CD81 Receptor Regions outside the Large Extracellular Loop Determine Hepatitis C Virus Entry into Hepatoma Cells


Abstract
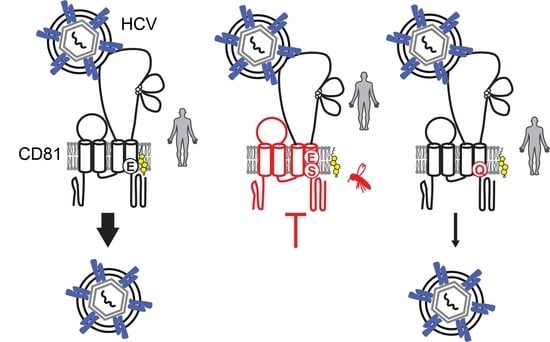
1. Introduction
2. Materials and Methods
2.1. Tetraspanin Chimera Construct Generation
2.2. Cell Culture, Ectopic Expression of Tetraspanin Variants in Lunet N#3 Cells and CHO745 Cells, HCV Replicons, HCV Pseudoparticles, and Cell Culture-Derived HCV (HCVcc)
2.3. Immunoblotting
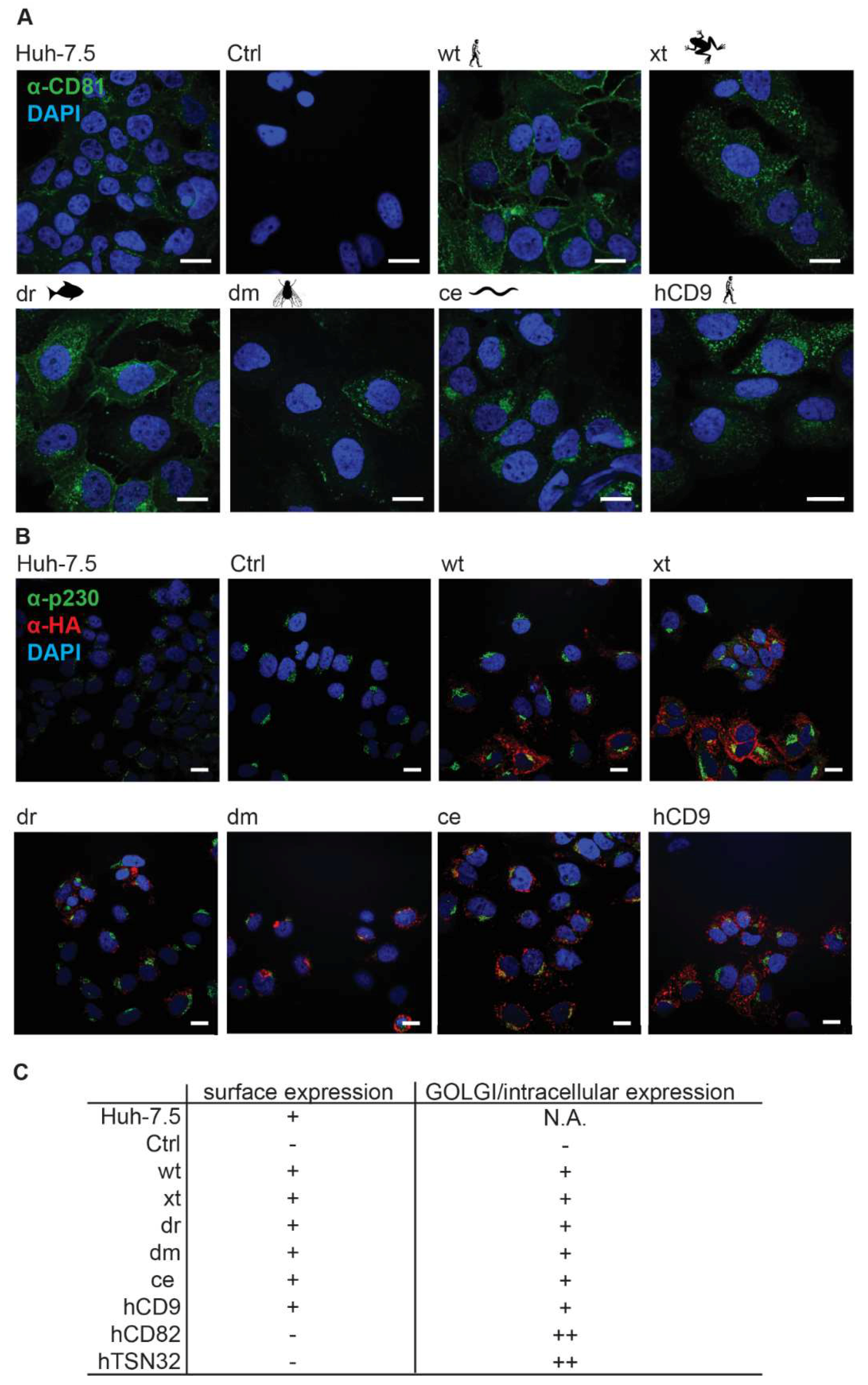
2.4. Immunofluorescence Analysis and Confocal Microscopy
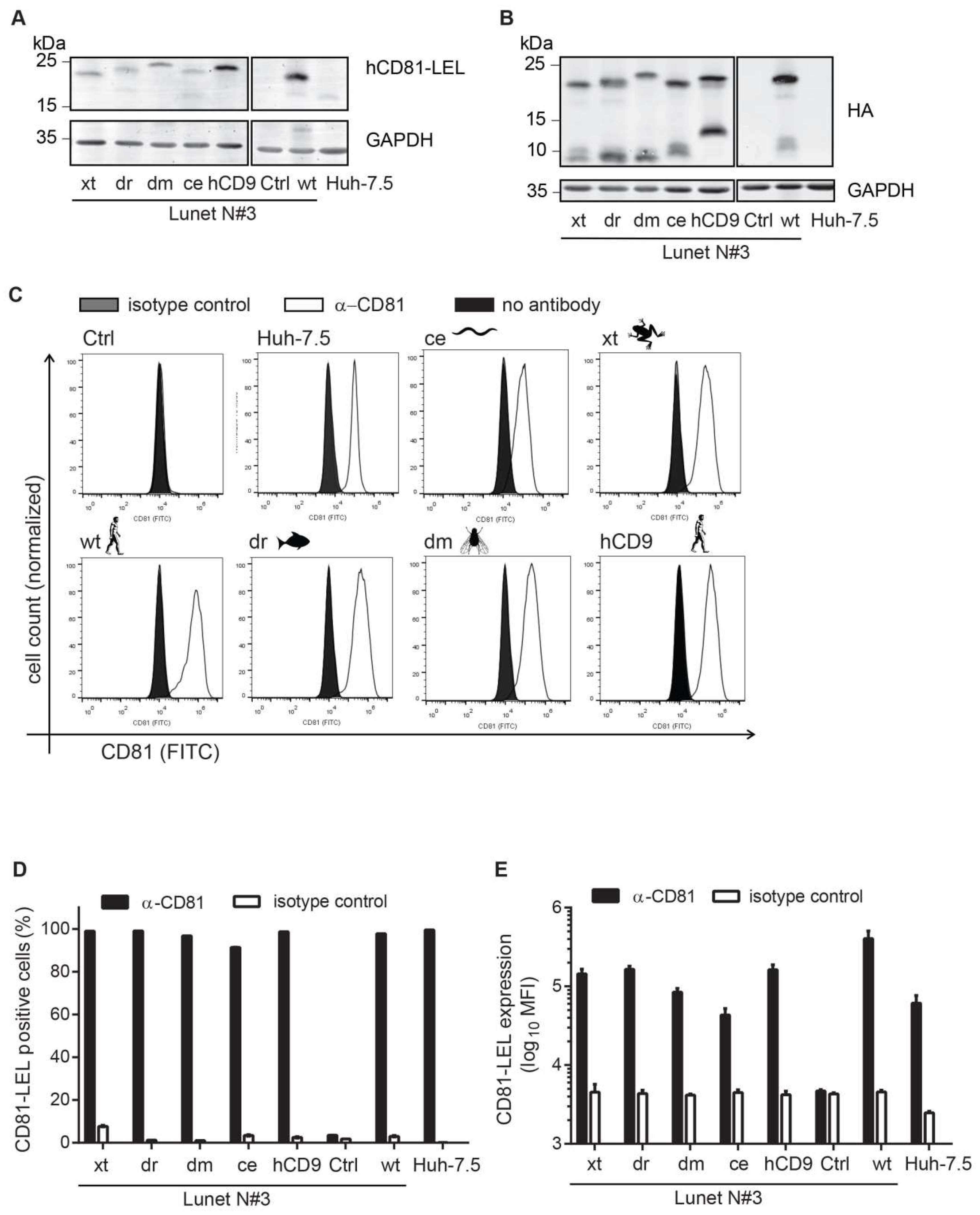
2.5. CD81 Surface Staining and Flow Cytometry
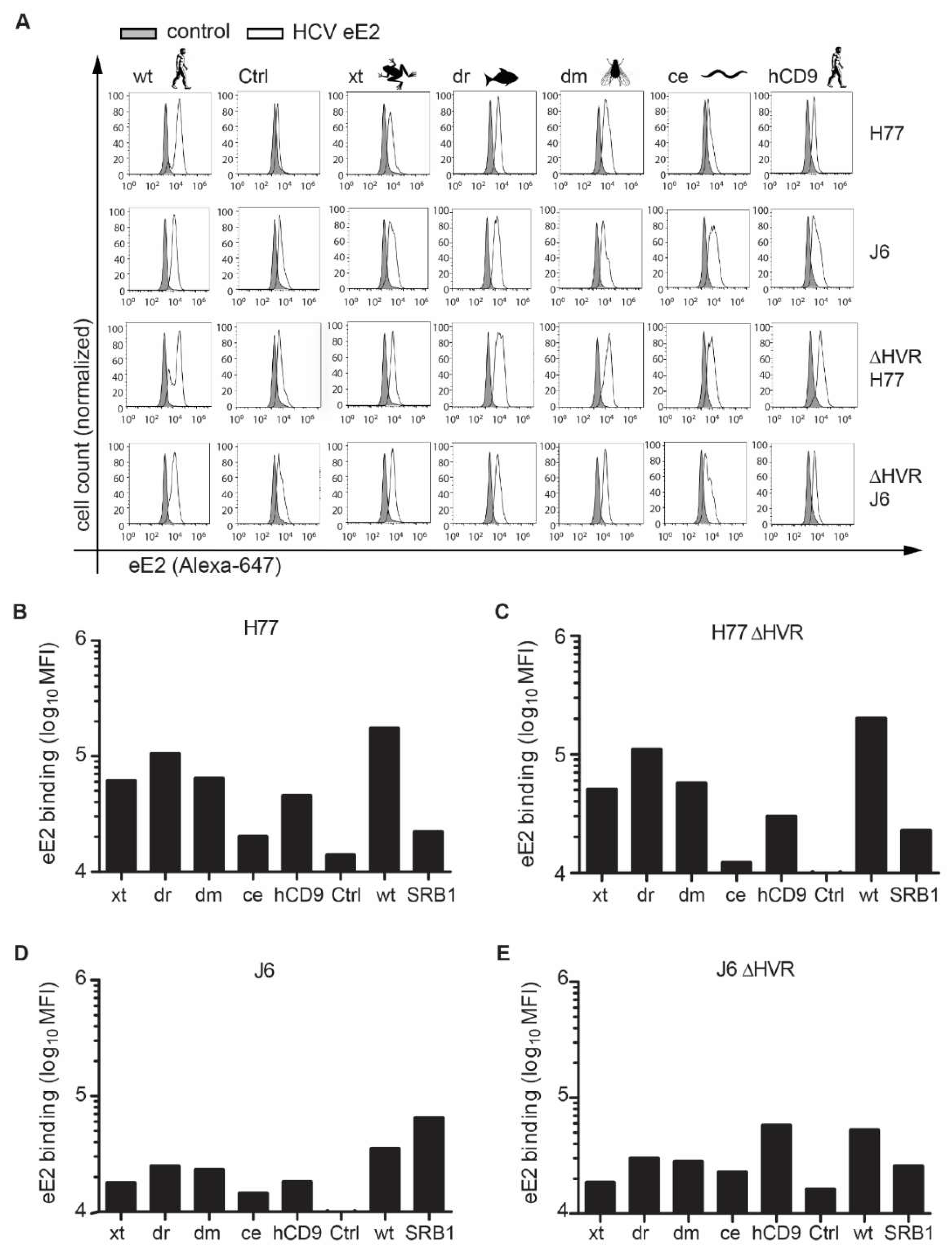
2.6. Soluble E2 Binding Assay
2.7. Pseudoparticle Infection
2.8. HCVcc, VSV and Coronavirus Infection
2.9. HCV RNA Quantification
2.10. HCV Plasma Membrane Fusion Assay
2.11. Alignments, Computational and Statistical Analyses
3. Results
3.1. CD81 Backbone Chimeras Bind to HCV E2 Glycoprotein
3.2. CD81-Tetraspanin Chimeras Are Expressed on the Surface of Human Hepatoma Cells
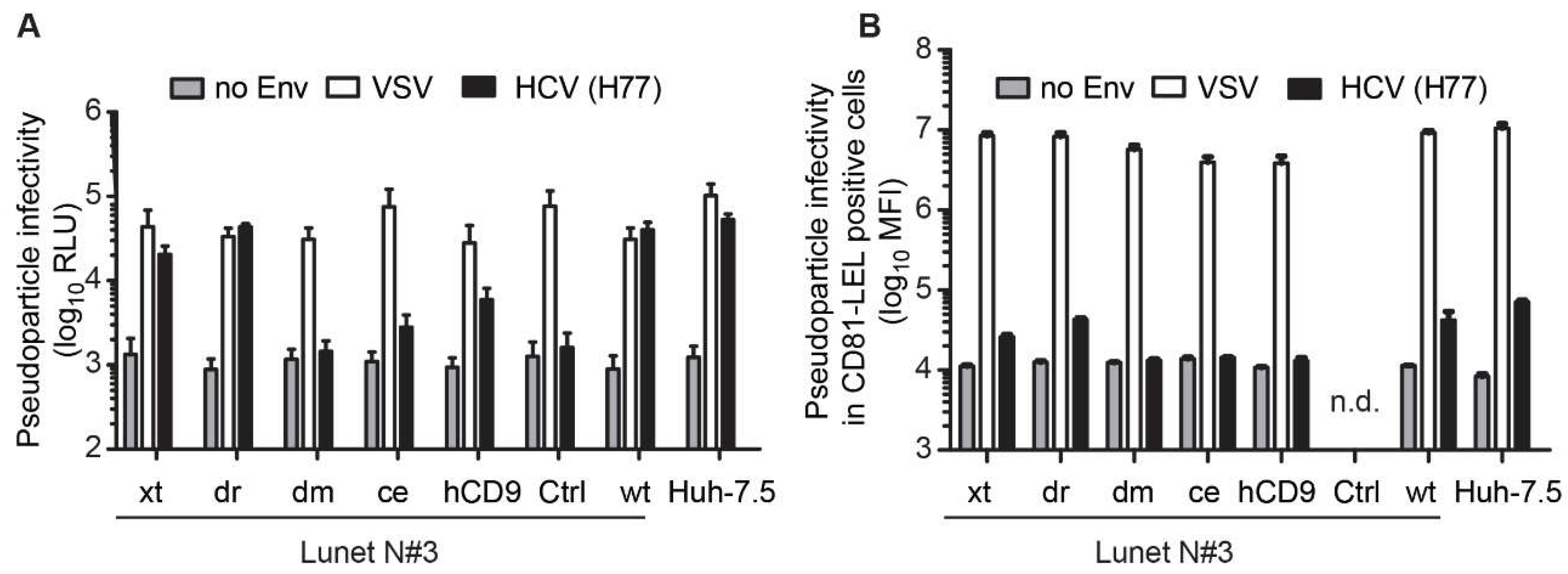
3.3. The hCD81 Backbone Influences HCV Pseudoparticle Infectivity
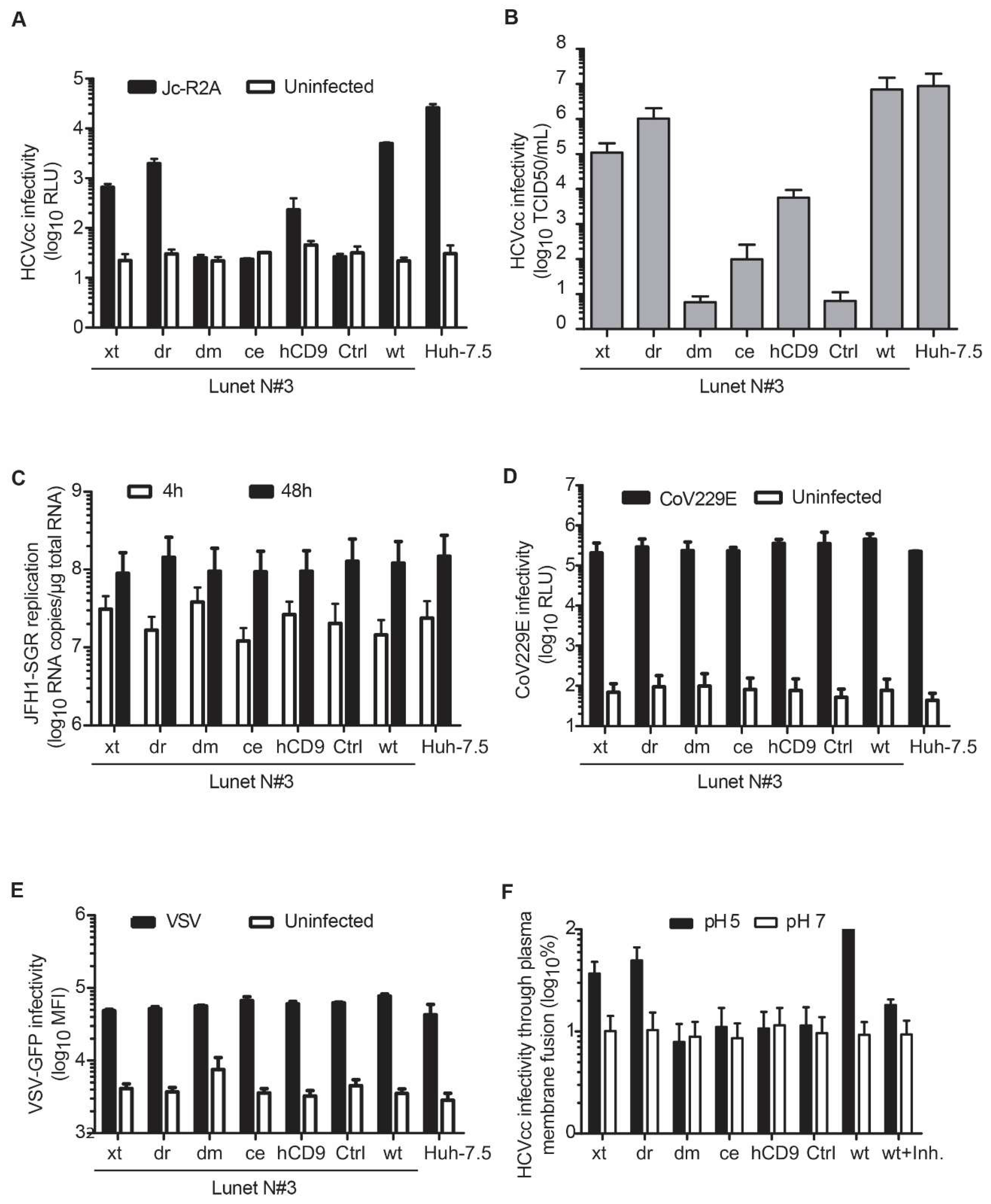
3.4. Entry of HCV Cell Culture Virus Depends on the hCD81 Backbone
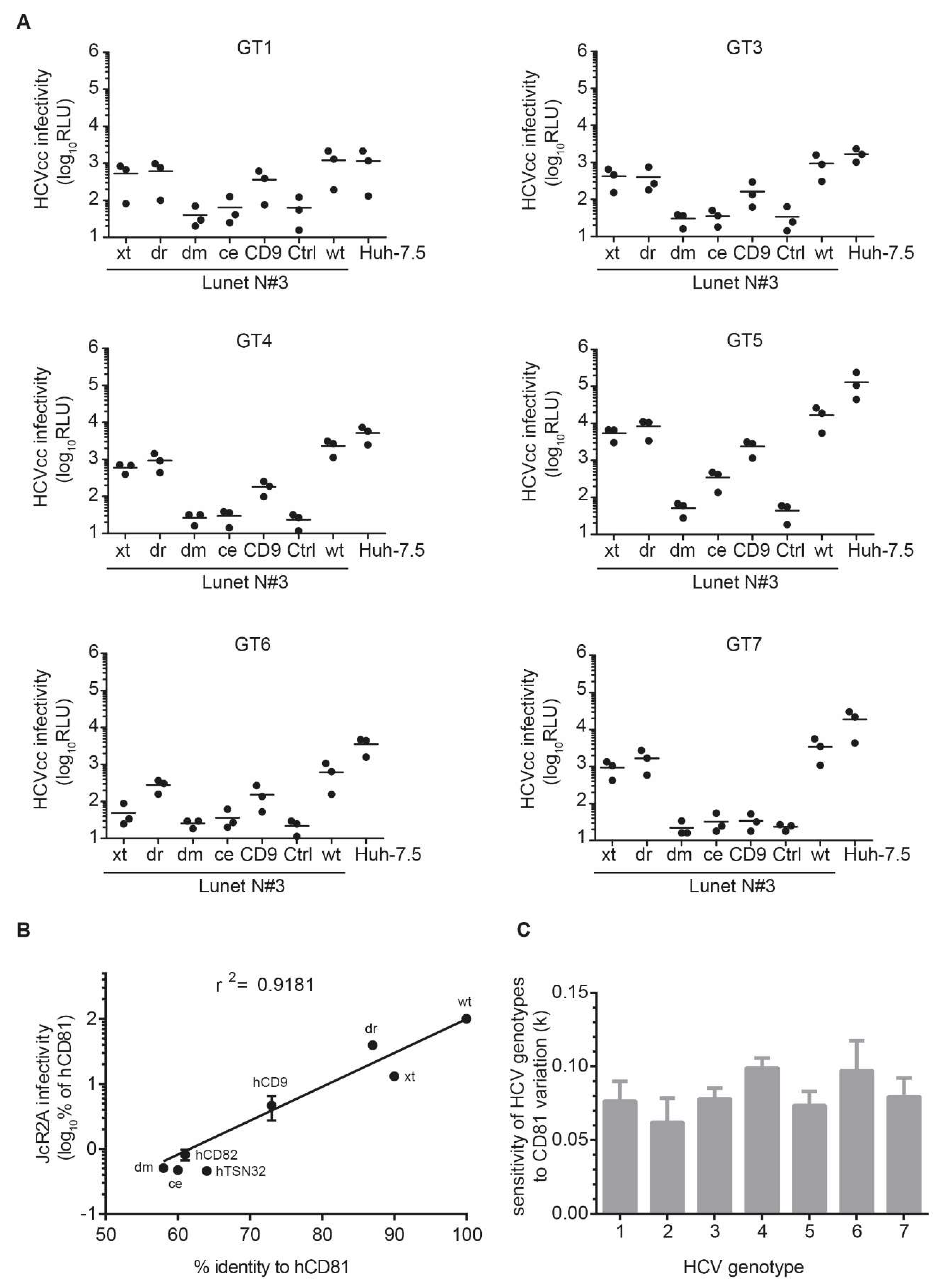
3.5. The Seven HCV Genotypes Depend Similarly on the hCD81 Backbone
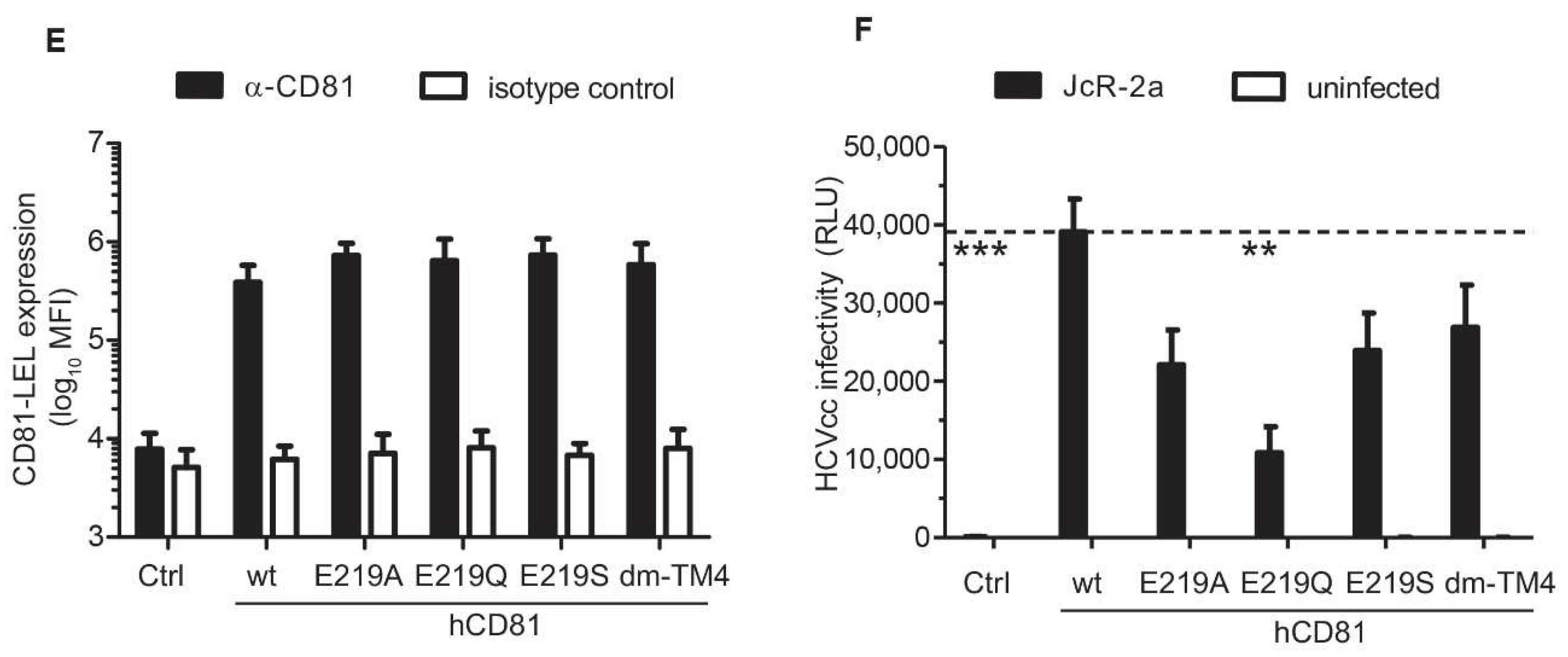
3.6. The hCD81 Glutamate Residue 219 Contributes to HCV Susceptibility
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mercer, J.; Schelhaas, M.; Helenius, A. Virus entry by endocytosis. Annu. Rev. Biochem. 2010, 79, 803–833. [Google Scholar] [CrossRef] [PubMed]
- Gerold, G.; Bruening, J.; Pietschmann, T. Decoding protein networks during virus entry by quantitative proteomics. Virus Res. 2016, 218, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Dubuisson, J.; Penin, F.; Moradpour, D. Interaction of hepatitis C virus proteins with host cell membranes and lipids. Trends Cell Biol. 2002, 12, 517–523. [Google Scholar] [CrossRef]
- Alvisi, G.; Madan, V.; Bartenschlager, R. Hepatitis C virus and host cell lipids: An intimate connection. RNA Biol. 2011, 8, 258–269. [Google Scholar] [CrossRef] [PubMed]
- Gerold, G.; Bruening, J.; Weigel, B.; Pietschmann, T. Protein Interactions during the Flavivirus and Hepacivirus Life Cycle. Mol. Cell Proteom. 2017, 16, S75–S91. [Google Scholar] [CrossRef] [PubMed]
- Vos, T.; Barber, R.M.; Bell, B.; Bertozzi-Villa, A.; Biryukov, S.; Bolliger, I.; Charlson, F.; Davis, A.; Degenhardt, L.; Dicker, D. Global Burden of Disease Study 2013 Collaborators Global, regional, and national incidence, prevalence, and years lived with disability for 301 acute and chronic diseases and injuries in 188 countries, 1990–2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet 2015, 386, 743–800. [Google Scholar]
- Cohen, J. New report halves the number of people infected with hepatitis C worldwide. Science 2017. [Google Scholar] [CrossRef]
- Pileri, P.; Uematsu, Y.; Campagnoli, S.; Galli, G.; Falugi, F.; Petracca, R.; Weiner, A.J.; Houghton, M.; Rosa, D.; Grandi, G.; et al. Binding of hepatitis C virus to CD81. Science 1998, 282, 938–941. [Google Scholar] [CrossRef] [PubMed]
- Ploss, A.; Evans, M.J.; Gaysinskaya, V.A.; Panis, M.; You, H.; de Jong, Y.P.; Rice, C.M. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 2009, 457, 882–886. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.J.; von Hahn, T.; Tscherne, D.M.; Syder, A.J.; Panis, M.; Wölk, B.; Hatziioannou, T.; McKeating, J.A.; Bieniasz, P.D.; Rice, C.M. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 2007, 446, 801–805. [Google Scholar] [CrossRef] [PubMed]
- Scarselli, E.; Ansuini, H.; Cerino, R.; Roccasecca, R.M.; Acali, S.; Filocamo, G.; Traboni, C.; Nicosia, A.; Cortese, R.; Vitelli, A. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J. 2002, 21, 5017–5025. [Google Scholar] [CrossRef] [PubMed]
- Ujino, S.; Nishitsuji, H.; Hishiki, T.; Sugiyama, K.; Takaku, H.; Shimotohno, K. Hepatitis C virus utilizes VLDLR as a novel entry pathway. Proc. Natl. Acad. Sci. USA 2016, 113, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Albecka, A.; Belouzard, S.; Op de Beeck, A.; Descamps, V.; Goueslain, L.; Bertrand-Michel, J.; Tercé, F.; Duverlie, G.; Rouillé, Y.; Dubuisson, J. Role of low-density lipoprotein receptor in the hepatitis C virus life cycle. Hepatology 2012, 55, 998–1007. [Google Scholar] [CrossRef] [PubMed]
- Brazzoli, M.; Bianchi, A.; Filippini, S.; Weiner, A.; Zhu, Q.; Pizza, M.; Crotta, S. CD81 is a central regulator of cellular events required for hepatitis C virus infection of human hepatocytes. J. Virol. 2008, 82, 8316–8329. [Google Scholar] [CrossRef] [PubMed]
- Farquhar, M.J.; Hu, K.; Harris, H.J.; Davis, C.; Brimacombe, C.L.; Fletcher, S.J.; Baumert, T.F.; Rappoport, J.Z.; Balfe, P.; McKeating, J.A. Hepatitis C virus induces CD81 and claudin-1 endocytosis. J. Virol. 2012, 86, 4305–4316. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.R.; Mateu, G.; Dreux, M.; Grakoui, A.; Cosset, F.L.; Melikyan, G.B. Hepatitis C virus is primed by CD81 protein for low pH-dependent fusion. J. Biol. Chem. 2011, 286, 30361–30376. [Google Scholar] [CrossRef] [PubMed]
- Charrin, S.; Manié, S.; Thiele, C.; Billard, M.; Gerlier, D.; Boucheix, C.; Rubinstein, E. A physical and functional link between cholesterol and tetraspanins. Eur. J. Immunol. 2003, 33, 2479–2489. [Google Scholar] [CrossRef] [PubMed]
- Delandre, C.; Penabaz, T.R.; Passarelli, A.L.; Chapes, S.K.; Clem, R.J. Mutation of juxtamembrane cysteines in the tetraspanin CD81 affects palmitoylation and alters interaction with other proteins at the cell surface. Exp. Cell Res. 2009, 315, 1953–1963. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, B.; Kelly, B.; McMillan, B.J.; Seegar, T.C.; Dror, R.O.; Kruse, A.C.; Blacklow, S.C. Crystal Structure of a Full-Length Human Tetraspanin Reveals a Cholesterol-Binding Pocket. Cell 2016, 167, 1041–1051. [Google Scholar] [CrossRef] [PubMed]
- Levy, S.; Shoham, T. Protein-protein interactions in the tetraspanin web. Physiology (Bethesda) 2005, 20, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.; Harris, H.J.; Hu, K.; Drummer, H.E.; McKeating, J.A.; Mullins, J.G.; Balfe, P. In silico directed mutagenesis identifies the CD81/claudin-1 hepatitis C virus receptor interface. Cell Microbiol. 2012, 14, 1892–1903. [Google Scholar] [CrossRef] [PubMed]
- Zona, L.; Lupberger, J.; Sidahmed-Adrar, N.; Thumann, C.; Harris, H.J.; Barnes, A.; Florentin, J.; Tawar, R.G.; Xiao, F.; Turek, M.; et al. HRas signal transduction promotes hepatitis C virus cell entry by triggering assembly of the host tetraspanin receptor complex. Cell Host Microbe 2013, 13, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Gerold, G.; Meissner, F.; Bruening, J.; Welsch, K.; Perin, P.M.; Baumert, T.F.; Vondran, F.W.; Kaderali, L.; Marcotrigiano, J.; Khan, A.G.; et al. Quantitative Proteomics Identifies Serum Response Factor Binding Protein 1 as a Host Factor for Hepatitis C Virus Entry. Cell Rep. 2015, 12, 864–878. [Google Scholar] [CrossRef] [PubMed]
- Flint, M.; von Hahn, T.; Zhang, J.; Farquhar, M.; Jones, C.T.; Balfe, P.; Rice, C.M.; McKeating, J.A. Diverse CD81 proteins support hepatitis C virus infection. J. Virol. 2006, 80, 11331–11342. [Google Scholar] [CrossRef] [PubMed]
- Rajesh, S.; Sridhar, P.; Tews, B.A.; Fénéant, L.; Cocquerel, L.; Ward, D.G.; Berditchevski, F.; Overduin, M. Structural basis of ligand interactions of the large extracellular domain of tetraspanin CD81. J. Virol. 2012, 86, 9606–9616. [Google Scholar] [CrossRef] [PubMed]
- Potel, J.; Rassam, P.; Montpellier, C.; Kaestner, L.; Werkmeister, E.; Tews, B.A.; Couturier, C.; Popescu, C.I.; Baumert, T.F.; Rubinstein, E.; et al. EWI-2wint promotes CD81 clustering that abrogates Hepatitis C Virus entry. Cell Microbiol. 2013, 15, 1234–1252. [Google Scholar] [CrossRef] [PubMed]
- Montpellier, C.; Tews, B.A.; Poitrimole, J.; Rocha-Perugini, V.; D’Arienzo, V.; Potel, J.; Zhang, X.A.; Rubinstein, E.; Dubuisson, J.; Cocquerel, L. Interacting regions of CD81 and two of its partners, EWI-2 and EWI-2wint, and their effect on hepatitis C virus infection. J. Biol. Chem. 2011, 286, 13954–13965. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Tang, Z.Y.; Klumpp, B.; Wolff-Vorbeck, G.; Barth, H.; Levy, S.; von Weizsäcker, F.; Blum, H.E.; Baumert, T.F. Primary hepatocytes of Tupaia belangeri as a potential model for hepatitis C virus infection. J. Clin. Investig. 2002, 109, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.C.; Riezu-Boj, J.I.; Lasarte, J.J.; Guillen, J.; Su, J.H.; Civeira, M.P.; Prieto, J. Transmission of hepatitis C virus infection to tree shrews. Virology 1998, 244, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Amako, Y.; Tsukiyama-Kohara, K.; Katsume, A.; Hirata, Y.; Sekiguchi, S.; Tobita, Y.; Hayashi, Y.; Hishima, T.; Funata, N.; Yonekawa, H.; et al. Pathogenesis of hepatitis C virus infection in Tupaia belangeri. J. Virol. 2010, 84, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Scull, M.A.; Shi, C.; de Jong, Y.P.; Gerold, G.; Ries, M.; von Schaewen, M.; Donovan, B.M.; Labitt, R.N.; Horwitz, J.A.; Gaska, J.M.; et al. Hepatitis C virus infects rhesus macaque hepatocytes and simianized mice. Hepatology 2015, 62, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Vogt, A.; Scull, M.A.; Friling, T.; Horwitz, J.A.; Donovan, B.M.; Dorner, M.; Gerold, G.; Labitt, R.N.; Rice, C.M.; Ploss, A. Recapitulation of the hepatitis C virus life-cycle in engineered murine cell lines. Virology 2013, 444, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Dorner, M.; Horwitz, J.A.; Donovan, B.M.; Labitt, R.N.; Budell, W.C.; Friling, T.; Vogt, A.; Catanese, M.T.; Satoh, T.; Kawai, T.; et al. Completion of the entire hepatitis C virus life cycle in genetically humanized mice. Nature 2013, 501, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Dorner, M.; Horwitz, J.A.; Robbins, J.B.; Barry, W.T.; Feng, Q.; Mu, K.; Jones, C.T.; Schoggins, J.W.; Catanese, M.T.; Burton, D.R.; et al. A genetically humanized mouse model for hepatitis C virus infection. Nature 2011, 474, 208–211. [Google Scholar] [CrossRef] [PubMed]
- McKeating, J.A.; Zhang, L.Q.; Logvinoff, C.; Flint, M.; Zhang, J.; Yu, J.; Butera, D.; Ho, D.D.; Dustin, L.B.; Rice, C.M.; et al. Diverse hepatitis C virus glycoproteins mediate viral infection in a CD81-dependent manner. J. Virol. 2004, 78, 8496–8505. [Google Scholar] [CrossRef] [PubMed]
- UniProt Consortium. UniProt: A hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [Google Scholar]
- Cserzö, M.; Eisenhaber, F.; Eisenhaber, B.; Simon, I. On filtering false positive transmembrane protein predictions. Protein Eng. 2002, 15, 745–752. [Google Scholar] [CrossRef] [PubMed]
- Blight, K.J.; McKeating, J.A.; Rice, C.M. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J. Virol. 2002, 76, 13001–13014. [Google Scholar] [CrossRef] [PubMed]
- Bitzegeio, J.; Bankwitz, D.; Hueging, K.; Haid, S.; Brohm, C.; Zeisel, M.B.; Herrmann, E.; Iken, M.; Ott, M.; Baumert, T.F.; et al. Adaptation of hepatitis C virus to mouse CD81 permits infection of mouse cells in the absence of human entry factors. PLoS Pathog. 2010, 6, e1000978. [Google Scholar] [CrossRef] [PubMed]
- Esko, J.D.; Stewart, T.E.; Taylor, W.H. Animal cell mutants defective in glycosaminoglycan biosynthesis. Proc. Natl. Acad. Sci. USA 1985, 82, 3197–3201. [Google Scholar] [CrossRef] [PubMed]
- Bankwitz, D.; Steinmann, E.; Bitzegeio, J.; Ciesek, S.; Friesland, M.; Herrmann, E.; Zeisel, M.B.; Baumert, T.F.; Keck, Z.Y.; Foung, S.K.; et al. Hepatitis C virus hypervariable region 1 modulates receptor interactions, conceals the CD81 binding site, and protects conserved neutralizing epitopes. J. Virol. 2010, 84, 5751–5763. [Google Scholar] [CrossRef] [PubMed]
- Bartosch, B.; Dubuisson, J.; Cosset, F.L. Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J. Exp. Med. 2003, 197, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Haid, S.; Windisch, M.P.; Bartenschlager, R.; Pietschmann, T. Mouse-specific residues of claudin-1 limit hepatitis C virus genotype 2a infection in a human hepatocyte cell line. J. Virol. 2010, 84, 964–975. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.G.; Whidby, J.; Miller, M.T.; Scarborough, H.; Zatorski, A.V.; Cygan, A.; Price, A.A.; Yost, S.A.; Bohannon, C.D.; Jacob, J.; et al. Structure of the core ectodomain of the hepatitis C virus envelope glycoprotein 2. Nature 2014, 509, 381–384. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Wu, Y.J.; Gerber, M.; Berger-Rentsch, M.; Heimrich, B.; Schwemmle, M.; Zimmer, G. Fusion-active glycoprotein G mediates the cytotoxicity of vesicular stomatitis virus M mutants lacking host shut-off activity. J. Gen. Virol. 2010, 91, 2782–2793. [Google Scholar] [CrossRef] [PubMed]
- Pfefferle, S.; Schöpf, J.; Kögl, M.; Friedel, C.C.; Müller, M.A.; Carbajo-Lozoya, J.; Stellberger, T.; von Dall’Armi, E.; Herzog, P.; Kallies, S.; et al. The SARS-coronavirus-host interactome: Identification of cyclophilins as target for pan-coronavirus inhibitors. PLoS Pathog. 2011, 7, e1002331. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P. SSE: A nucleotide and amino acid sequence analysis platform. BMC Res. Notes 2012, 5, 50. [Google Scholar] [CrossRef] [PubMed]
- Kärber, G. Beitrag zur kollektiven Behandlung pharmakologischer Reihenversuche. Arch. Exp. Pathol. Pharmakol. 1931, 162, 480–487. [Google Scholar] [CrossRef]
- Spearman, C. The method of “right and wrong cases” (“constant stimuli”) without Gauss’s formulae. Br. J. Psychol. 1908, 2, 227–242. [Google Scholar] [CrossRef]
- Bankwitz, D.; Vieyres, G.; Hueging, K.; Bitzegeio, J.; Doepke, M.; Chhatwal, P.; Haid, S.; Catanese, M.T.; Zeisel, M.B.; Nicosia, A.; et al. Role of hypervariable region 1 for the interplay of hepatitis C virus with entry factors and lipoproteins. J. Virol. 2014, 88, 12644–12655. [Google Scholar] [CrossRef] [PubMed]
- Roccasecca, R.; Ansuini, H.; Vitelli, A.; Meola, A.; Scarselli, E.; Acali, S.; Pezzanera, M.; Ercole, B.B.; McKeating, J.; Yagnik, A.; et al. Binding of the hepatitis C virus E2 glycoprotein to CD81 is strain specific and is modulated by a complex interplay between hypervariable regions 1 and 2. J. Virol. 2003, 77, 1856–1867. [Google Scholar] [CrossRef] [PubMed]
- Brimacombe, C.L.; Wilson, G.K.; Hübscher, S.G.; McKeating, J.A.; Farquhar, M.J. A role for CD81 and hepatitis C virus in hepatoma mobility. Viruses 2014, 6, 1454–1472. [Google Scholar] [CrossRef] [PubMed]
- Levy, S.; Todd, S.C.; Maecker, H.T. CD81 (TAPA-1): A molecule involved in signal transduction and cell adhesion in the immune system. Annu. Rev. Immunol. 1998, 16, 89–109. [Google Scholar] [CrossRef] [PubMed]
- Fénéant, L.; Levy, S.; Cocquerel, L. CD81 and hepatitis C virus (HCV) infection. Viruses 2014, 6, 535–572. [Google Scholar] [CrossRef] [PubMed]
- Perin, P.M.; Haid, S.; Brown, R.J.; Doerrbecker, J.; Schulze, K.; Zeilinger, C.; von Schaewen, M.; Heller, B.; Vercauteren, K.; Luxenburger, E.; et al. Flunarizine prevents hepatitis C virus membrane fusion in a genotype-dependent manner by targeting the potential fusion peptide within E1. Hepatology 2016, 63, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Holmes, E.C.; Cha, T.A.; Chan, S.W.; McOmish, F.; Irvine, B.; Beall, E.; Yap, P.L.; Kolberg, J.; Urdea, M.S. Classification of hepatitis C virus into six major genotypes and a series of subtypes by phylogenetic analysis of the NS-5 region. J. Gen. Virol. 1993, 74 Pt 11, 2391–2399. [Google Scholar] [CrossRef]
- Haid, S.; Grethe, C.; Dill, M.T.; Heim, M.; Kaderali, L.; Pietschmann, T. Isolate-dependent use of claudins for cell entry by hepatitis C virus. Hepatology 2014, 59, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Sourisseau, M.; Michta, M.L.; Zony, C.; Israelow, B.; Hopcraft, S.E.; Narbus, C.M.; Parra Martín, A.; Evans, M.J. Temporal analysis of hepatitis C virus cell entry with occludin directed blocking antibodies. PLoS Pathog. 2013, 9, e1003244. [Google Scholar] [CrossRef] [PubMed]
- Drummer, H.E.; Wilson, K.A.; Poumbourios, P. Determinants of CD81 dimerization and interaction with hepatitis C virus glycoprotein E2. Biochem. Biophys. Res. Commun. 2005, 328, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Lavillette, D.; Tarr, A.W.; Voisset, C.; Donot, P.; Bartosch, B.; Bain, C.; Patel, A.H.; Dubuisson, J.; Ball, J.K.; Cosset, F.L. Characterization of host-range and cell entry properties of the major genotypes and subtypes of hepatitis C virus. Hepatology 2005, 41, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Randall, G.; Higginbottom, A.; Monk, P.; Rice, C.M.; McKeating, J.A. CD81 is required for hepatitis C virus glycoprotein-mediated viral infection. J. Virol. 2004, 78, 1448–1455. [Google Scholar] [CrossRef] [PubMed]
- Koutsoudakis, G.; Herrmann, E.; Kallis, S.; Bartenschlager, R.; Pietschmann, T. The level of CD81 cell surface expression is a key determinant for productive entry of hepatitis C virus into host cells. J. Virol. 2007, 81, 588–598. [Google Scholar] [CrossRef] [PubMed]
- Sainz, B.; Barretto, N.; Martin, D.N.; Hiraga, N.; Imamura, M.; Hussain, S.; Marsh, K.A.; Yu, X.; Chayama, K.; Alrefai, W.A.; et al. Identification of the Niemann-Pick C1-like 1 cholesterol absorption receptor as a new hepatitis C virus entry factor. Nat. Med. 2012, 18, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Kapadia, S.B.; Barth, H.; Baumert, T.; McKeating, J.A.; Chisari, F.V. Initiation of hepatitis C virus infection is dependent on cholesterol and cooperativity between CD81 and scavenger receptor B type I. J. Virol. 2007, 81, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Ciczora, Y.; Callens, N.; Penin, F.; Pécheur, E.I.; Dubuisson, J. Transmembrane domains of hepatitis C virus envelope glycoproteins: Residues involved in E1E2 heterodimerization and involvement of these domains in virus entry. J. Virol. 2007, 81, 2372–2381. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Fukuhara, T.; Ono, C.; Uemura, K.; Kawachi, Y.; Shiokawa, M.; Mori, H.; Wada, M.; Shima, R.; Okamoto, T.; et al. Lipoprotein Receptors Redundantly Participate in Entry of Hepatitis C Virus. PLoS Pathog. 2016, 12, e1005610. [Google Scholar] [CrossRef] [PubMed]
- Lupberger, J.; Zeisel, M.B.; Xiao, F.; Thumann, C.; Fofana, I.; Zona, L.; Davis, C.; Mee, C.J.; Turek, M.; Gorke, S.; et al. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat. Med. 2011, 17, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Harris, H.J.; Farquhar, M.J.; Mee, C.J.; Davis, C.; Reynolds, G.M.; Jennings, A.; Hu, K.; Yuan, F.; Deng, H.; Hubscher, S.G.; et al. CD81 and claudin 1 coreceptor association: Role in hepatitis C virus entry. J. Virol. 2008, 82, 5007–5020. [Google Scholar] [CrossRef] [PubMed]
- Carloni, V.; Mazzocca, A.; Ravichandran, K.S. Tetraspanin CD81 is linked to ERK/MAPKinase signaling by Shc in liver tumor cells. Oncogene 2004, 23, 1566–1574. [Google Scholar] [CrossRef] [PubMed]
- Spoden, G.; Kühling, L.; Cordes, N.; Frenzel, B.; Sapp, M.; Boller, K.; Florin, L.; Schelhaas, M. Human papillomavirus types 16, 18, and 31 share similar endocytic requirements for entry. J. Virol. 2013, 87, 7765–7773. [Google Scholar] [CrossRef] [PubMed]
- Scheffer, K.D.; Gawlitza, A.; Spoden, G.A.; Zhang, X.A.; Lambert, C.; Berditchevski, F.; Florin, L. Tetraspanin CD151 mediates papillomavirus type 16 endocytosis. J. Virol. 2013, 87, 3435–3446. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Kovalenko, O.V.; Tang, W.; Claas, C.; Stipp, C.S.; Hemler, M.E. Palmitoylation supports assembly and function of integrin-tetraspanin complexes. J. Cell Biol. 2004, 167, 1231–1240. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.Z.; Luo, Y.; Cao, M.M.; Liu, Y.; Liu, X.Q.; Wang, W.; Wu, D.G.; Guan, M.; Xu, Q.Q.; Ren, H.; et al. Significance of palmitoylation of CD81 on its association with tetraspanin-enriched microdomains and mediating hepatitis C virus cell entry. Virology 2012, 429, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Stiles, K.M.; Kielian, M. Role of TSPAN9 in Alphavirus Entry and Early Endosomes. J. Virol. 2016, 90, 4289–4297. [Google Scholar] [CrossRef] [PubMed]
- Ooi, Y.S.; Stiles, K.M.; Liu, C.Y.; Taylor, G.M.; Kielian, M. Genome-wide RNAi screen identifies novel host proteins required for alphavirus entry. PLoS Pathog. 2013, 9, e1003835. [Google Scholar] [CrossRef] [PubMed]
- Gordón-Alonso, M.; Yañez-Mó, M.; Barreiro, O.; Alvarez, S.; Muñoz-Fernández, M.A.; Valenzuela-Fernández, A.; Sánchez-Madrid, F. Tetraspanins CD9 and CD81 modulate HIV-1-induced membrane fusion. J. Immunol. 2006, 177, 5129–5137. [Google Scholar] [CrossRef] [PubMed]
- Hochdorfer, D.; Florin, L.; Sinzger, C.; Lieber, D. Tetraspanin CD151 promotes initial events in human cytomegalovirus infection. J. Virol. 2016, 90, 6430–6442. [Google Scholar] [CrossRef] [PubMed]
- Frey, E.A.; Miller, D.S.; Jahr, T.G.; Sundan, A.; Bazil, V.; Espevik, T.; Finlay, B.B.; Wright, S.D. Soluble CD14 participates in the response of cells to lipopolysaccharide. J. Exp. Med. 1992, 176, 1665–1671. [Google Scholar] [CrossRef] [PubMed]
- Merz, A.; Long, G.; Hiet, M.S.; Brügger, B.; Chlanda, P.; Andre, P.; Wieland, F.; Krijnse-Locker, J.; Bartenschlager, R. Biochemical and morphological properties of hepatitis C virus particles and determination of their lipidome. J. Biol. Chem. 2011, 286, 3018–3032. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.U.; Bassendine, M.F.; Burt, A.D.; Martin, C.; Pumeechockchai, W.; Toms, G.L. Association between hepatitis C virus and very-low-density lipoprotein (VLDL)/LDL analyzed in iodixanol density gradients. J. Virol. 2006, 80, 2418–2428. [Google Scholar] [PubMed]
- Thomssen, R.; Bonk, S.; Propfe, C.; Heermann, K.H.; Kochel, H.G.; Uy, A. Association of hepatitis C virus in human sera with beta-lipoprotein. Med. Microbiol. Immunol. 1992, 181, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Gastaminza, P.; Dryden, K.A.; Boyd, B.; Wood, M.R.; Law, M.; Yeager, M.; Chisari, F.V. Ultrastructural and biophysical characterization of hepatitis C virus particles produced in cell culture. J. Virol. 2010, 84, 10999–11009. [Google Scholar] [CrossRef] [PubMed]
- Catanese, M.T.; Uryu, K.; Kopp, M.; Edwards, T.J.; Andrus, L.; Rice, W.J.; Silvestry, M.; Kuhn, R.J.; Rice, C.M. Ultrastructural analysis of hepatitis C virus particles. Proc. Natl. Acad. Sci. USA 2013, 110, 9505–9510. [Google Scholar] [CrossRef] [PubMed]
- Lussignol, M.; Kopp, M.; Molloy, K.; Vizcay-Barrena, G.; Fleck, R.A.; Dorner, M.; Bell, K.L.; Chait, B.T.; Rice, C.M.; Catanese, M.T. Proteomics of HCV virions reveals an essential role for the nucleoporin Nup98 in virus morphogenesis. Proc. Natl. Acad. Sci. USA 2016, 113, 2484–2489. [Google Scholar] [CrossRef] [PubMed]
- Monazahian, M.; Bohme, I.; Bonk, S.; Koch, A.; Scholz, C.; Grethe, S.; Thomssen, R. Low density lipoprotein receptor as a candidate receptor for hepatitis C virus. J. Med. Virol. 1999, 57, 223–229. [Google Scholar] [CrossRef]
- Molina, S.; Castet, V.; Fournier-Wirth, C.; Pichard-Garcia, L.; Avner, R.; Harats, D.; Roitelman, J.; Barbaras, R.; Graber, P.; Ghersa, P.; et al. The low-density lipoprotein receptor plays a role in the infection of primary human hepatocytes by hepatitis C virus. J. Hepatol. 2007, 46, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P. The origin of hepatitis C virus. Curr. Top. Microbiol. Immunol. 2013, 369, 1–15. [Google Scholar] [PubMed]
- Gerold, G.; Pietschmann, T. The HCV life cycle: In vitro tissue culture systems and therapeutic targets. Dig. Dis. 2014, 32, 525–537. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Length | Identity with hCD81 (aa) | hCD81-LEL Chimera (w/o HA-Tag) | Length | Identity with hCD81 (aa) |
|---|---|---|---|---|---|
| hCD81 | 236 | 100% | hCD81 | 236 | 100% |
| xtCD81 | 237 | 72% | xt | 235 | 90% |
| drCD81 | 236 | 64% | dr | 237 | 87% |
| hCD9 | 228 | 45% | h | 233 | 73% |
| dmTSP96F | 268 | 27% | dm | 231 | 58% |
| hCD82 | 267 | 26% | hCD82 | 238 | 61% |
| ceTSP9 | 233 | 25% | ce | 242 | 60% |
| hTSN32 | 320 | 15% | hTSN32 | 317 | 64% |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Banse, P.; Moeller, R.; Bruening, J.; Lasswitz, L.; Kahl, S.; Khan, A.G.; Marcotrigiano, J.; Pietschmann, T.; Gerold, G. CD81 Receptor Regions outside the Large Extracellular Loop Determine Hepatitis C Virus Entry into Hepatoma Cells. Viruses 2018, 10, 207. https://doi.org/10.3390/v10040207
Banse P, Moeller R, Bruening J, Lasswitz L, Kahl S, Khan AG, Marcotrigiano J, Pietschmann T, Gerold G. CD81 Receptor Regions outside the Large Extracellular Loop Determine Hepatitis C Virus Entry into Hepatoma Cells. Viruses. 2018; 10(4):207. https://doi.org/10.3390/v10040207
Chicago/Turabian StyleBanse, Pia, Rebecca Moeller, Janina Bruening, Lisa Lasswitz, Sina Kahl, Abdul G. Khan, Joseph Marcotrigiano, Thomas Pietschmann, and Gisa Gerold. 2018. "CD81 Receptor Regions outside the Large Extracellular Loop Determine Hepatitis C Virus Entry into Hepatoma Cells" Viruses 10, no. 4: 207. https://doi.org/10.3390/v10040207
APA StyleBanse, P., Moeller, R., Bruening, J., Lasswitz, L., Kahl, S., Khan, A. G., Marcotrigiano, J., Pietschmann, T., & Gerold, G. (2018). CD81 Receptor Regions outside the Large Extracellular Loop Determine Hepatitis C Virus Entry into Hepatoma Cells. Viruses, 10(4), 207. https://doi.org/10.3390/v10040207