Visualizing Viral Infection In Vivo by Multi-Photon Intravital Microscopy

Abstract
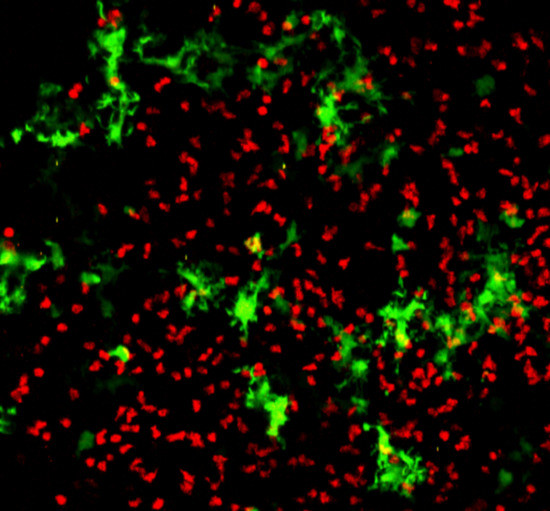
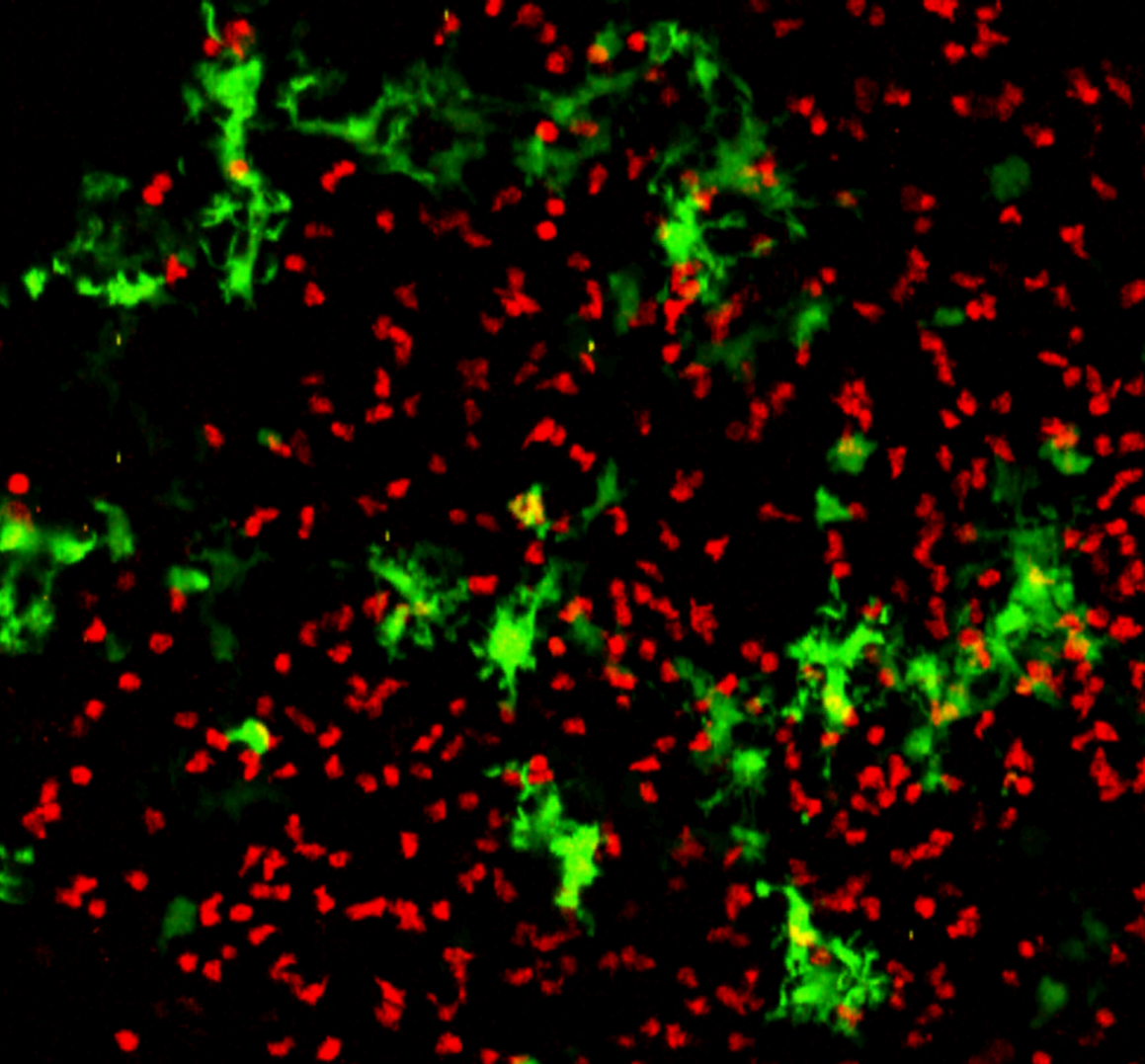{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Multi-Photon Excitation Microscopy
3. Experimental Systems for Imaging Viral Infections
4. Challenges for Intravital Imaging
4.1. Animal Preparation for Short- and Long-Term Intravital Imaging
4.2. Labeling Strategies for Intravital Imaging
5. MP-IVM Studies of Virus Infection
5.1. HIV-Infected Cells Can Form Syncytia In Vivo and Contribute to Systemic Spread
5.2. HIV Nef Interferes with T Cell Diapedesis for Lymph Node Homing In Vivo
5.3. MLV Establishes Infection in Secondary Lymphoid Tissue by Trans-Infection and Exploits Virological Synapses for Cell-To-Cell Transmission
5.4. Contact-Dependent Transmission Contributes to Local Tissue Spread of HIV for Multi-Copy Transmission of Viral Genomes
5.5. PRV-Induced Changes in Ca2+ Signaling Causes Peripheral Neuropathy
6. Combinations of MP-IVM
6.1. Optogenetic Manipulations of Protein Function and Cell Dynamics Using MP-IVM
6.2. In Vivo Imaging and Transcription Analysis of Single Cells within Spatially Confined Tissue Using Photoactivation
7. Summary and Future Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Hulme, A.E.; Hope, T.J. Live Cell Imaging of Retroviral Entry. Annu. Rev. Virol. 2014, 1, 501–515. [Google Scholar] [CrossRef] [PubMed]
- Campbell, E.M.; Hope, T.J. Live Cell Imaging of the HIV-1 Life Cycle. Trends Microbiol. 2008, 16, 580–587. [Google Scholar] [CrossRef] [PubMed]
- Yaniv, M. Small DNA Tumour Viruses and Their Contributions to Our Understanding of Transcription Control. Virology 2009, 384, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Marsh, M.; Helenius, A. Virus Entry: Open Sesame. Cell 2006, 124, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Pelkmans, L.; Kartenbeck, J.; Helenius, A. Caveolar Endocytosis of Simian Virus 40 Reveals a New Two-Step Vesicular-Transport Pathway to the Er. Nat. Cell Biol. 2001, 3, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Parton, R.G.; Richards, A.A. Lipid Rafts and Caveolae as Portals for Endocytosis: New Insights and Common Mechanisms. Traffic 2003, 4, 724–738. [Google Scholar] [CrossRef] [PubMed]
- Van Engelenburg, S.B.; Shtengel, G.; Sengupta, P.; Waki, K.; Jarnik, M.; Ablan, S.D.; Freed, E.O.; Hess, H.F.; Lippincott-Schwartz, J. Distribution of Escrt Machinery at HIV Assembly Sites Reveals Virus Scaffolding of Escrt Subunits. Science 2014, 343, 653–656. [Google Scholar] [CrossRef] [PubMed]
- Hanne, J.; Zila, V.; Heilemann, M.; Muller, B.; Krausslich, H.G. Super-Resolved Insights into Human Immunodeficiency Virus Biology. FEBS Lett. 2016, 590, 1858–1876. [Google Scholar] [CrossRef] [PubMed]
- Risco, C.; de Castro, I.F.; Sanz-Sanchez, L.; Narayan, K.; Grandinetti, G.; Subramaniam, S. Three-Dimensional Imaging of Viral Infections. Annu. Rev. Virol. 2014, 1, 453–473. [Google Scholar] [CrossRef] [PubMed]
- Romero-Brey, I.; Bartenschlager, R. Viral Infection at High Magnification: 3d Electron Microscopy Methods to Analyze the Architecture of Infected Cells. Viruses 2015, 7, 6316–6345. [Google Scholar] [CrossRef] [PubMed]
- Cyrklaff, M.; Frischknecht, F.; Kudryashev, M. Functional Insights into Pathogen Biology from 3d Electron Microscopy. FEMS Microbiol. Rev. 2017, 41, 828–853. [Google Scholar] [CrossRef] [PubMed]
- Mattei, S.; Glass, B.; Hagen, W.J.; Krausslich, H.G.; Briggs, J.A. The Structure and Flexibility of Conical HIV-1 Capsids Determined within Intact Virions. Science 2016, 354, 1434–1437. [Google Scholar] [CrossRef] [PubMed]
- Schur, F.K.; Obr, M.; Hagen, W.J.; Wan, W.; Jakobi, A.J.; Kirkpatrick, J.M.; Sachse, C.; Krausslich, H.G.; Briggs, J.A. An Atomic Model of HIV-1 Capsid-Sp1 Reveals Structures Regulating Assembly and Maturation. Science 2016, 353, 506–508. [Google Scholar] [CrossRef] [PubMed]
- Munro, J.B.; Gorman, J.; Ma, X.; Zhou, Z.; Arthos, J.; Burton, D.R.; Koff, W.C.; Courter, J.R.; Smith, A.B., 3rd; Kwong, P.D.; et al. Conformational Dynamics of Single HIV-1 Envelope Trimers on the Surface of Native Virions. Science 2014, 346, 759–763. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Lu, M.; Gorman, J.; Terry, D.S.; Hong, X.; Zhou, Z.; Zhao, H.; Altman, R.B.; Arthos, J.; Blanchard, S.C.; et al. HIV-1 Env Trimer Opens through an Asymmetric Intermediate in Which Individual Protomers Adopt Distinct Conformations. eLife 2018, 7, e34271. [Google Scholar] [CrossRef] [PubMed]
- Contag, C.H.; Spilman, S.D.; Contag, P.R.; Oshiro, M.; Eames, B.; Dennery, P.; Stevenson, D.K.; Benaron, D.A. Visualizing Gene Expression in Living Mammals Using a Bioluminescent Reporter. Photochem. Photobiol. 1997, 66, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Bhaumik, S.; Gambhir, S.S. Optical Imaging of Renilla Luciferase Reporter Gene Expression in Living Mice. Proc. Natl. Acad. Sci. USA 2002, 99, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Tran, V.; Moser, L.A.; Poole, D.S.; Mehle, A. Highly Sensitive Real-Time in vivo Imaging of an Influenza Reporter Virus Reveals Dynamics of Replication and Spread. J. Virol. 2013, 87, 13321–13329. [Google Scholar] [CrossRef] [PubMed]
- Luker, G.D.; Bardill, J.P.; Prior, J.L.; Pica, C.M.; Piwnica-Worms, D.; Leib, D.A. Noninvasive Bioluminescence Imaging of Herpes Simplex Virus Type 1 Infection and Therapy in Living Mice. J. Virol. 2002, 76, 12149–12161. [Google Scholar] [CrossRef] [PubMed]
- Mehle, A. Fiat Luc: Bioluminescence Imaging Reveals in vivo Viral Replication Dynamics. PLoS Pathog. 2015, 11, e1005081. [Google Scholar] [CrossRef] [PubMed]
- Contag, P.R.; Olomu, I.N.; Stevenson, D.K.; Contag, C.H. Bioluminescent Indicators in Living Mammals. Nat. Med. 1998, 4, 245–247. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.Y.; Johnson, M.; Sato, M.; Berger, F.; Gambhir, S.S.; Carey, M.; Iruela-Arispe, M.L.; Wu, L. Visualization of Advanced Human Prostate Cancer Lesions in Living Mice by a Targeted Gene Transfer Vector and Optical Imaging. Nat. Med. 2002, 8, 891–897. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, T.J.; Mailander, V.; Tucker, A.A.; Olomu, A.B.; Zhang, W.; Cao, Y.; Negrin, R.S.; Contag, C.H. Visualizing the Kinetics of Tumor-Cell Clearance in Living Animals. Proc. Natl. Acad. Sci. USA 1999, 96, 12044–12049. [Google Scholar] [CrossRef] [PubMed]
- Edinger, M.; Sweeney, T.J.; Tucker, A.A.; Olomu, A.B.; Negrin, R.S.; Contag, C.H. Noninvasive Assessment of Tumor Cell Proliferation in Animal Models. Neoplasia 1999, 1, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Kuchimaru, T.; Iwano, S.; Kiyama, M.; Mitsumata, S.; Kadonosono, T.; Niwa, H.; Maki, S.; Kizaka-Kondoh, S. A Luciferin Analogue Generating near-Infrared Bioluminescence Achieves Highly Sensitive Deep-Tissue Imaging. Nat. Commun. 2016, 7, 11856. [Google Scholar] [CrossRef] [PubMed]
- Iwano, S.; Sugiyama, M.; Hama, H.; Watakabe, A.; Hasegawa, N.; Kuchimaru, T.; Tanaka, K.Z.; Takahashi, M.; Ishida, Y.; Hata, J.; et al. Single-Cell Bioluminescence Imaging of Deep Tissue in Freely Moving Animals. Science 2018, 359, 935–939. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.S.; Chaurette, J.P.; Adams, S.T., Jr.; Reddy, G.R.; Paley, M.A.; Aronin, N.; Prescher, J.A.; Miller, S.C. A Synthetic Luciferin Improves Bioluminescence Imaging in Live Mice. Nat. Methods 2014, 11, 393–395. [Google Scholar] [CrossRef] [PubMed]
- Germain, R.N.; Robey, E.A.; Cahalan, M.D. A Decade of Imaging Cellular Motility and Interaction Dynamics in the Immune System. Science 2012, 336, 1676–1681. [Google Scholar] [CrossRef] [PubMed]
- Cahalan, M.D.; Parker, I. Choreography of Cell Motility and Interaction Dynamics Imaged by Two-Photon Microscopy in Lymphoid Organs. Ann. Rev. Immunol. 2008, 26, 585–626. [Google Scholar] [CrossRef] [PubMed]
- Sewald, X.; Gonzalez, D.G.; Haberman, A.M.; Mothes, W. In vivo Imaging of Virological Synapses. Nat. Commun. 2012, 3, 1320. [Google Scholar] [CrossRef] [PubMed]
- Sewald, X.; Ladinsky, M.S.; Uchil, P.D.; Beloor, J.; Pi, R.; Herrmann, C.; Motamedi, N.; Murooka, T.T.; Brehm, M.A.; Greiner, D.L.; et al. Retroviruses Use CD169-Mediated Trans-Infection of Permissive Lymphocytes to Establish Infection. Science 2015, 350, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Testa, I.; Urban, N.T.; Jakobs, S.; Eggeling, C.; Willig, K.I.; Hell, S.W. Nanoscopy of Living Brain Slices with Low Light Levels. Neuron 2012, 75, 992–1000. [Google Scholar] [CrossRef] [PubMed]
- Berning, S.; Willig, K.I.; Steffens, H.; Dibaj, P.; Hell, S.W. Nanoscopy in a Living Mouse Brain. Science 2012, 335, 551. [Google Scholar] [CrossRef] [PubMed]
- Göppert-Mayer, M. Über Elementarakte mit zwei Quantensprüngen. Ann. Phys. 1931, 3, 273–294. [Google Scholar] [CrossRef]
- Kaiser, W.; Garrett, C.G.B. Two-Photon Excitation in Caf2: Eu2+. Phys. Rev. Lett. 1961, 7, 229–231. [Google Scholar] [CrossRef]
- Denk, W.; Strickler, J.H.; Webb, W.W. Two-Photon Laser Scanning Fluorescence Microscopy. Science 1990, 248, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Dawson, J.B.; Barker, D.J.; Ellis, D.J.; Grassam, E.; Cotterill, J.A.; Fisher, G.W.; Feather, J.W. A Theoretical and Experimental Study of Light Absorption and Scattering by in vivo Skin. Phys. Med. Biol. 1980, 25, 695–709. [Google Scholar] [CrossRef] [PubMed]
- Weissleder, R.; Ntziachristos, V. Shedding Light onto Live Molecular Targets. Nat. Med. 2003, 9, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Konig, K. Multiphoton Microscopy in Life Sciences. J. Microsc. 2000, 200, 83–104. [Google Scholar] [CrossRef] [PubMed]
- Zipfel, W.R.; Williams, R.M.; Christie, R.; Nikitin, A.Y.; Hyman, B.T.; Webb, W.W. Live Tissue Intrinsic Emission Microscopy Using Multiphoton-Excited Native Fluorescence and Second Harmonic Generation. Proc. Natl. Acad. Sci. USA 2003, 100, 7075–7080. [Google Scholar] [CrossRef] [PubMed]
- Skala, M.C.; Riching, K.M.; Gendron-Fitzpatrick, A.; Eickhoff, J.; Eliceiri, K.W.; White, J.G.; Ramanujam, N. In vivo Multiphoton Microscopy of Nadh and Fad Redox States, Fluorescence Lifetimes, and Cellular Morphology in Precancerous Epithelia. Proc. Natl. Acad. Sci. USA 2007, 104, 19494–19499. [Google Scholar] [CrossRef] [PubMed]
- Piston, D.W.; Masters, B.R.; Webb, W.W. Three-Dimensionally Resolved Nad(P)H Cellular Metabolic Redox Imaging of the in Situ Cornea with Two-Photon Excitation Laser Scanning Microscopy. J. Microsc. 1995, 178, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Bennett, B.D.; Jetton, T.L.; Ying, G.; Magnuson, M.A.; Piston, D.W. Quantitative Subcellular Imaging of Glucose Metabolism within Intact Pancreatic Islets. J. Biol. Chem. 1996, 271, 3647–3651. [Google Scholar] [CrossRef] [PubMed]
- Roth, S.; Freund, I. Optical Second-Harmonic Scattering in Rat-Tail Tendon. Biopolymers 1981, 20, 1271–1290. [Google Scholar] [CrossRef] [PubMed]
- Campagnola, P.J.; Millard, A.C.; Terasaki, M.; Hoppe, P.E.; Malone, C.J.; Mohler, W.A. Three-Dimensional High-Resolution Second-Harmonic Generation Imaging of Endogenous Structural Proteins in Biological Tissues. Biophys. J. 2002, 82, 493–508. [Google Scholar] [CrossRef]
- Friedl, P.; Wolf, K.; von Andrian, U.H.; Harms, G. Biological Second and Third Harmonic Generation Microscopy. Curr. Protoc. Cell Biol. 2007, 4, 15. [Google Scholar] [PubMed]
- Sherer, N.M.; Lehmann, M.J.; Jimenez-Soto, L.F.; Horensavitz, C.; Pypaert, M.; Mothes, W. Retroviruses Can Establish Filopodial Bridges for Efficient Cell-to-Cell Transmission. Nat. Cell Biol. 2007, 9, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Sherer, N.M.; Heidecker, G.; Derse, D.; Mothes, W. Assembly of the Murine Leukemia Virus Is Directed Towards Sites of Cell-Cell Contact. PLoS Biol. 2009, 7, e1000163. [Google Scholar] [CrossRef] [PubMed]
- Hubner, W.; McNerney, G.P.; Chen, P.; Dale, B.M.; Gordon, R.E.; Chuang, F.Y.; Li, X.D.; Asmuth, D.M.; Huser, T.; Chen, B.K. Quantitative 3d Video Microscopy of HIV Transfer across T Cell Virological Synapses. Science 2009, 323, 1743–1747. [Google Scholar] [CrossRef] [PubMed]
- Baumgartel, V.; Ivanchenko, S.; Dupont, A.; Sergeev, M.; Wiseman, P.W.; Krausslich, H.G.; Brauchle, C.; Muller, B.; Lamb, D.C. Live-Cell Visualization of Dynamics of HIV Budding Site Interactions with an Escrt Component. Nat. Cell Biol. 2011, 13, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Jouvenet, N.; Zhadina, M.; Bieniasz, P.D.; Simon, S.M. Dynamics of Escrt Protein Recruitment during Retroviral Assembly. Nat. Cell Biol. 2011, 13, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.P.; Kobiler, O.; Enquist, L.W. Alphaherpesvirus Axon-to-Cell Spread Involves Limited Virion Transmission. Proc. Natl. Acad. Sci. USA 2012, 109, 17046–17051. [Google Scholar] [CrossRef] [PubMed]
- Glushakova, S.; Baibakov, B.; Margolis, L.B.; Zimmerberg, J. Infection of Human Tonsil Histocultures: A Model for HIV Pathogenesis. Nat. Med. 1995, 1, 1320–1322. [Google Scholar] [CrossRef] [PubMed]
- Penn, M.L.; Grivel, J.C.; Schramm, B.; Goldsmith, M.A.; Margolis, L. Cxcr4 Utilization Is Sufficient to Trigger Cd4+ T Cell Depletion in HIV-1-Infected Human Lymphoid Tissue. Proc. Natl. Acad. Sci. USA 1999, 96, 663–668. [Google Scholar] [CrossRef] [PubMed]
- McConkey, C.A.; Delorme-Axford, E.; Nickerson, C.A.; Kim, K.S.; Sadovsky, Y.; Boyle, J.P.; Coyne, C.B. A Three-Dimensional Culture System Recapitulates Placental Syncytiotrophoblast Development and Microbial Resistance. Sci. Adv. 2016, 2, e1501462. [Google Scholar] [CrossRef] [PubMed]
- Drummond, C.G.; Nickerson, C.A.; Coyne, C.B. A Three-Dimensional Cell Culture Model to Study Enterovirus Infection of Polarized Intestinal Epithelial Cells. mSphere 2016, 1, e00030-15. [Google Scholar] [CrossRef] [PubMed]
- Bramley, J.C.; Drummond, C.G.; Lennemann, N.J.; Good, C.A.; Kim, K.S.; Coyne, C.B. A Three-Dimensional Cell Culture System to Model Rna Virus Infections at the Blood-Brain Barrier. mSphere 2017, 2, e000206-17. [Google Scholar] [CrossRef] [PubMed]
- Lanik, W.E.; Mara, M.A.; Mihi, B.; Coyne, C.B.; Good, M. Stem Cell-Derived Models of Viral Infections in the Gastrointestinal Tract. Viruses 2018, 10, 124. [Google Scholar] [CrossRef] [PubMed]
- Saxena, K.; Blutt, S.E.; Ettayebi, K.; Zeng, X.L.; Broughman, J.R.; Crawford, S.E.; Karandikar, U.C.; Sastri, N.P.; Conner, M.E.; Opekun, A.R.; et al. Human Intestinal Enteroids: A New Model to Study Human Rotavirus Infection, Host Restriction, and Pathophysiology. J. Virol. 2016, 90, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Ettayebi, K.; Crawford, S.E.; Murakami, K.; Broughman, J.R.; Karandikar, U.; Tenge, V.R.; Neill, F.H.; Blutt, S.E.; Zeng, X.L.; Qu, L.; et al. Replication of Human Noroviruses in Stem Cell-Derived Human Enteroids. Science 2016, 353, 1387–1393. [Google Scholar] [CrossRef] [PubMed]
- Drummond, C.G.; Bolock, A.M.; Ma, C.; Luke, C.J.; Good, M.; Coyne, C.B. Enteroviruses Infect Human Enteroids and Induce Antiviral Signaling in a Cell Lineage-Specific Manner. Proc. Natl. Acad. Sci. USA 2017, 114, 1672–1677. [Google Scholar] [CrossRef] [PubMed]
- Murooka, T.T.; Deruaz, M.; Marangoni, F.; Vrbanac, V.D.; Seung, E.; von Andrian, U.H.; Tager, A.M.; Luster, A.D.; Mempel, T.R. HIV-Infected T Cells Are Migratory Vehicles for Viral Dissemination. Nature 2012, 490, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Stieh, D.J.; Matias, E.; Xu, H.; Fought, A.J.; Blanchard, J.L.; Marx, P.A.; Veazey, R.S.; Hope, T.J. Th17 Cells Are Preferentially Infected Very Early after Vaginal Transmission of Siv in Macaques. Cell Host Microbe 2016, 19, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Wilen, C.B.; Lee, S.; Hsieh, L.L.; Orchard, R.C.; Desai, C.; Hykes, B.L., Jr.; McAllaster, M.R.; Balce, D.R.; Feehley, T.; Brestoff, J.R.; et al. Tropism for Tuft Cells Determines Immune Promotion of Norovirus Pathogenesis. Science 2018, 360, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Tomov, V.T.; Palko, O.; Lau, C.W.; Pattekar, A.; Sun, Y.; Tacheva, R.; Bengsch, B.; Manne, S.; Cosma, G.L.; Eisenlohr, L.C.; et al. Differentiation and Protective Capacity of Virus-Specific Cd8(+) T Cells Suggest Murine Norovirus Persistence in an Immune-Privileged Enteric Niche. Immunity 2017, 47, 723.e5–738.e5. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Wilen, C.B.; Orvedahl, A.; McCune, B.T.; Kim, K.W.; Orchard, R.C.; Peterson, S.T.; Nice, T.J.; Baldridge, M.T.; Virgin, H.W. Norovirus Cell Tropism Is Determined by Combinatorial Action of a Viral Non-Structural Protein and Host Cytokine. Cell Host Microbe 2017, 22, 449.e4–459.e4. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.P.; Enquist, L.W. Axonal Spread of Neuroinvasive Viral Infections. Trends Microbiol. 2015, 23, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Symeonides, M.; Murooka, T.T.; Bellfy, L.N.; Roy, N.H.; Mempel, T.R.; Thali, M. HIV-1-Induced Small T Cell Syncytia Can Transfer Virus Particles to Target Cells through Transient Contacts. Viruses 2015, 7, 6590–6603. [Google Scholar] [CrossRef] [PubMed]
- Li, J.L.; Goh, C.C.; Keeble, J.L.; Qin, J.S.; Roediger, B.; Jain, R.; Wang, Y.; Chew, W.K.; Weninger, W.; Ng, L.G. Intravital Multiphoton Imaging of Immune Responses in the Mouse Ear Skin. Nat. Protoc. 2012, 7, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Dorand, R.D.; Barkauskas, D.S.; Evans, T.A.; Petrosiute, A.; Huang, A.Y. Comparison of Intravital Thinned Skull and Cranial Window Approaches to Study CNS Immunobiology in the Mouse Cortex. Intravital 2014, 3, e29728. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.T.; Pan, F.; Yang, G.; Gan, W.B. Choice of Cranial Window Type for in vivo Imaging Affects Dendritic Spine Turnover in the Cortex. Nat. Neurosci. 2007, 10, 549–551. [Google Scholar] [CrossRef] [PubMed]
- Presson, R.G., Jr.; Brown, M.B.; Fisher, A.J.; Sandoval, R.M.; Dunn, K.W.; Lorenz, K.S.; Delp, E.J.; Salama, P.; Molitoris, B.A.; Petrache, I. Two-Photon Imaging within the Murine Thorax without Respiratory and Cardiac Motion Artifact. Am. J. Pathol. 2011, 179, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Vinegoni, C.; Lee, S.; Feruglio, P.F.; Weissleder, R. Advanced Motion Compensation Methods for Intravital Optical Microscopy. IEEE J. Sel. Top. Quantum Electron. 2014, 20, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Looney, M.R.; Bhattacharya, J. Live Imaging of the Lung. Ann. Rev. Phys. 2014, 76, 431–445. [Google Scholar] [CrossRef] [PubMed]
- Vinegoni, C.; Lee, S.; Gorbatov, R.; Weissleder, R. Motion Compensation Using a Suctioning Stabilizer for Intravital Microscopy. Intravital 2012, 1, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Looney, M.R.; Thornton, E.E.; Sen, D.; Lamm, W.J.; Glenny, R.W.; Krummel, M.F. Stabilized Imaging of Immune Surveillance in the Mouse Lung. Nat. Methods 2011, 8, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Lochhead, K.M.; Zager, R.A. Fluorinated Anesthetic Exposure Activates the Renal Cortical Sphingomyelinase Cascade. Kidney Int. 1998, 54, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Pena, J.R.; Wolska, B.M. Differential Effects of Isoflurane and Ketamine/Inactin Anesthesia on Camp and Cardiac Function in Fvb/N Mice during Basal State and Beta-Adrenergic Stimulation. Basic Res. Cardiol. 2005, 100, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Caro, A.C.; Hankenson, F.C.; Marx, J.O. Comparison of Thermoregulatory Devices Used During Anesthesia of C57bl/6 Mice and Correlations between Body Temperature and Physiologic Parameters. J. Am. Assoc. Lab. Anim. Sci. 2013, 52, 577–583. [Google Scholar] [PubMed]
- Yeh, A.T.; Nassif, N.; Zoumi, A.; Tromberg, B.J. Selective Corneal Imaging Using Combined Second-Harmonic Generation and Two-Photon Excited Fluorescence. Opt. Lett. 2002, 27, 2082–2084. [Google Scholar] [CrossRef] [PubMed]
- Zinselmeyer, B.H.; Lynch, J.N.; Zhang, X.; Aoshi, T.; Miller, M.J. Video-Rate Two-Photon Imaging of Mouse Footpad—A Promising Model for Studying Leukocyte Recruitment Dynamics During Inflammation. Inflamm. Res. 2008, 57, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Peters, N.C.; Egen, J.G.; Secundino, N.; Debrabant, A.; Kimblin, N.; Kamhawi, S.; Lawyer, P.; Fay, M.P.; Germain, R.N.; Sacks, D. In vivo Imaging Reveals an Essential Role for Neutrophils in Leishmaniasis Transmitted by Sand Flies. Science 2008, 321, 970–974. [Google Scholar] [CrossRef] [PubMed]
- Alieva, M.; Ritsma, L.; Giedt, R.J.; Weissleder, R.; van Rheenen, J. Imaging Windows for Long-Term Intravital Imaging: General Overview and Technical Insights. Intravital 2014, 3, e29917. [Google Scholar] [CrossRef] [PubMed]
- Ritsma, L.; Steller, E.J.; Beerling, E.; Loomans, C.J.; Zomer, A.; Gerlach, C.; Vrisekoop, N.; Seinstra, D.; van Gurp, L.; Schafer, R.; et al. Intravital Microscopy through an Abdominal Imaging Window Reveals a Pre-Micrometastasis Stage during Liver Metastasis. Sci. Trans. Med. 2012, 4, 158ra145. [Google Scholar] [CrossRef] [PubMed]
- Ritsma, L.; Steller, E.J.; Ellenbroek, S.I.; Kranenburg, O.; Borel Rinkes, I.H.; van Rheenen, J. Surgical Implantation of an Abdominal Imaging Window for Intravital Microscopy. Nat. Protoc. 2013, 8, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Kedrin, D.; Gligorijevic, B.; Wyckoff, J.; Verkhusha, V.V.; Condeelis, J.; Segall, J.E.; van Rheenen, J. Intravital Imaging of Metastatic Behavior through a Mammary Imaging Window. Nat. Methods 2008, 5, 1019–1021. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Smith, B.R.; Parashurama, N.; Yoon, J.K.; Song, S.Y.; Miething, C.; Mallick, P.; Lowe, S.; Gambhir, S.S. Unexpected Dissemination Patterns in Lymphoma Progression Revealed by Serial Imaging within a Murine Lymph Node. Cancer Res. 2012, 72, 6111–6118. [Google Scholar] [CrossRef] [PubMed]
- Meijer, E.F.J.; Jeong, H.S.; Pereira, E.R.; Ruggieri, T.A.; Blatter, C.; Vakoc, B.J.; Padera, T.P. Murine Chronic Lymph Node Window for Longitudinal Intravital Lymph Node Imaging. Nat. Protoc. 2017, 12, 1513–1520. [Google Scholar] [CrossRef] [PubMed]
- Friebel, M.; Helfmann, J.; Netz, U.; Meinke, M. Influence of Oxygen Saturation on the Optical Scattering Properties of Human Red Blood Cells in the Spectral Range 250 to 2,000 Nm. J. Biomed. Opt. 2009, 14, 034001. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, E.A.; Campbell, R.E.; Lin, J.Y.; Lin, M.Z.; Miyawaki, A.; Palmer, A.E.; Shu, X.; Zhang, J.; Tsien, R.Y. The Growing and Glowing Toolbox of Fluorescent and Photoactive Proteins. Trends Biochem. Sci. 2017, 42, 111–129. [Google Scholar] [CrossRef] [PubMed]
- Piatkevich, K.D.; Hulit, J.; Subach, O.M.; Wu, B.; Abdulla, A.; Segall, J.E.; Verkhusha, V.V. Monomeric Red Fluorescent Proteins with a Large Stokes Shift. Proc. Natl. Acad. Sci. USA 2010, 107, 5369–5374. [Google Scholar] [CrossRef] [PubMed]
- Kawano, H.; Kogure, T.; Abe, Y.; Mizuno, H.; Miyawaki, A. Two-Photon Dual-Color Imaging Using Fluorescent Proteins. Nat. Methods 2008, 5, 373–374. [Google Scholar] [CrossRef] [PubMed]
- Kogure, T.; Kawano, H.; Abe, Y.; Miyawaki, A. Fluorescence Imaging Using a Fluorescent Protein with a Large Stokes Shift. Methods 2008, 45, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Cranfill, P.J.; Sell, B.R.; Baird, M.A.; Allen, J.R.; Lavagnino, Z.; de Gruiter, H.M.; Kremers, G.J.; Davidson, M.W.; Ustione, A.; Piston, D.W. Quantitative Assessment of Fluorescent Proteins. Nat. Methods 2016, 13, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Costantini, L.M.; Fossati, M.; Francolini, M.; Snapp, E.L. Assessing the Tendency of Fluorescent Proteins to Oligomerize under Physiologic Conditions. Traffic 2012, 13, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Fujimori, T. Reporter Mouse Lines for Fluorescence Imaging. Dev. Growth Differ. 2013, 55, 390–405. [Google Scholar] [CrossRef] [PubMed]
- Okabe, M.; Ikawa, M.; Kominami, K.; Nakanishi, T.; Nishimune, Y. Green Mice as a Source of Ubiquitous Green Cells. FEBS Lett. 1997, 407, 313–319. [Google Scholar] [CrossRef]
- Fink, D.; Wohrer, S.; Pfeffer, M.; Tombe, T.; Ong, C.J.; Sorensen, P.H. Ubiquitous Expression of the Monomeric Red Fluorescent Protein Mcherry in Transgenic Mice. Genesis 2010, 48, 723–729. [Google Scholar] [CrossRef] [PubMed]
- Lindquist, R.L.; Shakhar, G.; Dudziak, D.; Wardemann, H.; Eisenreich, T.; Dustin, M.L.; Nussenzweig, M.C. Visualizing Dendritic Cell Networks in vivo. Nat. Immunol. 2004, 5, 1243–1250. [Google Scholar] [CrossRef] [PubMed]
- Geissmann, F.; Jung, S.; Littman, D.R. Blood Monocytes Consist of Two Principal Subsets with Distinct Migratory Properties. Immunity 2003, 19, 71–82. [Google Scholar] [CrossRef]
- Faust, N.; Varas, F.; Kelly, L.M.; Heck, S.; Graf, T. Insertion of Enhanced Green Fluorescent Protein into the Lysozyme Gene Creates Mice with Green Fluorescent Granulocytes and Macrophages. Blood 2000, 96, 719–726. [Google Scholar] [PubMed]
- Denk, W.; Delaney, K.R.; Gelperin, A.; Kleinfeld, D.; Strowbridge, B.W.; Tank, D.W.; Yuste, R. Anatomical and Functional Imaging of Neurons Using 2-Photon Laser Scanning Microscopy. J. Neurosci. Methods 1994, 54, 151–162. [Google Scholar] [CrossRef]
- Bousso, P. T-Cell Activation by Dendritic Cells in the Lymph Node: Lessons from the Movies. Nat. Rev. Immunol. 2008, 8, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Cahalan, M.D.; Parker, I.; Wei, S.H.; Miller, M.J. Two-Photon Tissue Imaging: Seeing the Immune System in a Fresh Light. Nat. Rev. Immunol. 2002, 2, 872–880. [Google Scholar] [CrossRef] [PubMed]
- Hickman, H.D. New Insights into Antiviral Immunity Gained through Intravital Imaging. Curr. Opin. Virol. 2017, 22, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Hor, J.L.; Whitney, P.G.; Zaid, A.; Brooks, A.G.; Heath, W.R.; Mueller, S.N. Spatiotemporally Distinct Interactions with Dendritic Cell Subsets Facilitates Cd4+ and Cd8+ T Cell Activation to Localized Viral Infection. Immunity 2015, 43, 554–565. [Google Scholar] [CrossRef] [PubMed]
- Hickman, H.D.; Takeda, K.; Skon, C.N.; Murray, F.R.; Hensley, S.E.; Loomis, J.; Barber, G.N.; Bennink, J.R.; Yewdell, J.W. Direct Priming of Antiviral Cd8+ T Cells in the Peripheral Interfollicular Region of Lymph Nodes. Nat. Immunol. 2008, 9, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Hickman, H.D.; Reynoso, G.V.; Ngudiankama, B.F.; Cush, S.S.; Gibbs, J.; Bennink, J.R.; Yewdell, J.W. Cxcr3 Chemokine Receptor Enables Local Cd8(+) T Cell Migration for the Destruction of Virus-Infected Cells. Immunity 2015, 42, 524–537. [Google Scholar] [CrossRef] [PubMed]
- Iannacone, M.; Moseman, E.A.; Tonti, E.; Bosurgi, L.; Junt, T.; Henrickson, S.E.; Whelan, S.P.; Guidotti, L.G.; von Andrian, U.H. Subcapsular Sinus Macrophages Prevent CNS Invasion on Peripheral Infection with a Neurotropic Virus. Nature 2010, 465, 1079–1083. [Google Scholar] [CrossRef] [PubMed]
- Fackler, O.T.; Murooka, T.T.; Imle, A.; Mempel, T.R. Adding New Dimensions: Towards an Integrative Understanding of HIV-1 Spread. Nat. Rev. Microbiol. 2014, 12, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Sewald, X.; Motamedi, N.; Mothes, W. Viruses Exploit the Tissue Physiology of the Host to Spread in vivo. Curr. Opin. Cell Biol. 2016, 41, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Nieva, J.L.; Madan, V.; Carrasco, L. Viroporins: Structure and Biological Functions. Nat. Rev. Microbiol. 2012, 10, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.X.; Fung, T.S.; Chong, K.K.; Shukla, A.; Hilgenfeld, R. Accessory Proteins of Sars-Cov and Other Coronaviruses. Antivir. Res. 2014, 109, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Kirchhoff, F. Immune Evasion and Counteraction of Restriction Factors by HIV-1 and Other Primate Lentiviruses. Cell Host Microbe 2010, 8, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.V.; Miller, A.D. Serine Phosphorylation-Independent Downregulation of Cell-Surface Cd4 by Nef. Nature 1991, 350, 508–511. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, O.; Marechal, V.; Le Gall, S.; Lemonnier, F.; Heard, J.M. Endocytosis of Major Histocompatibility Complex Class I Molecules Is Induced by the HIV-1 Nef Protein. Nat. Med. 1996, 2, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Pereira, E.A.; daSilva, L.L. HIV-1 Nef: Taking Control of Protein Trafficking. Traffic 2016, 17, 976–996. [Google Scholar] [CrossRef] [PubMed]
- Rosa, A.; Chande, A.; Ziglio, S.; De Sanctis, V.; Bertorelli, R.; Goh, S.L.; McCauley, S.M.; Nowosielska, A.; Antonarakis, S.E.; Luban, J.; et al. HIV-1 Nef Promotes Infection by Excluding Serinc5 from Virion Incorporation. Nature 2015, 526, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Usami, Y.; Wu, Y.; Gottlinger, H.G. Serinc3 and Serinc5 Restrict HIV-1 Infectivity and Are Counteracted by Nef. Nature 2015, 526, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Janardhan, A.; Swigut, T.; Hill, B.; Myers, M.P.; Skowronski, J. HIV-1 Nef Binds the Dock2-Elmo1 Complex to Activate Rac and Inhibit Lymphocyte Chemotaxis. PLoS Biol. 2004, 2, e6. [Google Scholar] [CrossRef] [PubMed]
- Choe, E.Y.; Schoenberger, E.S.; Groopman, J.E.; Park, I.W. HIV Nef Inhibits T Cell Migration. J. Biol. Chem. 2002, 277, 46079–46084. [Google Scholar] [CrossRef] [PubMed]
- Stolp, B.; Reichman-Fried, M.; Abraham, L.; Pan, X.; Giese, S.I.; Hannemann, S.; Goulimari, P.; Raz, E.; Grosse, R.; Fackler, O.T. HIV-1 Nef Interferes with Host Cell Motility by Deregulation of Cofilin. Cell Host Microbe 2009, 6, 174–186. [Google Scholar] [CrossRef] [PubMed]
- Stolp, B.; Imle, A.; Coelho, F.M.; Hons, M.; Gorina, R.; Lyck, R.; Stein, J.V.; Fackler, O.T. HIV-1 Nef Interferes with T-Lymphocyte Circulation through Confined Environments in vivo. Proc. Natl. Acad. Sci. USA 2012, 109, 18541–18546. [Google Scholar] [CrossRef] [PubMed]
- Zhong, P.; Agosto, L.M.; Munro, J.B.; Mothes, W. Cell-to-Cell Transmission of Viruses. Curr. Opin. Virol. 2013, 3, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Sattentau, Q. Avoiding the Void: Cell-to-Cell Spread of Human Viruses. Nature reviews. Microbiology 2008, 6, 815–826. [Google Scholar] [PubMed]
- McDonald, D.; Wu, L.; Bohks, S.M.; KewalRamani, V.N.; Unutmaz, D.; Hope, T.J. Recruitment of HIV and Its Receptors to Dendritic Cell-T Cell Junctions. Science 2003, 300, 1295–1297. [Google Scholar] [CrossRef] [PubMed]
- Cameron, P.U.; Freudenthal, P.S.; Barker, J.M.; Gezelter, S.; Inaba, K.; Steinman, R.M. Dendritic Cells Exposed to Human Immunodeficiency Virus Type-1 Transmit a Vigorous Cytopathic Infection to Cd4+ T Cells. Science 1992, 257, 383–387. [Google Scholar] [CrossRef] [PubMed]
- Jolly, C.; Kashefi, K.; Hollinshead, M.; Sattentau, Q.J. HIV-1 Cell to Cell Transfer across an Env-Induced, Actin-Dependent Synapse. J. Exp. Med. 2004, 199, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Del Portillo, A.; Tripodi, J.; Najfeld, V.; Wodarz, D.; D.Levy, N.; Chen, B.K. Multiploid Inheritance of HIV-1 During Cell-to-Cell Infection. J. Virol. 2011, 85, 7169–7176. [Google Scholar] [CrossRef] [PubMed]
- Zhong, P.; Agosto, L.M.; Ilinskaya, A.; Dorjbal, B.; Truong, R.; Derse, D.; Uchil, P.D.; Heidecker, G.; Mothes, W. Cell-to-Cell Transmission Can Overcome Multiple Donor and Target Cell Barriers Imposed on Cell-Free HIV. PLoS ONE 2013, 8, e53138. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.A.; Martin, N.; Mitar, I.; Jones, E.; Sattentau, Q.J. Multiple Proviral Integration Events after Virological Synapse-Mediated HIV-1 Spread. Virology 2013, 443, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Agosto, L.M.; Zhong, P.; Munro, J.; Mothes, W. Highly Active Antiretroviral Therapies Are Effective against HIV-1 Cell-to-Cell Transmission. PLoS Pathog. 2014, 10, e1003982. [Google Scholar] [CrossRef] [PubMed]
- Titanji, B.K.; Pillay, D.; Jolly, C. Combination Antiretroviral Therapy and Cell-Cell Spread of Wild-Type and Drug-Resistant Human Immunodeficiency Virus-1. J. Gen. Virol. 2017, 98, 821–834. [Google Scholar] [CrossRef] [PubMed]
- Abela, I.A.; Berlinger, L.; Schanz, M.; Reynell, L.; Gunthard, H.F.; Rusert, P.; Trkola, A. Cell-Cell Transmission Enables HIV-1 to Evade Inhibition by Potent Cd4bs Directed Antibodies. PLoS Pathog. 2012, 8, e1002634. [Google Scholar] [CrossRef] [PubMed]
- Duncan, C.J.; Williams, J.P.; Schiffner, T.; Gartner, K.; Ochsenbauer, C.; Kappes, J.; Russell, R.A.; Frater, J.; Sattentau, Q.J. High-Multiplicity HIV-1 Infection and Neutralizing Antibody Evasion Mediated by the Macrophage-T Cell Virological Synapse. J. Virol. 2014, 88, 2025–2034. [Google Scholar] [CrossRef] [PubMed]
- Malbec, M.; Porrot, F.; Rua, R.; Horwitz, J.; Klein, F.; Halper-Stromberg, A.; Scheid, J.F.; Eden, C.; Mouquet, H.; Nussenzweig, M.C.; et al. Broadly Neutralizing Antibodies That Inhibit HIV-1 Cell to Cell Transmission. J. Exp. Med. 2013, 210, 2813–2821. [Google Scholar] [CrossRef] [PubMed]
- Law, K.M.; Komarova, N.L.; Yewdall, A.W.; Lee, R.K.; Herrera, O.L.; Wodarz, D.; Chen, B.K. In vivo HIV-1 Cell-to-Cell Transmission Promotes Multicopy Micro-Compartmentalized Infection. Cell Rep. 2016, 15, 2771–2783. [Google Scholar] [CrossRef] [PubMed]
- Compton, A.A.; Schwartz, O. They Might Be Giants: Does Syncytium Formation Sink or Spread HIV Infection? PLoS Pathog. 2017, 13, e1006099. [Google Scholar] [CrossRef] [PubMed]
- Ekstrand, M.I.; Enquist, L.W.; Pomeranz, L.E. The Alpha-Herpesviruses: Molecular Pathfinders in Nervous System Circuits. Trends Mol. Med. 2008, 14, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Brittle, E.E.; Reynolds, A.E.; Enquist, L.W. Two Modes of Pseudorabies Virus Neuroinvasion and Lethality in Mice. J. Virol. 2004, 78, 12951–12963. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.A.; Gross, S.P.; Enquist, L.W. Herpesviruses Use Bidirectional Fast-Axonal Transport to Spread in Sensory Neurons. Proc. Natl. Acad. Sci. USA 2001, 98, 3466–3470. [Google Scholar] [CrossRef] [PubMed]
- Kramer, T.; Greco, T.M.; Taylor, M.P.; Ambrosini, A.E.; Cristea, I.M.; Enquist, L.W. Kinesin-3 Mediates Axonal Sorting and Directional Transport of Alphaherpesvirus Particles in Neurons. Cell Host Microbe 2012, 12, 806–814. [Google Scholar] [CrossRef] [PubMed]
- Granstedt, A.E.; Szpara, M.L.; Kuhn, B.; Wang, S.S.; Enquist, L.W. Fluorescence-Based Monitoring of in vivo Neural Activity Using a Circuit-Tracing Pseudorabies Virus. PLoS ONE 2009, 4, e6923. [Google Scholar] [CrossRef] [PubMed]
- Granstedt, A.E.; Bosse, J.B.; Thiberge, S.Y.; Enquist, L.W. In vivo Imaging of Alphaherpesvirus Infection Reveals Synchronized Activity Dependent on Axonal Sorting of Viral Proteins. Proc. Natl. Acad. Sci. USA 2013, 110, e3516-25. [Google Scholar] [CrossRef] [PubMed]
- Karreman, M.A.; Hyenne, V.; Schwab, Y.; Goetz, J.G. Intravital Correlative Microscopy: Imaging Life at the Nanoscale. Trends Cell Biol. 2016, 26, 848–863. [Google Scholar] [CrossRef] [PubMed]
- Boyden, E.S.; Zhang, F.; Bamberg, E.; Nagel, G.; Deisseroth, K. Millisecond-Timescale, Genetically Targeted Optical Control of Neural Activity. Nat. Neurosci. 2005, 8, 1263–1268. [Google Scholar] [CrossRef] [PubMed]
- Guglielmi, G.; Falk, H.J.; De Renzis, S. Optogenetic Control of Protein Function: From Intracellular Processes to Tissue Morphogenesis. Trends Cell Biol. 2016, 26, 864–874. [Google Scholar] [CrossRef] [PubMed]
- Weitzman, M.; Hahn, K.M. Optogenetic Approaches to Cell Migration and Beyond. Curr. Opin. Cell Biol. 2014, 30, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Bromley, S.K.; Mempel, T.R.; Luster, A.D. Orchestrating the Orchestrators: Chemokines in Control of T Cell Traffic. Nat. Immunol. 2008, 9, 970–980. [Google Scholar] [CrossRef] [PubMed]
- Krummel, M.F.; Bartumeus, F.; Gerard, A. T Cell Migration, Search Strategies and Mechanisms. Nat. Rev. Immunol. 2016, 16, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Paluch, E.K.; Aspalter, I.M.; Sixt, M. Focal Adhesion-Independent Cell Migration. Ann. Rev. Cell Dev. Biol. 2016, 32, 469–490. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.I.; Frey, D.; Lungu, O.I.; Jaehrig, A.; Schlichting, I.; Kuhlman, B.; Hahn, K.M. A Genetically Encoded Photoactivatable Rac Controls the Motility of Living Cells. Nature 2009, 461, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Yazawa, M.; Sadaghiani, A.M.; Hsueh, B.; Dolmetsch, R.E. Induction of Protein-Protein Interactions in Live Cells Using Light. Nat. Biotechnol. 2009, 27, 941–945. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; He, L.; Wu, Y.I.; Hahn, K.M.; Montell, D.J. Light-Mediated Activation Reveals a Key Role for Rac in Collective Guidance of Cell Movement in vivo. Nat. Cell Biol. 2010, 12, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.K.; Deng, Q.; Cavnar, P.J.; Wu, Y.I.; Hahn, K.M.; Huttenlocher, A. Differential Regulation of Protrusion and Polarity by Pi3k During Neutrophil Motility in Live Zebrafish. Dev. Cell 2010, 18, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Levskaya, A.; Weiner, O.D.; Lim, W.A.; Voigt, C.A. Spatiotemporal Control of Cell Signalling Using a Light-Switchable Protein Interaction. Nature 2009, 461, 997–1001. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.W.; Otomo, C.; Chory, J.; Rosen, M.K. Genetically Encoded Photoswitching of Actin Assembly through the Cdc42-Wasp-Arp2/3 Complex Pathway. Proc. Natl. Acad. Sci. USA 2008, 105, 12797–12802. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Park, H.; Kyung, T.; Kim, N.Y.; Kim, S.; Kim, J.; Heo, W.D. Reversible Protein Inactivation by Optogenetic Trapping in Cells. Nat. Methods 2014, 11, 633–636. [Google Scholar] [CrossRef] [PubMed]
- Bugaj, L.J.; Choksi, A.T.; Mesuda, C.K.; Kane, R.S.; Schaffer, D.V. Optogenetic Protein Clustering and Signaling Activation in Mammalian Cells. Nat. Methods 2013, 10, 249–252. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.X.; Chung, H.K.; Lam, A.J.; Lin, M.Z. Optical Control of Protein Activity by Fluorescent Protein Domains. Science 2012, 338, 810–814. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.V.; Chu, P.H.; Hahn, K.M.; Zaidel-Bar, R. An Optogenetic Tool for the Activation of Endogenous Diaphanous-Related Formins Induces Thickening of Stress Fibers without an Increase in Contractility. Cytoskeleton 2013, 70, 394–407. [Google Scholar] [CrossRef] [PubMed]
- Medaglia, C.; Giladi, A.; Stoler-Barak, L.; De Giovanni, M.; Salame, T.M.; Biram, A.; David, E.; Li, H.; Iannacone, M.; Shulman, Z.; et al. Spatial Reconstruction of Immune Niches by Combining Photoactivatable Reporters and Scrna-Seq. Science 2017, 358, 1622–1626. [Google Scholar] [CrossRef] [PubMed]
- Sagoo, P.; Garcia, Z.; Breart, B.; Lemaitre, F.; Michonneau, D.; Albert, M.L.; Levy, Y.; Bousso, P. In vivo Imaging of Inflammasome Activation Reveals a Subcapsular Macrophage Burst Response That Mobilizes Innate and Adaptive Immunity. Nat. Med. 2016, 22, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Breart, B.; Lemaitre, F.; Celli, S.; Bousso, P. Two-Photon Imaging of Intratumoral Cd8+ T Cell Cytotoxic Activity During Adoptive T Cell Therapy in Mice. J. Clin. Investig. 2008, 118, 1390–1397. [Google Scholar] [CrossRef] [PubMed]
- Garrod, K.R.; Moreau, H.D.; Garcia, Z.; Lemaitre, F.; Bouvier, I.; Albert, M.L.; Bousso, P. Dissecting T Cell Contraction in vivo Using a Genetically Encoded Reporter of Apoptosis. Cell Rep. 2012, 2, 1438–1447. [Google Scholar] [CrossRef] [PubMed]
- Zeng, M.; Smith, A.J.; Wietgrefe, S.W.; Southern, P.J.; Schacker, T.W.; Reilly, C.S.; Estes, J.D.; Burton, G.F.; Silvestri, G.; Lifson, J.D.; et al. Cumulative Mechanisms of Lymphoid Tissue Fibrosis and T Cell Depletion in HIV-1 and Siv Infections. J. Clin. Investig. 2011, 121, 998–1008. [Google Scholar] [CrossRef] [PubMed]
- Zeng, M.; Southern, P.J.; Reilly, C.S.; Beilman, G.J.; Chipman, J.G.; Schacker, T.W.; Haase, A.T. Lymphoid Tissue Damage in HIV-1 Infection Depletes Naive T Cells and Limits T Cell Reconstitution after Antiretroviral Therapy. PLoS Pathog. 2012, 8, e1002437. [Google Scholar] [CrossRef] [PubMed]
- Azar, G.A.; Lemaitre, F.; Robey, E.A.; Bousso, P. Subcellular Dynamics of T Cell Immunological Synapses and Kinapses in Lymph Nodes. Proc. Natl. Acad. Sci. USA 2010, 107, 3675–3680. [Google Scholar] [CrossRef] [PubMed]
- Melichar, H.J.; Li, O.; Herzmark, P.; Padmanabhan, R.K.; Oliaro, J.; Ludford-Menting, M.J.; Bousso, P.; Russell, S.M.; Roysam, B.; Robey, E.A. Quantifying Subcellular Distribution of Fluorescent Fusion Proteins in Cells Migrating within Tissues. Immunol. Cell Biol. 2011, 89, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Thestrup, T.; Litzlbauer, J.; Bartholomaus, I.; Mues, M.; Russo, L.; Dana, H.; Kovalchuk, Y.; Liang, Y.; Kalamakis, G.; Laukat, Y.; et al. Optimized Ratiometric Calcium Sensors for Functional in vivo Imaging of Neurons and T Lymphocytes. Nat. Methods 2014, 11, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Lodygin, D.; Odoardi, F.; Schlager, C.; Korner, H.; Kitz, A.; Nosov, M.; van den Brandt, J.; Reichardt, H.M.; Haberl, M.; Flugel, A. A Combination of Fluorescent Nfat and H2b Sensors Uncovers Dynamics of T Cell Activation in Real Time During CNS Autoimmunity. Nat. Med. 2013, 19, 784–790. [Google Scholar] [CrossRef] [PubMed]
- Marangoni, F.; Murooka, T.T.; Manzo, T.; Kim, E.Y.; Carrizosa, E.; Elpek, N.M.; Mempel, T.R. The Transcription Factor Nfat Exhibits Signal Memory during Serial T Cell Interactions with Antigen-Presenting Cells. Immunity 2013, 38, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Pesic, M.; Bartholomaus, I.; Kyratsous, N.I.; Heissmeyer, V.; Wekerle, H.; Kawakami, N. 2-Photon Imaging of Phagocyte-Mediated T Cell Activation in the CNS. J. Clin. Investig. 2013, 123, 1192–1201. [Google Scholar] [CrossRef] [PubMed]
- Len, A.C.L.; Starling, S.; Shivkumar, M.; Jolly, C. HIV-1 Activates T Cell Signaling Independently of Antigen to Drive Viral Spread. Cell Rep. 2017, 18, 1062–1074. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sewald, X. Visualizing Viral Infection In Vivo by Multi-Photon Intravital Microscopy. Viruses 2018, 10, 337. https://doi.org/10.3390/v10060337
Sewald X. Visualizing Viral Infection In Vivo by Multi-Photon Intravital Microscopy. Viruses. 2018; 10(6):337. https://doi.org/10.3390/v10060337
Chicago/Turabian StyleSewald, Xaver. 2018. "Visualizing Viral Infection In Vivo by Multi-Photon Intravital Microscopy" Viruses 10, no. 6: 337. https://doi.org/10.3390/v10060337
APA StyleSewald, X. (2018). Visualizing Viral Infection In Vivo by Multi-Photon Intravital Microscopy. Viruses, 10(6), 337. https://doi.org/10.3390/v10060337