Enterovirus Transmission by Secretory Autophagy


{kind=link}
Abstract
1. Introduction
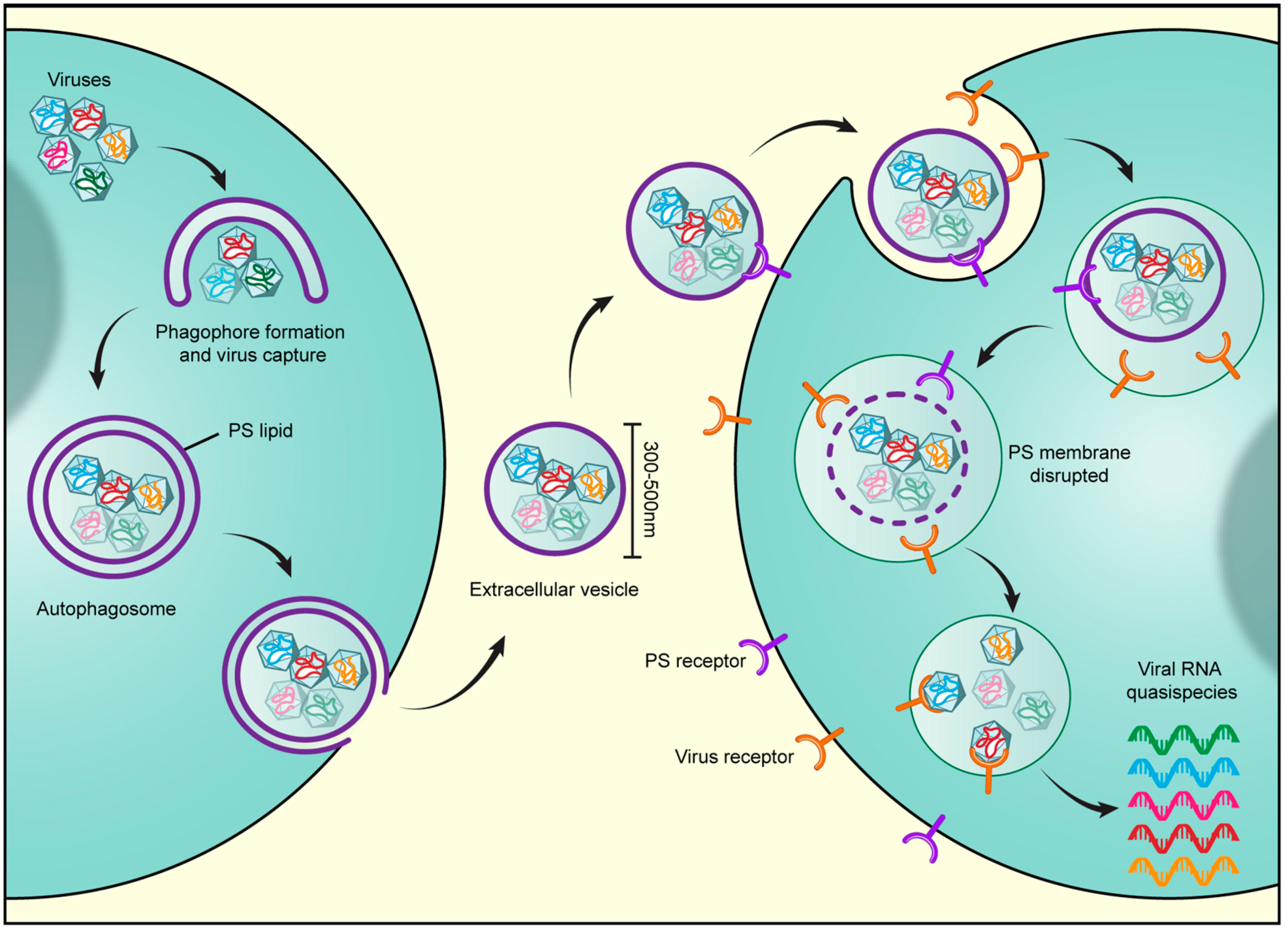
2. Secretory Autophagosomes Help Viruses Get Out of Cells
3. Advantages of Harnessing Secretory Autophagy for Viral Transmission
Conflicts of Interest
References
- Ohsumi, Y. Historical landmarks of autophagy research. Cell Res. 2014, 24, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B. Autophagy in mammalian development and differentiation. Nat. Cell Biol. 2010, 12, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [PubMed]
- Mattera, R.; Park, S.Y.; de Pace, R.; Guardia, C.M.; Bonifacino, J.S. AP-4 mediates export of ATG9A from the trans-Golgi network to promote autophagosome formation. Proc. Natl. Acad. Sci. USA 2017, 114, E10697–E10706. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Moreau, K.; Jahreiss, L.; Puri, C.; Rubinsztein, D.C. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat. Cell Biol. 2010, 12, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Hamasaki, M.; Furuta, N.; Matsuda, A.; Nezu, A.; Yamamoto, A.; Fujita, N.; Oomori, H.; Noda, T.; Haraguchi, T.; Hiraoka, Y.; et al. Autophagosomes form at ER-mitochondria contact sites. Nature 2013, 495, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Kishi-Itakura, C.; Koyama-Honda, I.; Itakura, E.; Mizushima, N. Ultrastructural analysis of autophagosome organization using mammalian autophagy-deficient cells. J. Cell Sci. 2014, 127, 4089–4102. [Google Scholar] [CrossRef] [PubMed]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Noda, T.; Yoshimori, T.; Tanaka, Y.; Ishii, T.; George, M.D.; Klionsky, D.J.; Ohsumi, M.; Ohsumi, Y. A protein conjugation system essential for autophagy. Nature 1998, 395, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, Y.; Kirisako, T.; Takao, T.; Satomi, Y.; Shimonishi, Y.; Ishihara, N.; Mizushima, N.; Tanida, I.; Kominami, E.; Ohsumi, M.; et al. A ubiquitin-like system mediates protein lipidation. Nature 2000, 408, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Diao, J.; Liu, R.; Rong, Y.; Zhao, M.; Zhang, J.; Lai, Y.; Zhou, Q.; Wilz, L.M.; Ji, J.; Vivona, S.; et al. ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature 2015, 520, 563–566. [Google Scholar] [CrossRef] [PubMed]
- Itakura, E.; Kishi-Itakura, C.; Mizushima, N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 2012, 151, 1256–1269. [Google Scholar] [CrossRef] [PubMed]
- Takats, S.; Nagy, P.; Varga, Á.; Pircs, K.; Kárpáti, M.; Varga, K.; Kovács, A.L.; Hegedűs, K.; Juhász, G. Autophagosomal Syntaxin17-dependent lysosomal degradation maintains neuronal function in Drosophila. J. Cell Biol. 2013, 201, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Jain, A.; Farzam, F.; Jia, J.; Gu, Y.; Choi, S.W.; Mudd, M.H.; Claude-Taupin, A.; Wester, M.J.; Lidke, K.A.; et al. Mechanism of Stx17 recruitment to autophagosomes via IRGM and mammalian Atg8 proteins. J. Cell Biol. 2018, 217, 997–1013. [Google Scholar] [CrossRef] [PubMed]
- Ponpuak, M.; Mandell, M.A.; Kimura, T.; Chauhan, S.; Cleyrat, C.; Deretic, V. Secretory autophagy. Curr. Opin. Cell Biol. 2015, 35, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Kenny, S.J.; Ge, L.; Xu, K.; Schekman, R. Translocation of interleukin-1beta into a vesicle intermediate in autophagy-mediated secretion. Elife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Bel, S.; Pendse, M.; Wang, Y.; Li, Y.; Ruhn, K.A.; Hassell, B.; Leal, T.; Winter, S.E.; Xavier, R.J.; Hooper, L.V. Paneth cells secrete lysozyme via secretory autophagy during bacterial infection of the intestine. Science 2017, 357, 1047–1052. [Google Scholar] [CrossRef] [PubMed]
- Mankelow, T.J.; Griffiths, R.E.; Trompeter, S.; Flatt, J.F.; Cogan, N.M.; Massey, E.J.; Anstee, D.J. Autophagic vesicles on mature human reticulocytes explain phosphatidylserine-positive red cells in sickle cell disease. Blood 2015, 126, 1831–1834. [Google Scholar] [CrossRef] [PubMed]
- Harris, J. Autophagy and IL-1 Family Cytokines. Front. Immunol. 2013, 4, 83. [Google Scholar] [CrossRef] [PubMed]
- Duran, J.M.; Anjard, C.; Stefan, C.; Loomis, W.F.; Malhotra, V. Unconventional secretion of Acb1 is mediated by autophagosomes. J. Cell Biol. 2010, 188, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Manjithaya, R.; Anjard, C.; Loomis, W.F.; Subramani, S. Unconventional secretion of Pichia pastoris Acb1 is dependent on GRASP protein, peroxisomal functions, and autophagosome formation. J. Cell Biol. 2010, 188, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Dengjel, J.; Schoor, O.; Fischer, R.; Reich, M.; Kraus, M.; Müller, M.; Kreymborg, K.; Altenberend, F.; Brandenburg, J.; Kalbacher, H.; et al. Autophagy promotes MHC class II presentation of peptides from intracellular source proteins. Proc. Natl. Acad. Sci. USA 2005, 102, 7922–7927. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Chauhan, S.; Jain, A.; Ponpuak, M.; Choi, S.W.; Mudd, M.; Peters, R.; Mandell, M.A.; Johansen, T.; Deretic, V. Galectins and TRIMs directly interact and orchestrate autophagic response to endomembrane damage. Autophagy 2017, 13, 1086–1087. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, S.; Kumar, S.; Jain, A.; Ponpuak, M.; Mudd, M.H.; Kimura, T.; Choi, S.W.; Peters, R.; Mandell, M.; Bruun, J.-A.; et al. TRIMs and Galectins Globally Cooperate and TRIM16 and Galectin-3 Co-direct Autophagy in Endomembrane Damage Homeostasis. Dev. Cell 2016, 39, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Evavold, C.L.; Ruan, J.; Tan, Y.; Xia, S.; Wu, H.; Kagan, J.C. The Pore-Forming Protein Gasdermin D Regulates Interleukin-1 Secretion from Living Macrophages. Immunity 2018, 48, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.H.; Du, W.; Hagemeijer, M.C.; Takvorian, P.M.; Pau, C.; Cali, A.; Brantner, C.A.; Stempinski, E.S.; Connelly, P.S.; Ma, H.-C.; et al. Phosphatidylserine vesicles enable efficient en bloc transmission of enteroviruses. Cell 2015, 160, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Bird, S.W.; Maynard, N.D.; Covert, M.W.; Kirkegaard, K. Nonlytic viral spread enhanced by autophagy components. Proc. Natl. Acad. Sci. USA 2014, 111, 13081–13086. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.M.; Tsueng, G.; Sin, J.; Mangale, V.; Rahawi, S.; McIntyre, L.L.; Williams, W.; Kha, N.; Cruz, C.; Hancock, B.M.; et al. Coxsackievirus B exits the host cell in shed microvesicles displaying autophagosomal markers. PLoS Pathog. 2014, 10, e1004045. [Google Scholar] [CrossRef] [PubMed]
- Jackson, W.T.; Giddings, T.H., Jr.; Taylor, M.P.; Mulinyawe, S.; Rabinovitch, M.; Kopito, R.R.; Kirkegaard, K. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 2005, 3, e156. [Google Scholar] [CrossRef] [PubMed]
- Hsu, N.Y.; Ilnytska, O.; Belov, G.; Santiana, M.; Chen, Y.-H.; Takvorian, P.M.; Pau, C.; van der Schaar, H.; Kaushik-Basu, N.; Balla, T.; et al. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell 2010, 141, 799–811. [Google Scholar] [CrossRef] [PubMed]
- Ilnytska, O.; Santiana, M.; Hsu, N.-Y.; Du, W.-L.; Chen, Y.-H.; Viktorova, E.G.; Belov, G.; Brinker, A.; Storch, J.; Moore, C.; et al. Enteroviruses harness the cellular endocytic machinery to remodel the host cell cholesterol landscape for effective viral replication. Cell Host Microbe 2013, 14, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Altan-Bonnet, N. Lipid Tales of Viral Replication and Transmission. Trends Cell Biol. 2017, 27, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Tucker, S.P.; Thornton, C.L.; Wimmer, E.; Compans, R.W. Vectorial release of poliovirus from polarized human intestinal epithelial cells. J. Virol. 1993, 67, 4274–4282. [Google Scholar] [PubMed]
- Dales, S.; Eggers, H.J.; Tamm, I.; Palade, G.E. Electron Microscopic Study of the Formation of Poliovirus. Virology 1965, 26, 379–389. [Google Scholar] [CrossRef]
- Ehrenfeld, E. Poliovirus-induced inhibition of host-cell protein synthesis. Cell 1982, 28, 435–436. [Google Scholar] [CrossRef]
- Fairn, G.D.; Schieber, N.L.; Ariotti, N.; Murphy, S.; Kuerschner, L.; Webb, R.I.; Grinstein, S.; Parton, R.G. High-resolution mapping reveals topologically distinct cellular pools of phosphatidylserine. J. Cell Biol. 2011, 194, 257–275. [Google Scholar] [CrossRef] [PubMed]
- Kay, J.G.; Koivusalo, M.; Ma, X.; Wohland, T.; Grinstein, S. Phosphatidylserine dynamics in cellular membranes. Mol. Biol. Cell 2012, 23, 2198–2212. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.; Torta, F.; Masai, K.; Lucast, L.; Czapla, H.; Tanner, L.B.; Narayanaswamy, P.; Wenk, M.R.; Nakatsu, F.; de Camilli, P. INTRACELLULAR TRANSPORT. PI4P/phosphatidylserine countertransport at ORP5- and ORP8-mediated ER-plasma membrane contacts. Science 2015, 349, 428–432. [Google Scholar] [CrossRef] [PubMed]
- Sohn, M.; Ivanova, P.; Brown, H.A.; Toth, D.J.; Varnai, P.; Kim, Y.J.; Balla, T. Lenz-Majewski mutations in PTDSS1 affect phosphatidylinositol 4-phosphate metabolism at ER-PM and ER-Golgi junctions. Proc. Natl. Acad. Sci. USA 2016, 113, 4314–4319. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, Q.; Li, M.; Mao, Q.; Chen, H.; Wu, W.; Jia, D.; Wei, T. Autophagy pathway induced by a plant virus facilitates viral spread and transmission by its insect vector. PLoS Pathog. 2017, 13, e1006727. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Duan, Y.; Han, C.; Yao, S.; Qi, X.; Gao, Y.; Maier, H.J.; Britton, P.; Chen, L.; Zhang, L.; et al. Infectious Bursal Disease Virus Subverts Autophagic Vacuoles To Promote Viral Maturation and Release. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.W.; Li, Z.L.; Yuan, S. The Role of Secretory Autophagy in Zika Virus Transfer through the Placental Barrier. Front. Cell Infect. Microbiol. 2016, 6, 206. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Parnell, L.A.; Diamond, M.S.; Mysorekar, I.U. Inhibition of autophagy limits vertical transmission of Zika virus in pregnant mice. J. Exp. Med. 2017, 214, 2303–2313. [Google Scholar] [CrossRef] [PubMed]
- Chiramel, A.I.; Best, S.M. Role of autophagy in Zika virus infection and pathogenesis. Virus Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Hamel, R.; Dejarnac, O.; Wichit, S.; Ekchariyawat, P.; Neyret, A.; Luplertlop, N.; Perera-Lecoin, M.; Surasombatpattana, P.; Talignani, L.; Thomas, F.; et al. Biology of Zika Virus Infection in Human Skin Cells. J. Virol. 2015, 89, 8880–8896. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Hensley, L.; McKnight, K.L.; Hu, F.; Madden, V.; Ping, L.; Jeong, S.-H.; Walker, C.; Lanford, R.E.; Lemon, S.M. A pathogenic picornavirus acquires an envelope by hijacking cellular membranes. Nature 2013, 496, 367–371. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Ambardekar, C.; Lu, Y.; Feng, Z. Distinct Entry Mechanisms for Nonenveloped and Quasi-Enveloped Hepatitis E Viruses. J. Virol. 2016, 90, 4232–4242. [Google Scholar] [CrossRef] [PubMed]
- Lemke, G. Phosphatidylserine Is the Signal for TAM Receptors and Their Ligands. Trends Biochem. Sci. 2017, 42, 738–748. [Google Scholar] [CrossRef] [PubMed]
- Amara, A.; Mercer, J. Viral apoptotic mimicry. Nat. Rev. Microbiol. 2015, 13, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Moller-Tank, S.; Maury, W. Phosphatidylserine receptors: Enhancers of enveloped virus entry and infection. Virology 2014, 468, 565–580. [Google Scholar] [CrossRef] [PubMed]
- Birge, R.B.; Boeltz, S.; Kumar, S.; Carlson, J.; Wanderley Calianese, J.; Calianese, D.; Barcinski, M.; Brekken, R.A.; Huang, X.; Hutchins, J.T.; et al. Phosphatidylserine is a global immunosuppressive signal in efferocytosis, infectious disease, and cancer. Cell Death Differ. 2016, 23, 962–978. [Google Scholar] [CrossRef] [PubMed]
- Altan-Bonnet, N.; Chen, Y.H. Intercellular Transmission of Viral Populations with Vesicles. J. Virol. 2015, 89, 12242–12244. [Google Scholar] [CrossRef] [PubMed]
- Andino, R.; Domingo, E. Viral quasispecies. Virology 2015, 479, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Borderia, A.V.; Isakov, O.; Moratorio, G.; Henningsson, R.; Agüera-González, S.; Organtini, L.; Gnädig, N.F.; Blanc, H.; Alcover, A.; Hafenstein, S.; et al. Group Selection and Contribution of Minority Variants during Virus Adaptation Determines Virus Fitness and Phenotype. PLoS Pathog. 2015, 11, e1004838. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mutsafi, Y.; Altan-Bonnet, N. Enterovirus Transmission by Secretory Autophagy. Viruses 2018, 10, 139. https://doi.org/10.3390/v10030139
Mutsafi Y, Altan-Bonnet N. Enterovirus Transmission by Secretory Autophagy. Viruses. 2018; 10(3):139. https://doi.org/10.3390/v10030139
Chicago/Turabian StyleMutsafi, Yael, and Nihal Altan-Bonnet. 2018. "Enterovirus Transmission by Secretory Autophagy" Viruses 10, no. 3: 139. https://doi.org/10.3390/v10030139
APA StyleMutsafi, Y., & Altan-Bonnet, N. (2018). Enterovirus Transmission by Secretory Autophagy. Viruses, 10(3), 139. https://doi.org/10.3390/v10030139