent bacteriophage PM1. Arch. Virol. 2014, 159, 2185–2187. [Google Scholar] [CrossRef] [PubMed]
- Born, Y.; Fieseler, L.; Marazzi, J.; Lurz, R.; Duffy, B.; Loessner, M.J. Novel virulent and broad host range Erwinia amylovora bacteriophages reveal a high degree of mosaicism and relationship to Enterobacteriaceae phages. Appl. Environ. Microbiol. 2011, 15, AEM-03022. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Dehbi, M.; Moeck, G.; Arhin, F.; Bauda, P.; Bergeron, D.; Callejo, M.; Ferretti, V.; Ha, N.; Kwan, T. Antimicrobial drug discovery through bacteriophage genomics. Nat. Biotechnol. 2004, 22, 185. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Non-Pathogen | Pathogen | ||||||
|---|---|---|---|---|---|---|---|
| Genus/Species/Subspecies of the Host Strain | # HER | Name of the Host Strain | ΦST32 | Genus/Species/Subspecies of the Host Strain | # HER | Name of the Host Strain | ΦST32 |
| Escherichia coli | 1022 | O44:K74 MUL-B37.2 | − | Escherichia coli | 1176 | N/A | +++ |
| Escherichia coli | 1024 | B (11303) | + | Escherichia coli | 1255 | O157:H7 C-8299-83 | − |
| Escherichia coli | 1025 | K12 C600 (λ) | + | Escherichia coli | 1256 | O157:H7 E318 | − |
| Escherichia coli | 1036 | C (13706) | ++++ | Escherichia coli | 1257 | O157:H7 A7793-B1 | − |
| Escherichia coli | 1037 | K12S | + | Escherichia coli | 1258 | O157:H7 C-8300-83 | − |
| Escherichia coli | 1040 | K12 (λ) Lederberg | + | Escherichia coli | 1259 | O157:H7 C-7685-84 | − |
| Escherichia coli | 1077 | W3350 | + | Escherichia coli | 1260 | O157:H7 CL40 | − |
| Escherichia coli | 1128 | MUL-B70.1 | − | Escherichia coli | 1261 | O157:H7 C-7111-85 | − |
| Escherichia coli | 1129 | O86:B7 MUL-B3.1 | − | Escherichia coli | 1262 | O157:H7 B1190-1 | − |
| Escherichia coli | 1139 | K12 65 | + | Escherichia coli | 1263 | O157:H7 B1328-C10 | − |
| Escherichia coli | 1144 | K12S Lederberg | − | Escherichia coli | 1264 | O157:H7 A8188-B3 | − |
| Escherichia coli | 1155 | K1 | ++++ | Escherichia coli | 1265 | O157:H7 C7420-85 | − |
| Escherichia coli | 1213 | JE-1 (N3) | − | Escherichia coli | 1266 | O157:H7 3283 | − |
| Escherichia coli | 1217 | JE-2(R62Rpilc) | + | Escherichia coli | 1267 | O157:H7 C-7140-85 | − |
| Escherichia coli | 1218 | J53(RIP69) | − | Escherichia coli | 1268 | O157:H7 5896 | − |
| Escherichia coli | 1219 | K12 J62-1(R997) | − | Escherichia coli | 1269 | O157:H7 C-7142-85 | − |
| Escherichia coli | 1221 | K12 J53-1(R15) | + | Escherichia coli | 1270 | O157:H7 C-91-84 | − |
| Escherichia coli | 1222 | JE-1 (RA1::TN5Sqr) | ++++ | Escherichia coli | H21 ST130 | ++++ | |
| Escherichia coli | 1240 | J62-1 (R27::TN7) | + | Escherichia coli | O165:H8 ST120 | − | |
| Escherichia coli | 1252 | 40 | + | Escherichia coli | O8:H16 ST110 | ++ | |
| Escherichia coli | 1253 | HM 8305 | − | Escherichia coli | H8 ST100 | ++ | |
| Escherichia coli | 1271 | K12 C600 (H-19J) | + | Escherichia coli | O153:H12 BW | − | |
| Escherichia coli | 1275 | K12 C600 | + | Shigella sonnei | 1043 | Y6R | + |
| Escherichia coli | 1290 | CSH39 | − | Shigella dysenteriae | 1031 | aSH | − |
| Escherichia coli | 1299 | K12 C600 (933-J) | + | Shigella dysenteriae | 1020 | SH(P2) | − |
| Escherichia coli | 1315 | F492 (O8:K27-:H-) | ++++ | Salmonella paratyphi | 1045 | B type 1 | − |
| Escherichia coli | 1337 | O103 2929 | + | Salmonella typhi | 1038 | ViA subtype Tananarive | − |
| Escherichia coli | 1366 | K12 MC4100 | + | Citrobacter freundii | 1518 | CF3 | − |
| Escherichia coli | 1374 | E69 O9:K30:H12 | − | Citrobacter freundii | CF4 | − | |
| Escherichia coli | 1375 | CWG 1028 | ++++ | Citrobacter freundii | 1516 | CF5 | − |
| Escherichia coli | 1382 | Ymel mel-1 supF58 | + | Citrobacter freundii | CF7 | − | |
| Escherichia coli | 1383 | Ymel (HK97) | + | Citrobacter freundii | CF8 | − | |
| Escherichia coli | 1392 | 0103 GVs | − | Citrobacter freundii | Sa1 | − | |
| Escherichia coli | 1393 | Rougier | − | Citrobacter freundii | Sa6 | − | |
| Escherichia coli | 1445 | TC4 | − | Citrobacter freundii | Sa59 | − | |
| Escherichia coli | 1446 | MB4 | − | ||||
| Escherichia coli | 1462 | C-3000 | + | ||||
| Escherichia coli | 1536 | SlyD | ++++ | ||||
| ORF | Strand | Start (pb) | End (pb) | Size (aa) | MW (kDa) | pI | SD Sequence (AGGAGGU) a | Predicted Protein Function | BLAST (Extent, % aa Identity) b | Aligned Protein Size (aa) | E Value | Accession Number |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | + | 673 | 2613 | 646 | 72.7 | 6.09 | TGGAGACttacaaATG | RNA polymerase | gp01 [Enterobacteria phage phiEcoM-GJ1] (598/646; 93%) | 648 | 0 | YP_001595396.1 |
| 2 | + | 2640 | 2846 | 68 | 7.89 | 4.51 | AGGATGGcattagTTG | gp02 [Enterobacteria phage phiEcoM-GJ1] (48/55; 87%) | 55 | 1 × 10−17 | YP_001595397.1 | |
| 3 | + | 3959 | 4177 | 72 | 4.14 | 8.15 | AGGAGAAtaaaATG | hypothetical protein [Klebsiella phage KP8] (23/73; 32%) | 71 | 8 × 10−4 | AVJ48916.1 | |
| 4 | + | 4217 | 4453 | 78 | 8.9 | 9.63 | CGGAGAGcagaaATG | gp04 [Enterobacteria phage phiEcoM-GJ1] (49/76; 64%) | 76 | 8 × 10−23 | YP_001595399.1 | |
| 5 | + | 4456 | 4644 | 62 | 7.5 | 4.53 | ACGAGGTtaatcATG | gp05 [Enterobacteria phage phiEcoM-GJ1] (61/62; 98%) | 62 | 1 × 10−37 | YP_001595400.1 | |
| 6 | + | 4641 | 4835 | 64 | 7.3 | 9.7 | TGGAGGCcaaATG | N/A | ||||
| 7 | + | 4832 | 5077 | 81 | 9.4 | 4.32 | AGGCGGGttggttGTG | N/A | ||||
| 8 | + | 5087 | 5260 | 57 | 6.7 | 9.25 | AGGAGTAttaaATG | gp07 [Enterobacteria phage phiEcoM-GJ1] (56/57; 98%) | 57 | 6 × 10−34 | YP_001595402.1 | |
| 9 | + | 5356 | 5667 | 103 | 11.6 | 4.5 | AGGTAATtaaATG | gp08 [Enterobacteria phage phiEcoM-GJ1] (84/99; 85%) | 99 | 2 × 10−54 | YP_001595404.1 | |
| 10 | + | 5683 | 5943 | 86 | 9.7 | 5.6 | GGGAGTTattATG | gp09 [Enterobacteria phage phiEcoM-GJ1] (79/86; 92%) | 87 | 6 × 10−53 | YP_001595404.1 | |
| 11 | + | 5936 | 6118 | 60 | 6.4 | 4.64 | TGGGAGTtctgtaccATG | N/A | ||||
| 12 | + | 6121 | 6348 | 75 | 8.4 | 5.24 | AGGATAAtcATG | gp10 [Enterobacteria phage phiEcoM-GJ1] (67/75; 89%) | 75 | 5 × 10−45 | YP_001595405.1 | |
| 13 | + | 6345 | 6575 | 76 | 8.6 | 9.58 | ACAAGGTttattgcaATG | gp11 [Enterobacteria phage phiEcoM-GJ1] (42/68; 62%) | 68 | 1 × 10−16 | YP_001595406.1 | |
| 14 | + | 6639 | 6929 | 96 | 10.7 | 9.47 | TGGAGCAtttATG | gp12 [Enterobacteria phage phiEcoM-GJ1] (87/96; 91%) | 96 | 7 × 10−59 | YP_001595407.1 | |
| 15 | + | 6922 | 7155 | 77 | 8.8 | 5.22 | AGAAGGTgaagcGTG | gp13 [Enterobacteria phage phiEcoM-GJ1] (68/77; 88%) | 77 | 5 × 10−44 | YP_001595408.1 | |
| 16 | + | 7152 | 7439 | 95 | 10.8 | 9.3 | TGGAGAAattaaagcaATG | gp14 [Enterobacteria phage phiEcoM-GJ1] (84/95; 88%) | 95 | 1 × 10−54 | YP_001595409.1 | |
| 17 | + | 7439 | 7636 | 65 | 7.82 | 9.81 | AGGTGATgtaATG | IME11_76 [Escherichia phage IME11] (29/65;45%) | 68 | 3 × 10−7 | YP_006990681.1 | |
| 18 | + | 7715 | 8197 | 160 | 18.8 | 8.79 | TGGAGGGcttATG | CBB_348 [Pectobacterium phage CBB] (72/160; 45%) | 161 | 2 × 10−36 | AMM43911.1 | |
| 19 | + | 8323 | 8700 | 125 | 14.2 | 5.78 | AAGAGAAtcttaatcATG | ssDNA-binding protein | gp15 [Enterobacteria phage phiEcoM-GJ1] (96/125; 77%) | 126 | 6 × 10−54 | YP_001595410.1 |
| 20 | + | 8823 | 9719 | 298 | 33.2 | 7.74 | GTGAGGAatatcATG | gp17 [Enterobacteria phage phiEcoM-GJ1] (159/229; 69%) | 229 | 5 × 10−109 | YP_001595412.1 | |
| 21 | + | 9775 | 10,125 | 116 | 13.1 | 6.07 | CGGAGCAtttATG | gp18 [Enterobacteria phage phiEcoM-GJ1] (114/116; 98%) | 116 | 2 × 10−80 | YP_001595413.1 | |
| 22 | + | 10,122 | 10,355 | 77 | 8.72 | 5.29 | AGGAAGTtaaATG | gp19 [Enterobacteria phage phiEcoM-GJ1] (56/77; 73%) | 77 | 5 × 10−32 | YP_001595414.1 | |
| 23 | + | 10,345 | 10,614 | 89 | 10.3 | 4.7 | AGGAAATccattccGTG | N/A | ||||
| 24 | + | 10,607 | 11,023 | 138 | 15.7 | 9.48 | AGGAGCTgaaaaATG | endolysin | gp21 [Enterobacteria phage phiEcoM-GJ1] (110/131; 84%) | 131 | 1 × 10−72 | YP_001595416.1 |
| 25 | + | 11,044 | 11,322 | 92 | 10.9 | 5.7 | TGGAGCAtccgATG | N/A | ||||
| 26 | + | 11,309 | 11,857 | 182 | 21.2 | 4.03 | GGGAGAAactcaATG | antirestriction protein | gp22 [Enterobacteria phage phiEcoM-GJ1] (145/181; 80%) | 181 | 2 × 10−100 | YP_001595417.1 |
| 27 | + | 11,850 | 12,089 | 79 | 9.2 | 6.9 | CGAAGGGatactattctcaaATG | gp23 [Enterobacteria phage phiEcoM-GJ1] (73/79; 92%) | 79 | 4 × 10−48 | YP_001595418.1 | |
| 28 | + | 13,092 | 13,295 | 67 | 7.5 | 4.58 | TGGAGAGttcctATG | gp25 [Enterobacteria phage phiEcoM-GJ1] (57/67; 85%) | 69 | 3 × 10−33 | YP_001595420.1 | |
| 29 | + | 13,292 | 13,582 | 96 | 11.3 | 9.1 | AGGAGCTgcaaaaATG | N/A | ||||
| 30 | + | 13,579 | 13,869 | 96 | 10.6 | 5.25 | CGGAGTTccattTTG | gp27 [Enterobacteria phage phiEcoM-GJ1] (93/96; 97%) | 97 | 3 × 10−59 | YP_001595422.1 | |
| 31 | + | 13,872 | 14,546 | 224 | 25.9 | 8.28 | ACAAGGCcactaaaaATG | gp28 [Enterobacteria phage phiEcoM-GJ1] (223/224; 99%) | 224 | 2 × 10−165 | YP_001595423.1 | |
| 32 | + | 14,670 | 15,008 | 112 | 12.3 | 4.47 | ATAAGGTatatacaaATG | gp29 [Enterobacteria phage phiEcoM-GJ1] (111/112; 99%) | 112 | 9 × 10−75 | YP_001595424.1 | |
| 33 | + | 15,395 | 15,532 | 45 | 5.1 | 8.99 | CGGAGCAataattaatTTG | N/A | ||||
| 34 | + | 15,547 | 15,789 | 80 | 8.8 | 9.24 | AGAAGCTatgccaatGTG | gp30 [Enterobacteria phage phiEcoM-GJ1] (77/80; 96%) | 80 | 1 × 10−50 | YP_001595425.1 | |
| 35 | + | 16,029 | 16,169 | 46 | 4.8 | 3.76 | TGGAGTCctcATG | N/A | ||||
| 36 | + | 16,178 | 16,414 | 78 | 8.5 | 9.05 | AGGTGATttATG | gp31 [Enterobacteria phage phiEcoM-GJ1] (77/78; 99%) | 78 | 1 × 10−44 | YP_001595426.1 | |
| 37 | + | 17,404 | 17,634 | 76 | 8.5 | 4.89 | TGGAGAGaaacATG | gp32 [Enterobacteria phage phiEcoM-GJ1] (74/76; 97%) | 76 | 2 × 10−44 | YP_001595427.1 | |
| 38 | + | 17,691 | 18,341 | 216 | 24.8 | 5.99 | CGGAGAGcaaATG | thymidylate synthase | gp33 [Enterobacteria phage phiEcoM-GJ1] (196/216; 91%) | 216 | 8 × 10−149 | YP_001595428.1 |
| 39 | + | 18,346 | 20,109 | 587 | 66 | 5.95 | ACCAGGAataaataaATG | helicase/primase | gp35 [Enterobacteria phage phiEcoM-GJ1] (560/587; 95%) | 587 | 0 | YP_001595430.1 |
| 40 | + | 20,175 | 22,109 | 644 | 74.6 | 6.41 | TGGAGCCatactGTG | DNA polymerase | gp37 [Enterobacteria phage phiEcoM-GJ1] (637/644; 99%) | 644 | 0 | YP_001595432.1 |
| 41 | + | 22,109 | 22,381 | 90 | 10.1 | 4.37 | CAGAGATtcactaATG | gp38 [Enterobacteria phage phiEcoM-GJ1] (86/90; 96%) | 90 | 2 × 10−54 | YP_001595433.1 | |
| 42 | + | 22,412 | 23,278 | 288 | 31 | 4.86 | AGGTACTcaaaATG | gp39 [Enterobacteria phage phiEcoM-GJ1] (288/288; 100%) | 288 | 0 | YP_001595434.1 | |
| 43 | + | 23,312 | 24,349 | 345 | 39.4 | 8.09 | GGGAGCCtttaattTTG | exonuclease | gp40 [Enterobacteria phage phiEcoM-GJ1] (342/345; 99%) | 345 | 0 | YP_001595435.1 |
| 44 | + | 24,361 | 24,885 | 174 | 20.1 | 9.46 | TGGAGTTggaATG | gp41 [Enterobacteria phage phiEcoM-GJ1] (173/174; 99%) | 174 | 3 × 10−124 | YP_001595436.1 | |
| 45 | + | 24,875 | 25,630 | 251 | 28.5 | 8.64 | AGAAAGAatcttaATG | DNA ligase | gp42 [Enterobacteria phage phiEcoM-GJ1] (233/251; 93%) | 251 | 6 × 10−175 | YP_001595437.1 |
| 46 | + | 25,623 | 26,249 | 208 | 23.5 | 6.38 | GTGAGGAaagttTTG | gp43 [Enterobacteria phage phiEcoM-GJ1] (203/208; 98%) | 208 | 1 × 10−147 | YP_001595438.1 | |
| 47 | + | 26,252 | 26,839 | 195 | 20.7 | 6.66 | ATCAAGTagagaaataatcATG | deoxyuridine 5’-triphosphate nucleotidylhydrolase | gp44 [Enterobacteria phage phiEcoM-GJ1] (164/195; 82%) | 199 | 4 × 10−105 | YP_001595439.1 |
| 48 | + | 26,858 | 27,064 | 68 | 8.2 | 4.32 | TGGAGCAtccATG | PP74_27 [Pectobacterium phage PP74] (37/68; 54%) | 73 | 3 × 10−17 | APD19639.1 | |
| 49 | + | 27,082 | 27,411 | 109 | 12.2 | 9.7 | TGGAACCtatctgaaATG | gp45 [Enterobacteria phage phiEcoM-GJ1] (109/109; 100%) | 109 | 4 × 10−74 | YP_001595440.1 | |
| 50 | + | 27,467 | 27,652 | 61 | 7 | 4.4 | CGGAGTCgcttATG | gp46 [Enterobacteria phage phiEcoM-GJ1] (61/61; 100%) | 61 | 1 × 10−37 | YP_001595441.1 | |
| 51 | + | 27,672 | 29,690 | 672 | 76 | 6.02 | AAGAGAAcgaatcaATG | large subunit terminase | gp48 [Enterobacteria phage phiEcoM-GJ1] (667/671; 99%) | 671 | 0 | YP_001595443.1 |
| 52 | + | 29,693 | 29,905 | 70 | 7.9 | 9.18 | TGGATGTaaatATG | gp49 [Enterobacteria phage phiEcoM-GJ1] (70/70; 100%) | 70 | 7 × 10−43 | YP_001595444.1 | |
| 53 | + | 29,905 | 31,221 | 438 | 49.1 | 8.16 | AGGAAGAaataATG | portal protein | gp50 [Enterobacteria phage phiEcoM-GJ1] (434/438; 99%) | 438 | 0 | YP_001595445.1 |
| 54 | + | 31,190 | 32,254 | 354 | 39 | 4.8 | AAAGGGTaacgcaaGTG | gp51 [Enterobacteria phage phiEcoM-GJ1] (353/354; 99%) | 354 | 0 | YP_001595446.1 | |
| 55 | + | 32,264 | 32,737 | 157 | 16.4 | 6.26 | ATAAGGTaagacaATG | gp52 [Enterobacteria phage phiEcoM-GJ1] (147/157; 94%) | 157 | 2 × 10−101 | YP_001595447.1 | |
| 56 | + | 32,818 | 33,030 | 70 | 7.3 | 6.06 | TGTAACTGTG | gp67 [Erwinia phage vB_EamM-Y2] (51/70; 73%) | 90 | 2 × 10−23 | YP_007004717.1 | |
| 57 | + | 33,086 | 34,093 | 335 | 36.7 | 5.17 | TGGATTAaattacATG | major capsid protein | gp53 [Enterobacteria phage phiEcoM-GJ1] (323/335; 96%) | 335 | 0 | YP_001595448.1 |
| 58 | + | 34,140 | 34,580 | 146 | 16.1 | 5.34 | AAGAGAAatagtaATG | gp54 [Enterobacteria phage phiEcoM-GJ1] (116/146; 79%) | 146 | 1 × 10−71 | YP_001595449.1 | |
| 59 | + | 34,581 | 34,970 | 129 | 14.6 | 4.48 | AGTTGGCgtaaATG | gp55 [Enterobacteria phage phiEcoM-GJ1] (124/129; 96%) | 129 | 2 × 10−86 | YP_001595450.1 | |
| 60 | + | 34,967 | 35,329 | 120 | 13.9 | 9.16 | GGGTCACagttTTG | gp56 [Enterobacteria phage phiEcoM-GJ1] (120/120; 100%) | 120 | 4 × 10−85 | YP_001595451.1 | |
| 61 | + | 35,326 | 35,838 | 170 | 19.3 | 4.98 | AGGAGTTagagaaATG | gp57 [Enterobacteria phage phiEcoM-GJ1] (167/170; 98%) | 170 | 2 × 10−120 | YP_001595452.1 | |
| 62 | + | 35,839 | 37,287 | 482 | 50.9 | 4.75 | AGGGAATctaaATG | gp58 [Enterobacteria phage phiEcoM-GJ1] (447/482; 93%) | 482 | 0 | YP_001595453.1 | |
| 63 | + | 37,298 | 37,753 | 151 | 16.5 | 6.55 | AGGTGCGataaGTG | gp59 [Enterobacteria phage phiEcoM-GJ1] (148/151; 98%) | 151 | 2 × 10−104 | YP_001595454.1 | |
| 64 | + | 37,765 | 38,223 | 152 | 17.3 | 5.1 | AGTAAGTATG | gp60 [Enterobacteria phage phiEcoM-GJ1] (152/152; 100%) | 152 | 4 × 10−107 | YP_001595455.1 | |
| 65 | + | 38,229 | 38,399 | 56 | 6.7 | 4.67 | CGGAGACagtttagtatccATG | gp61 [Enterobacteria phage phiEcoM-GJ1] (55/56; 98%) | 73 | 8 × 10−32 | YP_001595456.1 | |
| 66 | + | 38,383 | 42,108 | 1241 | 134.6 | 5.36 | AGAAACTcgaaccagtagATG | tail fiber | gp62 [Enterobacteria phage phiEcoM-GJ1] (946/1239; 76%) | 1239 | 0 | YP_001595457.1 |
| 67 | + | 42,182 | 43,291 | 369 | 40.7 | 5.13 | AATAGGTatatcgcaATG | gp63 [Enterobacteria phage phiEcoM-GJ1] (366/369; 99%) | 369 | 0 | YP_001595458.1 | |
| 68 | + | 43,291 | 44,178 | 295 | 31.2 | 5.98 | TGGAGTCattttaATG | gp64 [Enterobacteria phage phiEcoM-GJ1] (295/295; 100%) | 295 | 0 | YP_001595459.1 | |
| 69 | + | 44,175 | 44,537 | 120 | 13.7 | 5.07 | GGGACGTatcctATG | gp65 [Enterobacteria phage phiEcoM-GJ1] (120/120; 100%) | 120 | 2 × 10−84 | YP_001595460.1 | |
| 70 | + | 44,530 | 45,324 | 264 | 28.2 | 5.8 | AGAGTGTacttgaacGTG | baseplate assembly protein | gp66 [Enterobacteria phage phiEcoM-GJ1] (263/264; 99%) | 264 | 0 | YP_001595461.1 |
| 71 | + | 45,324 | 45,695 | 123 | 13.5 | 5.22 | ATGAAATaATG | gp67 [Enterobacteria phage phiEcoM-GJ1] (117/123; 95%) | 123 | 7 × 10−80 | YP_001595462.1 | |
| 72 | + | 45,671 | 46,828 | 385 | 41.2 | 4.55 | CGGAATTcttaacATG | gp68 [Enterobacteria phage phiEcoM-GJ1] (361/385; 94%) | 385 | 0 | YP_001595463.1 | |
| 73 | + | 46,830 | 47,471 | 213 | 23.5 | 5.82 | CAGATGTgacagtataatATG | gp69 [Enterobacteria phage phiEcoM-GJ1] (193/213; 91%) | 213 | 1 × 10−139 | YP_001595464.1 | |
| 74 | + | 47,471 | 48,619 | 382 | 42.3 | 5.49 | CGGAGAAataATG | gp70 [Enterobacteria phage phiEcoM-GJ1] (342/382; 90%) | 382 | 0 | YP_001595465.1 | |
| 75 | + | 48,619 | 50,016 | 465 | 50.2 | 8.17 | AGGCCATaATG | gp71 [Enterobacteria phage phiEcoM-GJ1] (314/465; 68%) | 465 | 0 | YP_001595466.1 | |
| 76 | + | 50,025 | 51,071 | 348 | 36.3 | 6.6 | AGGATTCaaaATG | tail fiber protein | gp72 [Enterobacteria phage phiEcoM-GJ1] (250/348; 72%) | 356 | 3 × 10−156 | YP_001595467.1 |
| 77 | + | 51,079 | 51,420 | 113 | 12.3 | 7.95 | AGGAACTcATG | holin | gp73 [Enterobacteria phage phiEcoM-GJ1] (110/113; 97%) | 113 | 8 × 10−74 | YP_001595468.1 |
| 78 | + | 51,438 | 51,992 | 184 | 20.7 | 9.57 | AGGAACTcgaATG | endolysin | gp74 [Enterobacteria phage phiEcoM-GJ1] (178/184; 97%) | 184 | 7 × 10−131 | YP_001595469.1 |
| 79 | + | 51,992 | 53,092 | 366 | 42.2 | 4.76 | AGGAAATctgtaATG | ribonucleotide reductase beta subunit | gp75 [Enterobacteria phage phiEcoM-GJ1] (340/366; 93%) | 372 | 0 | YP_001595470.1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, H.; Geagea, H.; Rousseau, G.M.; Labrie, S.J.; Tremblay, D.M.; Liu, X.; Moineau, S. Characterization of the Escherichia coli Virulent Myophage ST32. Viruses 2018, 10, 616. https://doi.org/10.3390/v10110616
Liu H, Geagea H, Rousseau GM, Labrie SJ, Tremblay DM, Liu X, Moineau S. Characterization of the Escherichia coli Virulent Myophage ST32. Viruses. 2018; 10(11):616. https://doi.org/10.3390/v10110616
Chicago/Turabian StyleLiu, Honghui, Hany Geagea, Geneviève M. Rousseau, Simon J. Labrie, Denise M. Tremblay, Xinchun Liu, and Sylvain Moineau. 2018. "Characterization of the Escherichia coli Virulent Myophage ST32" Viruses 10, no. 11: 616. https://doi.org/10.3390/v10110616
APA StyleLiu, H., Geagea, H., Rousseau, G. M., Labrie, S. J., Tremblay, D. M., Liu, X., & Moineau, S. (2018). Characterization of the Escherichia coli Virulent Myophage ST32. Viruses, 10(11), 616. https://doi.org/10.3390/v10110616