Prion Strain-Specific Structure and Pathology: A View from the Perspective of Glycobiology

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
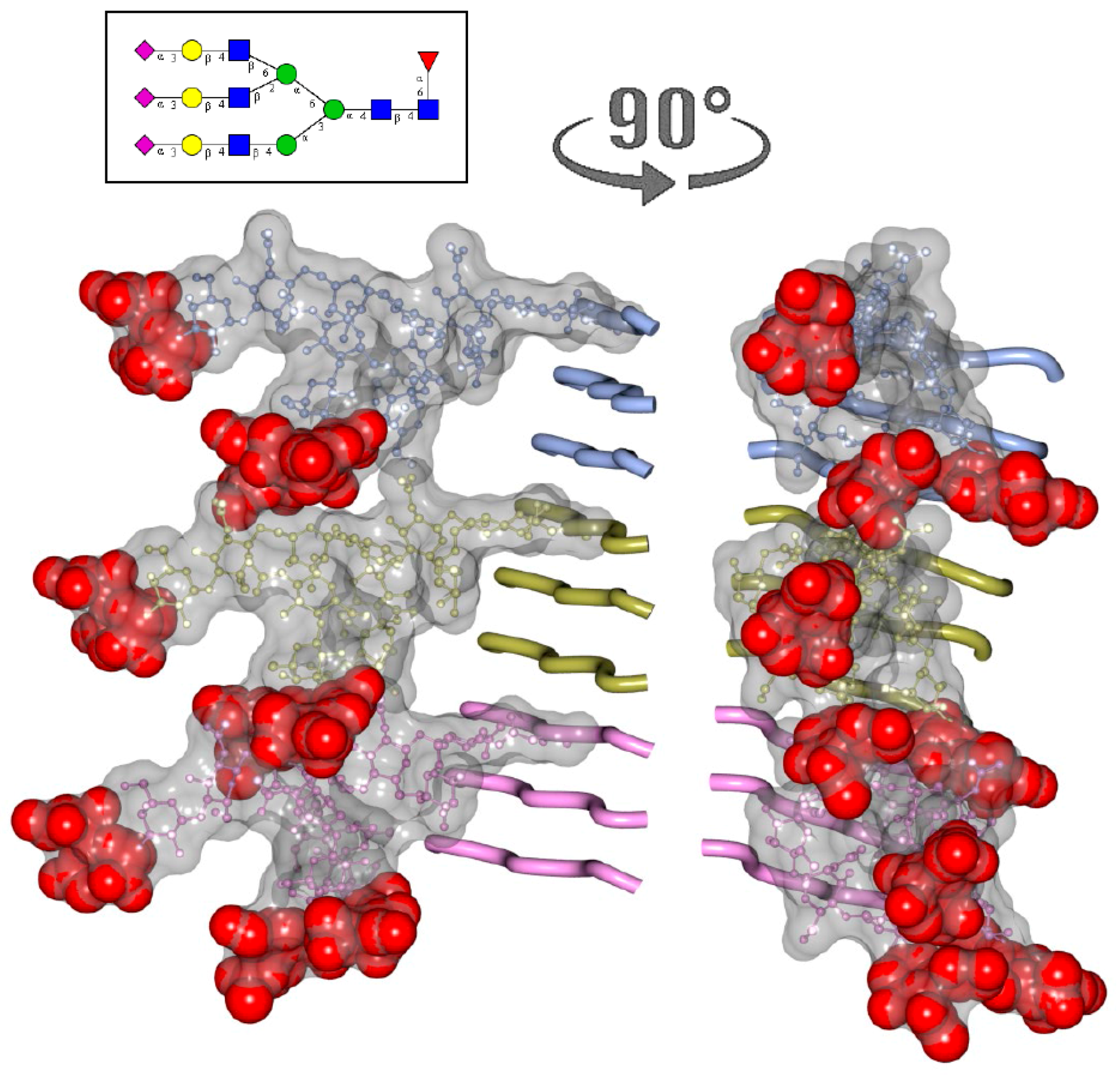
2. N-glycans Are Exposed on a Surface of PrPSc
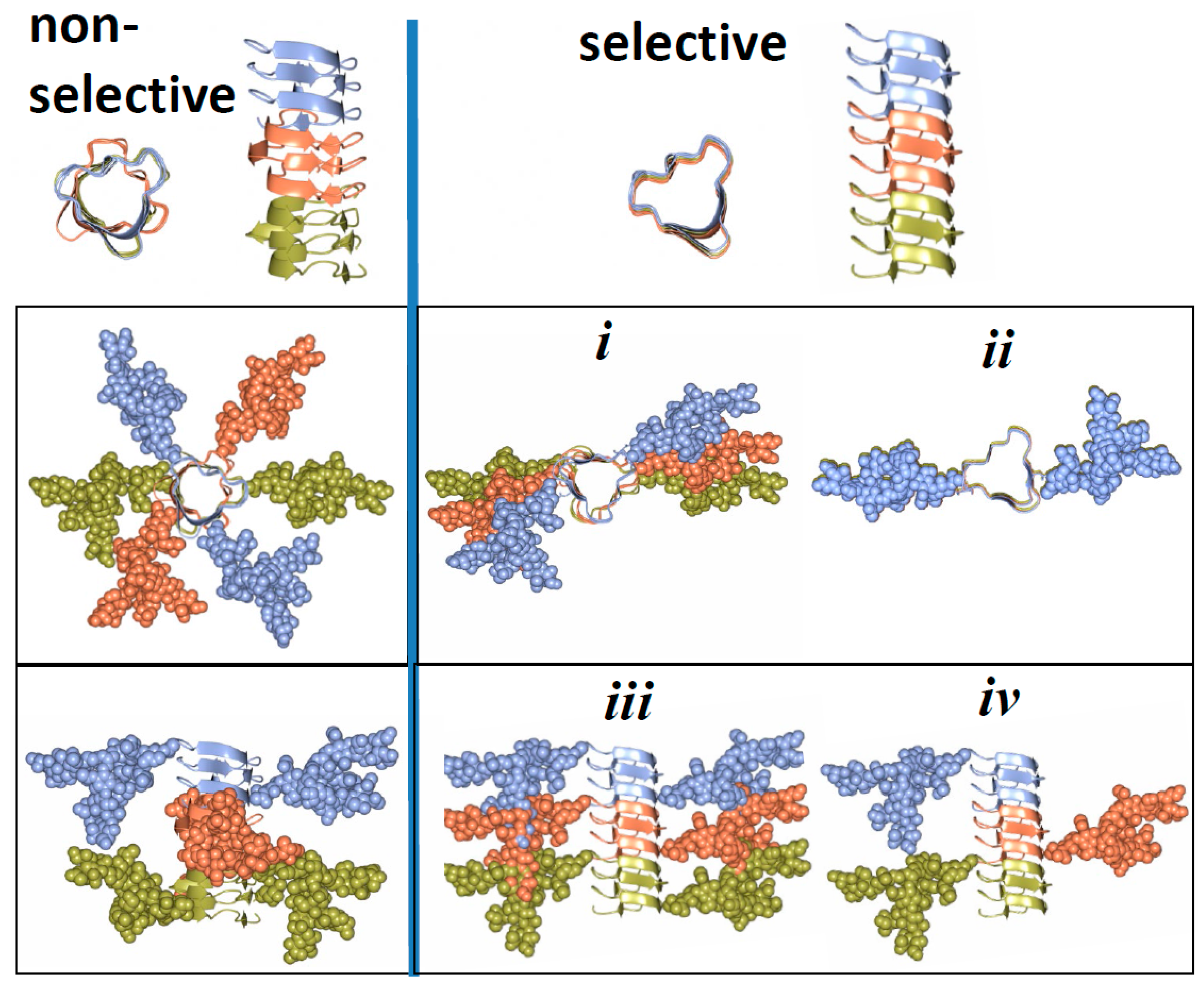
3. Structural Constraints Imposed by N-glycans
4. Two Alternative Views on Involvement of N-glycans
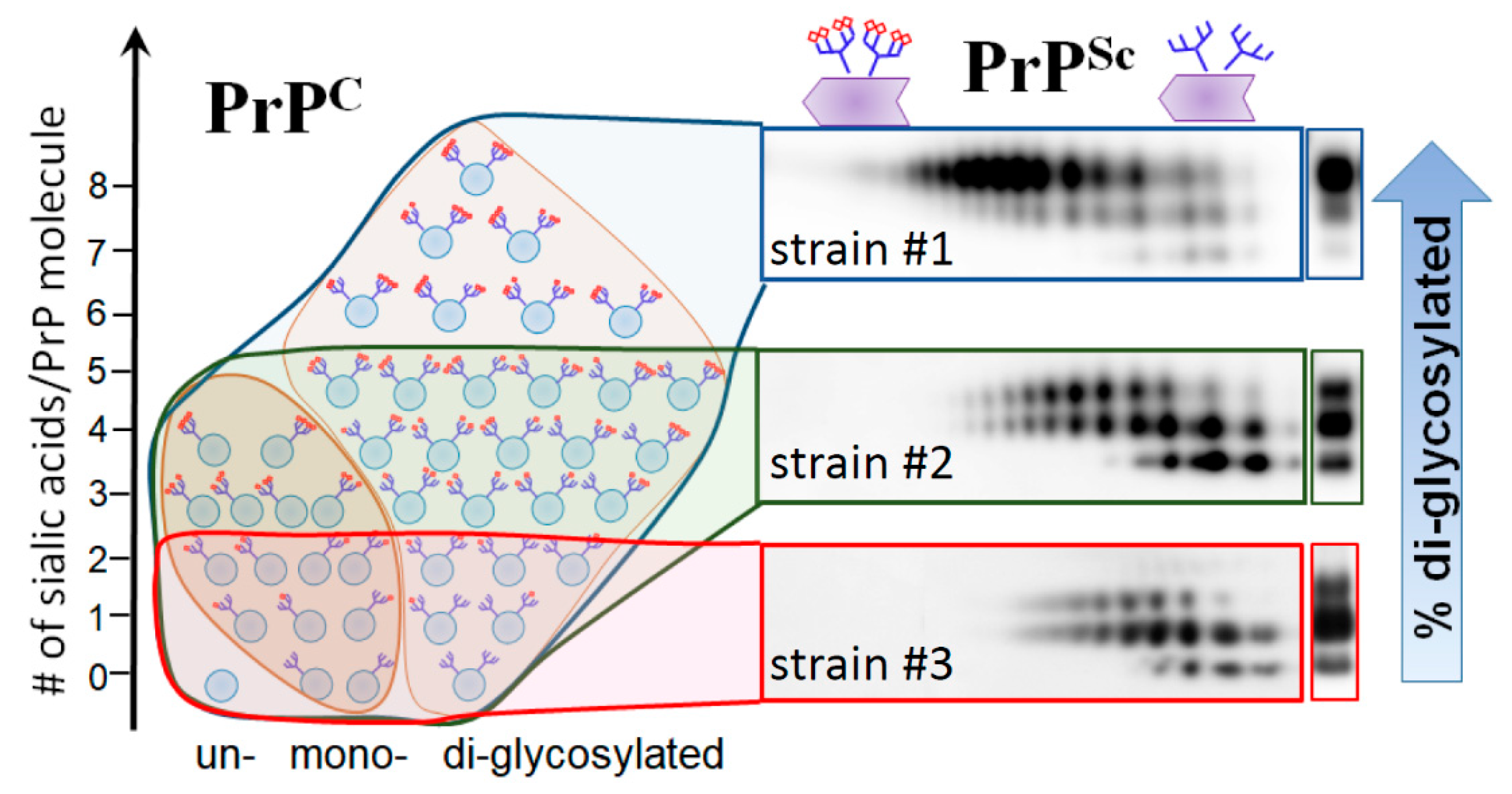
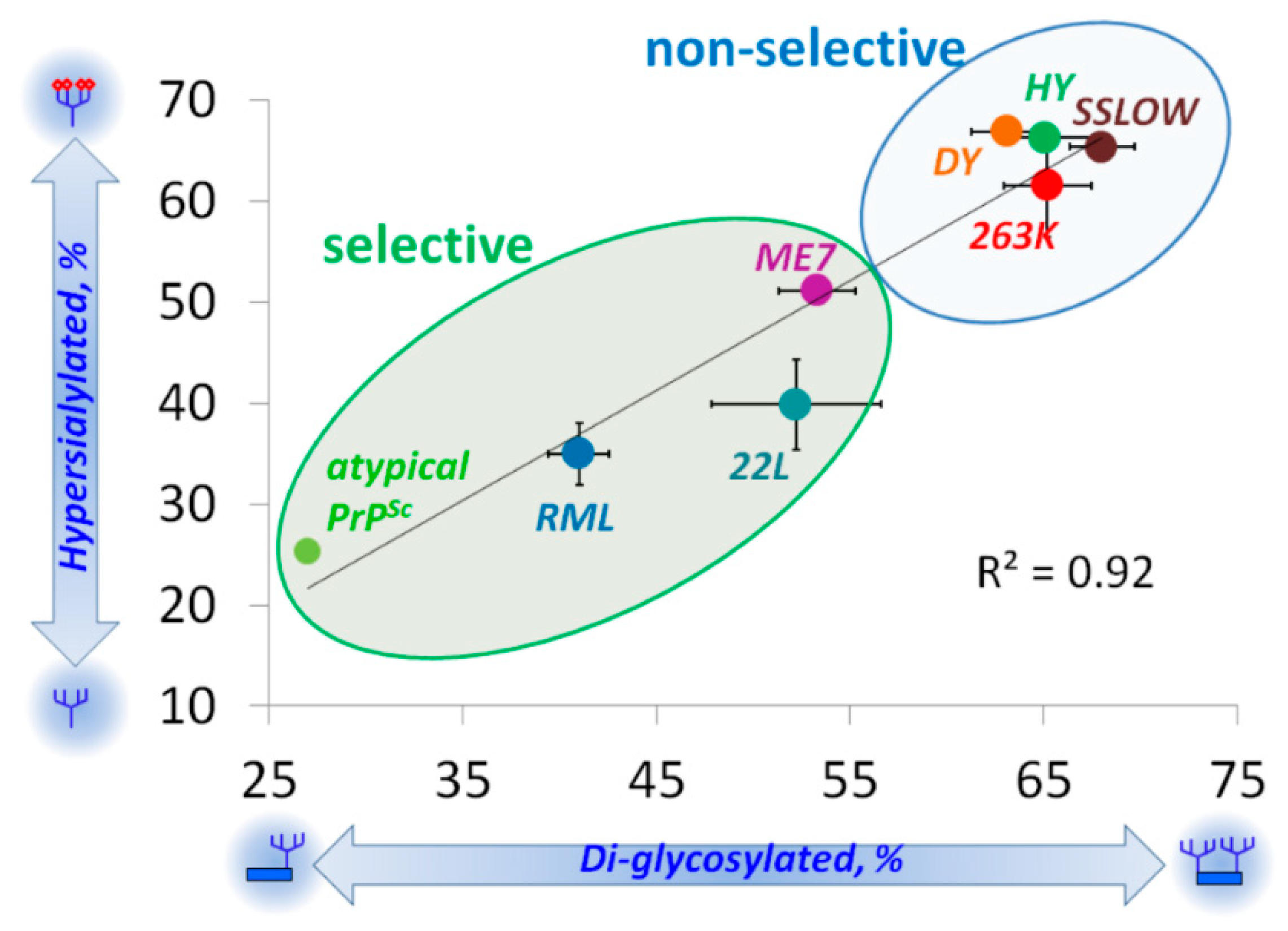
5. Relationship between Strain-Specific Structure and Selectivity for PrPC Sialoglycoforms
6. Role of N-glycans in Maintaining High Fidelity of Prion Replication
7. Role of Sialic Acid Residues and Electrostatic Repulsions in Prion Replication/Strain-Specific Properties
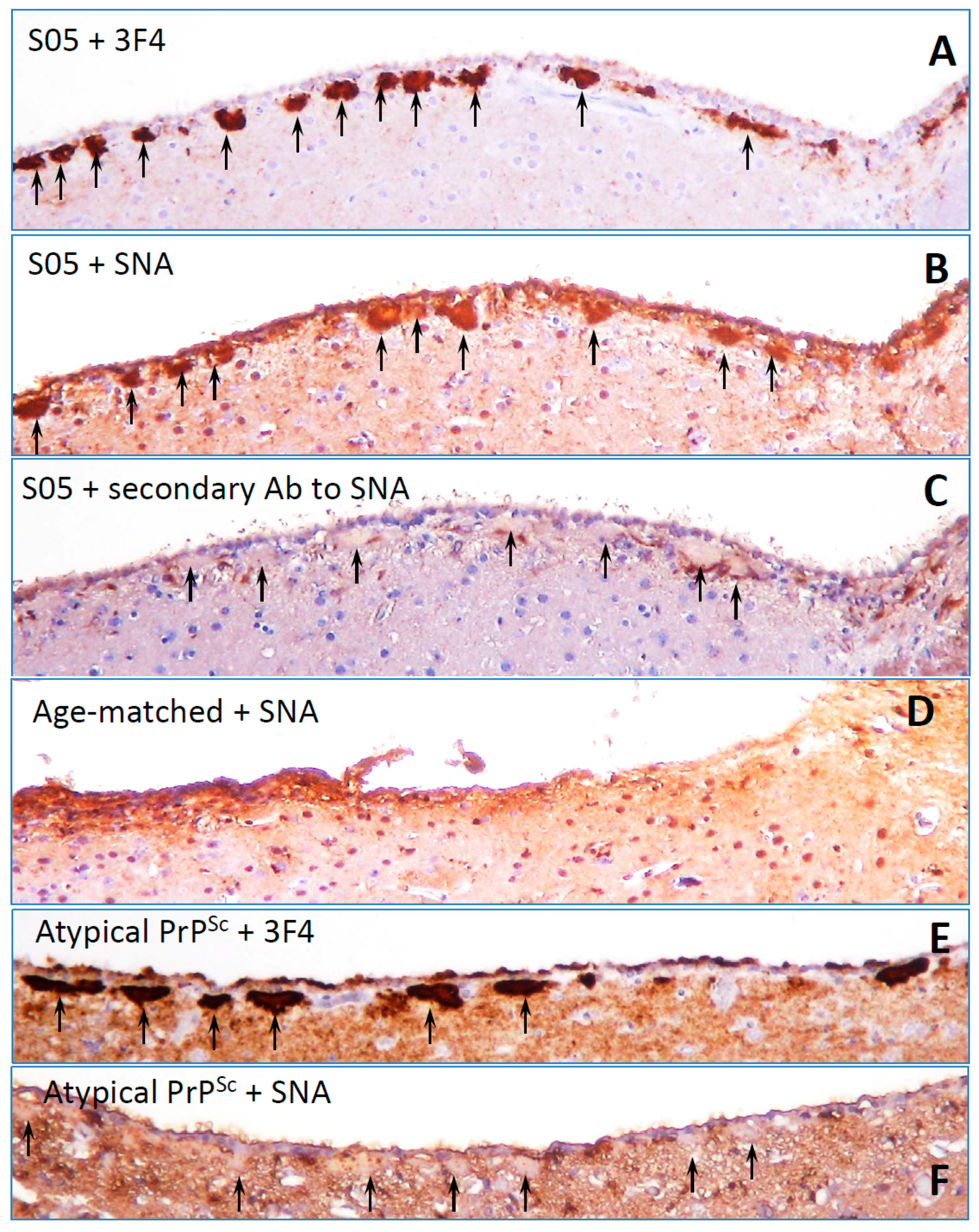
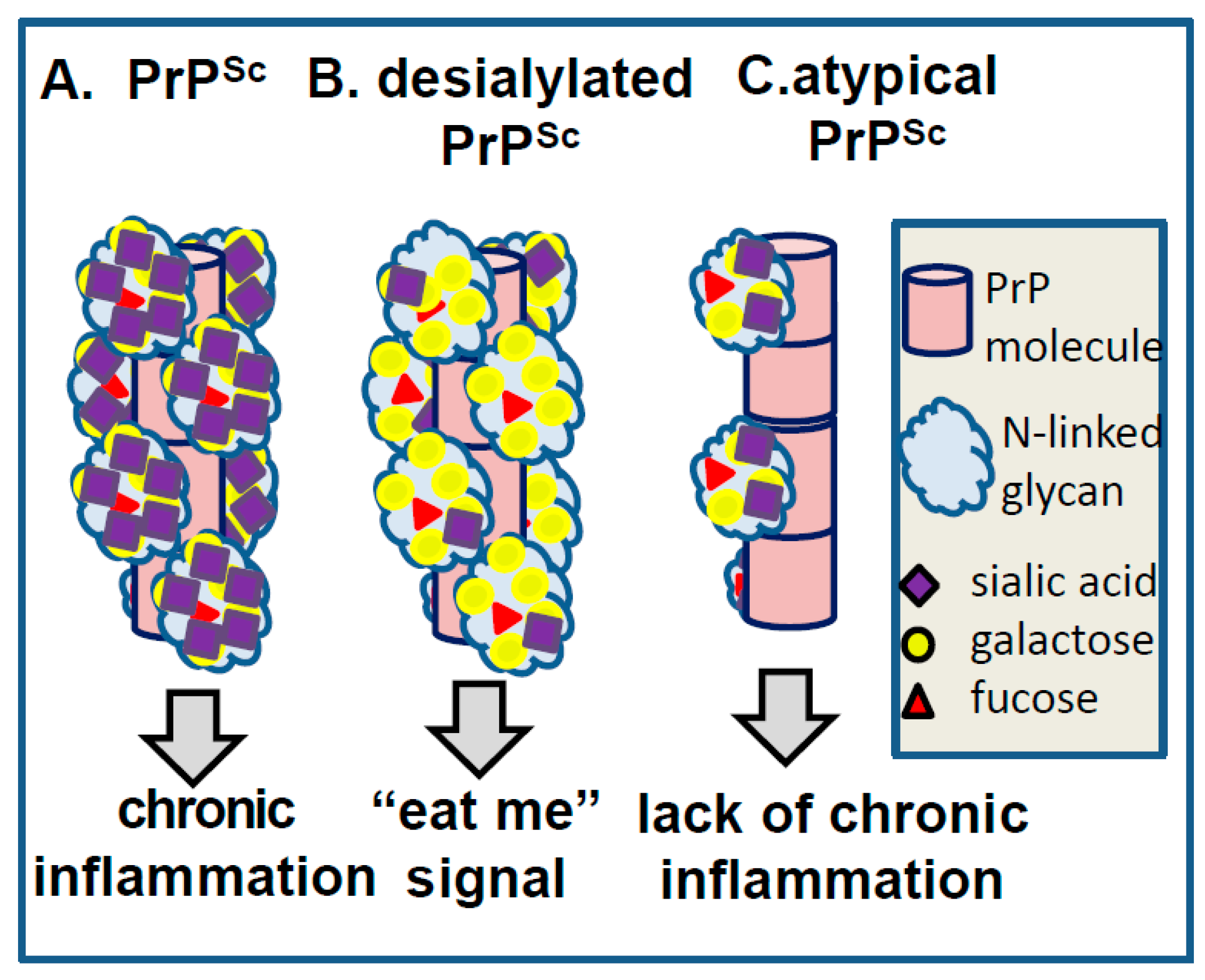
8. Surface Carbohydrate Epitopes of PrPSc as Molecular Cues for CNS
9. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Legname, G.; Baskakov, I.V.; Nguyen, H.O.B.; Riesner, D.; Cohen, F.E.; DeArmond, S.J.; Prusiner, S.B. Synthetic mammalian prions. Science 2004, 305, 673–676. [Google Scholar] [CrossRef] [PubMed]
- Cohen, F.E.; Prusiner, S.B. Pathologic conformations of prion proteins. Annu. Rev. Biochem. 1998, 67, 793–819. [Google Scholar] [CrossRef] [PubMed]
- Collinge, J.; Clarke, A.R. A General Model of Prion Strains and Their Pathogenicity. Science 2007, 318, 930–936. [Google Scholar] [CrossRef] [PubMed]
- Bessen, R.A.; Marsh, R.F. Identification of two biologically distinct strains of transmissible mink encephalopathy in hamsters. J. Gen. Virol. 1992, 73, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Peretz, D.; Scott, M.; Groth, D.; Williamson, A.; Burton, D.; Cohen, F.E.; Prusiner, S.B. Strain-specified relative conformational stability of the scrapie prion protein. Protein Sci. 2001, 10, 854–863. [Google Scholar] [CrossRef] [PubMed]
- Safar, J.; Wille, H.; Itri, V.; Groth, D.; Serban, H.; Torchia, M.; Cohen, F.E.; Prusiner, S.B. Eight prion strains have PrP Sc molecules with different conformations. Nat. Med. 1998, 4, 1157–1165. [Google Scholar] [CrossRef] [PubMed]
- Ayers, J.L.; Schutt, C.R.; Shikiya, R.A.; Aguzzi, A.; Kincaid, A.E.; Bartz, J.C. The strain-encoded relationship between PrP replication, stability and processing in neurons is predictive of the incubation period of disease. PLoS Pathog. 2011, 7, e1001317. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Montalban, N.; Makarava, N.; Savtchenko, R.; Baskakov, I.V. Relationship between Conformational Stability and Amplification Efficiency of Prions. Biochemistry 2011, 50, 7933–7940. [Google Scholar] [CrossRef] [PubMed]
- Klimova, N.; Makarava, N.; Baskakov, I.V. The diversity and relationship of prion protein self-replicating states. Virus Res. 2015, 207, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Caughey, B.; Raymond, G.J.; Bessen, R.A. Strain-dependent differences in b-sheet conformations of abnormal prion protein. J. Biol. Chem. 1998, 273, 32230–32235. [Google Scholar] [CrossRef] [PubMed]
- Spassov, S.; Beekes, M.; Naumann, D. Structural differences between TSEs strains investigated by FT-IR spectroscopy. Biochim. Biophys. Acta 2006, 1760, 1138–1149. [Google Scholar] [CrossRef] [PubMed]
- Thomzig, A.; Spassov, S.; Friedrich, M.; Naumann, D.; Beekes, M. Discriminating Scrapie and Bovine Spongiform Encephalopathy Isolates by Infrared Spectroscopy of Pathological Prion Protein. J. Biol. Chem. 2004, 279, 33847–33854. [Google Scholar] [CrossRef] [PubMed]
- Stahl, N.; Borchelt, D.R.; Hsiao, K.; Prusiner, S.B. Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell 1987, 51, 229–240. [Google Scholar] [CrossRef]
- Bolton, D.C.; Meyer, R.K.; Prusiner, S.B. Scrapie PrP 27-30 is a sialoglycoprotein. J. Virol. 1985, 53, 596–606. [Google Scholar] [PubMed]
- Turk, E.; Teplow, D.B.; Hood, L.E.; Prusiner, S.B. Purification and properties of the cellular and scrapie hamster prion proteins. Eur. J. Biochem. 1988, 176, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Stahl, N.; Baldwin, M.A.; Teplow, D.B.; Hood, L.; Gibson, B.W.; Burlingame, A.L.; Prusiner, S.B. Structural studies of the scrapie prion protein using mass spectrometry and amino acid sequencing. Biochemistry 1993, 32, 1991–2002. [Google Scholar] [CrossRef] [PubMed]
- Stahl, N.; Baldwin, M.A.; Hecker, R.; Pan, K.M.; Burlingame, A.L.; Prusiner, S.B. Glycosylinositol phospholipid anchors of the scrapie and cellular prion proteins contain sialic acid. Biochemistry 1992, 31, 5043–5053. [Google Scholar] [CrossRef] [PubMed]
- Rudd, P.M.; Endo, T.; Colominas, C.; Groth, D.; Wheeler, S.F.; Harvey, D.J.; Wormald, M.R.; Serban, H.; Prusiner, S.B.; Kobata, A.; et al. Glycosylation differences between the normal and pathogenic prion protein isoforms. Proc. Natl. Acad. Sci. USA 1999, 96, 13044–13049. [Google Scholar] [CrossRef] [PubMed]
- Endo, T.; Groth, D.; Prusiner, S.B.; Kobata, A. Diversity of oligosaccharide structures linked to asparagines of the scrapie prion protein. Biochemistry 1989, 28, 8380–8388. [Google Scholar] [CrossRef] [PubMed]
- Stimson, E.; Hope, J.; Chong, A.; Burlingame, A.L. Site-specific characterization of the N-linked glycans of murine prion protein by high-performance liquid chromatography/electrospray mass spectrometry and exoglycosidase digestions. Biochemistry 1999, 38, 4885–4895. [Google Scholar] [CrossRef] [PubMed]
- Katorcha, E.; Makarava, N.; Savtchenko, R.; D’Azzo, A.; Baskakov, I.V. Sialylation of prion protein controls the rate of prion amplification, the cross-species barrier, the ratio of PrPSc glycoform and prion infectivity. PLoS Pathog. 2014, 10, e1004366. [Google Scholar] [CrossRef] [PubMed]
- Katorcha, E.; Klimova, N.; Makarava, N.; Savtchenko, R.; Pan, X.; Annunziata, I.; Takahashi, K.; Miyagi, T.; Pshezhetsky, A.V.; d’Azzo, A.; et al. Knocking out of cellular neuraminidases Neu1, Neu3 or Neu4 does not affect sialylation status of the prion protein. PLoS ONE 2015, 10, e0143218. [Google Scholar]
- Wille, H.; Michelitsch, M.D.; Guenebaut, V.; Supattapone, S.; Serban, A.; Cohen, F.E.; Agard, D.A.; Prusiner, S.B. Structural studies of the scrapie prion protein by electron crystallography. Proc. Acad. Natl. Sci. USA 2002, 99, 3563–3568. [Google Scholar] [CrossRef] [PubMed]
- Govaerts, C.; Wille, H.; Prusiner, S.B.; Cohen, F.E. Evidance for assembly of prions with left-handed b-helices into trimers. Proc. Acad. Natl. Sci. USA 2004, 101, 8342–8347. [Google Scholar] [CrossRef] [PubMed]
- Requena, J.R.; Wille, H. The Structure of the infectious prion protein: Experimental data and molecular models. Prion 2014, 8, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Makarava, N.; Kovacs, G.G.; Savtchenko, R.; Alexeeva, I.; Ostapchenko, V.G.; Budka, H.; Rohwer, R.G.; Baskakov, I.V. A New Mechanism for Transmissible Prion Diseases. J. Neurosci. 2012, 32, 7345–7355. [Google Scholar] [CrossRef] [PubMed]
- Makarava, N.; Savtchenko, R.; Baskakov, I.V. Two alternative pathways for generating transmissible prion disease de novo. Acta Neuropathol. Commun. 2015, 3, 69. [Google Scholar] [CrossRef] [PubMed]
- Katorcha, E.; Baskakov, I.V. Analyses of N-linked glycans of PrPSc revealed predominantly 2,6-linked sialic acid residues. FEBS J. 2017, 284, 3727–3738. [Google Scholar] [CrossRef] [PubMed]
- Makarava, N.; Kovacs, G.G.; Bocharova, O.V.; Savtchenko, R.; Alexeeva, I.; Budka, H.; Rohwer, R.G.; Baskakov, I.V. Recombinant prion protein induces a new transmissible prion disease in wild type animals. Acta Neuropathol. 2010, 119, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Tycko, R.; Savtchenko, R.; Ostapchenko, V.G.; Makarava, N.; Baskakov, I.V. The a-Helical C-Terminal Domain of Full-Length Recombinant PrP Converts to an In-Register Parallel á-Sheet Structure in PrP Fibrils: Evidence from Solid State Nuclear Magnetic Resonance. Biochemistry 2010, 49, 9488–9497. [Google Scholar] [CrossRef] [PubMed]
- Cobb, N.J.; Sonnichsen, F.D.; McHaourab, H.; Surewicz, W. Molecular architecture of human prion protein amyloid: A parallel, in-register b-structure. Proc. Acad. Natl. Sci. USA 2007, 104, 18946–18951. [Google Scholar] [CrossRef] [PubMed]
- Groveman, B.R.; Dolan, M.A.; Taubner, L.M.; Kraus, A.; Wickner, R.B.; Caughey, B. Parallel in-register intermolecular β-sheet architectures for prion-seeded prion protein (PrP) amyloids. J. Biol. Chem. 2014, 289, 24129–24142. [Google Scholar] [CrossRef] [PubMed]
- Varquez-Fernandez, E.; Vos, M.R.; Afanasyev, P.; Cebey, L.; Sevillano, A.M.; Vidal, E.; Rosa, I.; Renault, L.; Ramos, A.; Peters, P.J.; et al. The Structural Architecture of an Infectious Mammalian Prion Using Electron Cryomicroscopy. PLoS Pathog. 2016, 12, e1005835. [Google Scholar]
- Baskakov, I.V.; Katorcha, E. Multifaceted role of sialylation in prion diseases. Front. Neurosci. 2016, 10, 358. [Google Scholar] [CrossRef] [PubMed]
- Lawson, V.A.; Collins, S.J.; Masters, C.L.; Hill, A.F. Prion protein glycosylation. J. Neurochem. 2005, 93, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Katorcha, E.; Makarava, N.; Savtchenko, R.; Baskakov, I.V. Sialylation of the prion protein glycans controls prion replication rate and glycoform ratio. Sci. Rep. 2015, 5, 16912. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Makarava, N.; Katorcha, E.; Savtchenko, R.; Brossmer, R.; Baskakov, I.V. Post-conversion sialylation of prions in lymphoid tissues. Proc. Acad. Natl. Sci. USA 2015, 112, E6654–E6662. [Google Scholar] [CrossRef] [PubMed]
- Katorcha, I.V.; Katorcha, E. Analysis of Covalent Modifications of Amyloidogenic Proteins Using Two-Dimensional Electrophoresis: Prion Protein and Its Sialylation. Methods Mol. Biol. 2018, 1779, 241–255. [Google Scholar] [PubMed]
- Makarava, N.; Kovacs, G.G.; Savtchenko, R.; Alexeeva, I.; Budka, H.; Rohwer, R.G.; Baskakov, I.V. Genesis of mammalian prions: From non-infectious amyloid fibrils to a transmissible prion disease. PLoS Pathog. 2011, 7, e1002419. [Google Scholar] [CrossRef] [PubMed]
- Nishina, K.; Deleault, N.R.; Mahal, S.; Baskakov, I.; Luhrs, T.; Riek, R.; Supattapone, S. The Stoichiometry of Host PrPC Glycoforms Modulates the Efficiency of PrPSc formation in vitro. Biochemistry 2006, 45, 14129–14139. [Google Scholar] [CrossRef] [PubMed]
- Piro, J.R.; Harris, B.T.; Nishina, K.; Soto, C.; Morales, R.; Rees, J.R.; Supattapone, S. Prion Protein Glycosylation Is Not Requiered for Strain-Specific Neurotropism. J. Virol. 2009, 83, 5321–5328. [Google Scholar] [CrossRef] [PubMed]
- Cancellotti, E.; Mahal, S.P.; Somerville, R.; Diack, A.; Brown, D.; Piccardo, P.; Weissmann, C.; Manson, J.C. Post-translational changes to PrP alter transmissible spongiform encephalopathy strain properties. EMBO J. 2013, 32, 756–769. [Google Scholar] [CrossRef] [PubMed]
- Wiseman, F.K.; Cancellotti, E.; Piccardo, P.; Iremonger, K.; Boyle, A.; Brown, D.; Ironside, J.W.; Manson, J.C.; Diack, A.B. The glycosylation status of PrPC is a key factor in determining transmissible spongiform encephalopathy transmission between species. J. Virol. 2015, 89, 4738–4747. [Google Scholar] [CrossRef] [PubMed]
- Stura, E.A.; Muller, B.H.; Bossus, M.; Michel, S.; Jolivet-Reynaud, C.; Ducancel, F. Crystal structure of human prostate-specific antigen in a sandwich antibody complex. J. Mol. Biol. 2011, 414, 530–544. [Google Scholar] [CrossRef] [PubMed]
- Rabinovich, G.A.; Croci, D.O. Regulatori Circuits Mediated by Lectin-Glycan Interaction in Autoimmunity and Cancer. Immunity 2012, 36, 322–335. [Google Scholar] [CrossRef] [PubMed]
- Linnartz, B.; Bodea, L.-G.; Neumann, H. Microglia carbohydrate-binding receptors for neural repair. Cell Tissue Res. 2012, 349, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Varki, A. Sialic acids in human health and disease. Trends Mol. Med. 2008, 14, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.C.; Neher, J.J. Microglial phagocytosis of live neurons. Nat. Rev. Neurosci. 2014, 15, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Aminoff, D.; Bruegge, W.F.; Bell, W.C.; Sarpolis, K.; Williams, R. Role of sialic acid in survival of erythrocytes in the circulation: Interaction of neuraminidase-treated and untreated erythrocytes with spleen and liver at the cellular level. Proc. Acad. Natl. Sci. USA 1977, 74, 1521–1524. [Google Scholar] [CrossRef]
- Jansen, A.J.G.; Josefsson, E.C.; Rumjantseva, V.; Liu, Q.P.; Falet, H.; Bergmeier, W.; Cifuni, S.; Sackstein, R.; von Andrian, U.H.; Wagner, D.D.; et al. Desialylation accelerates platelet clearance after refrigeration and initiates GPIba metalloproteinase-mediated cleavage in mice. Blood 2012, 119, 1263–1273. [Google Scholar] [CrossRef] [PubMed]
- Linnartz, B.; Kopatz, J.; Tenner, A.J.; Neumann, H. Sialic acid on the neuronal glycocalyx prevents complement C1 binding and complement receptor-3-mediated removal by microglia. J. Neurosci. 2012, 32, 946–952. [Google Scholar] [CrossRef] [PubMed]
- Linnartz-Gerlach, B.; Mathews, M.; Neumann, H. Sensing the neuronal glycocalyx by glial sialic acid binding immunoglobulin-like lectins. Neuroscience 2014, 275, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Linnartz-Gerlach, B.; Schuy, C.; Shahraz, A.; Tenner, A.J.; Neumann, H. Sialylation of neurites inhibits complement-mediated macrophage removal in a human macrophage-neuron Co-Culture System. Glia 2016, 64, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Katorcha, E.; Daus, M.L.; Gonzalez-Montalban, N.; Makarava, N.; Lasch, P.; Beekes, M.; Baskakov, I.V. Reversible off and on switching of prion infectivity via removing and reinstalling prion sialylation. Sci. Rep. 2016, 6, 33119. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Katorcha, E.; Daus, M.L.; Lasch, P.; Beekes, M.; Baskakov, I.V. Sialylation controls prion fate in vivo. J. Biol. Chem. 2017, 292, 2359–2368. [Google Scholar] [CrossRef] [PubMed]
- Castro-Seoane, R.; Hummerich, H.; Sweeting, T.; Tattum, M.H.; Linehan, J.M.; Fernandez de Marco, M.; Brandner, S.; Collinge, J.; Klohn, P.C. Plasmacytoid dendritic cells sequester high prion titres at early stages of prion infection. PLoS Pathog. 2012, 8, e1002538. [Google Scholar] [CrossRef] [PubMed]
- Michel, B.; Meyerett-Reid, C.; Johnson, T.; Ferguson, A.; Wyckoff, C.; Pulford, B.; Bender, H.; Avery, A.; Telling, G.; Dow, S.; et al. Incunabular Imminological Events in Prion Trafficing. Sci. Rep. 2012, 2, 440. [Google Scholar] [CrossRef] [PubMed]
- McCulloch, L.; Brown, K.L.; Bradford, B.M.; Hopkins, J.; Bailey, M.; Rajewsky, K.; Manson, J.C.; Mabbott, N.A. Follicular dendritic cell-specific prion protein (PrP) expression alone is sufficient to sustain prion infection in the spleen. PLoS Pathog. 2011, 7, e1002402. [Google Scholar] [CrossRef] [PubMed]
- Kujala, P.; Raymond, C.R.; Romeijn, M.; Godsave, S.F.; van Kasteren, S.I.; Wille, H.; Prusiner, S.B.; Mabbott, N.A.; Peters, P.J. Prion Uptake in the Gut: Identification of the First Uptake and Replication Sites. PLoS Pathog. 2011, 7, e1002449. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.L.; Stewart, K.; Ritchie, D.L.; Mabbott, N.A.; Williams, A.; Fraser, H.; Morrison, W.I.; Bruce, M.E. Scrapie replication in lyphoid tissues depends on prion protein-expressing follicular dendritic cells. Nat. Med. 1999, 5, 1308–1312. [Google Scholar] [CrossRef] [PubMed]
- Montrasio, F.; Frigg, R.; Glatzel, M.; Klein, M.A.; Mackay, F.; Aguzzi, A.; Weissmann, C. Impaired prion replication in spleens of mice lacking functional follicular dendritic cells. Science 2000, 288, 1257–1259. [Google Scholar] [CrossRef] [PubMed]
- Mabbott, N.A.; Mackay, F.; Minns, F.; Bruce, M.E. Temporal inactivation of follicular dendritic cells delays neuroinvasion of scrapie. Nat. Med. 2000, 6, 719–720. [Google Scholar] [CrossRef] [PubMed]
- Prinz, M.; Heikenwalder, M.; Junt, T.; Schwarz, P.; Glatzel, M.; Heppner, F.L.; Fu, Y.X.; Lipp, M.; Aguzzi, A. Positioning of follicular dendritic cells within the spleen controls prion neuroinvasion. Nature 2003, 425, 957–962. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Katorcha, E.; Makarava, N.; Barrett, J.P.; Loane, D.J.; Baskakov, I.V. Inflammatory response of microglia to prions is controlled by sialylation of PrPSc. Sci. Rep. 2018, 8, e11326. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.G.; Makarava, N.; Savtchenko, R.; Baskakov, I.V. Atypical and classical forms of the disease-associated state of the prion protein exhibit distinct neuronal tropism, deposition patterns, and lesion profiles. Am. J. Pathol. 2013, 183, 1539–1547. [Google Scholar] [CrossRef] [PubMed]
- Makarava, N.; Savtchenko, R.; Alexeeva, I.; Rohwer, R.G.; Baskakov, I.V. New Molecular Insight into Mechanism of Evolution of Mammalian Synthetic Prions. Am. J. Pathol. 2016, 186, 1006–1014. [Google Scholar] [CrossRef] [PubMed]
- Makarava, N.; Savtchenko, R.; Baskakov, I.V. Selective amplification of classical and atypical prions using modified protein misfolding cyclic amplification. J. Biol. Chem. 2013, 288, 33–41. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baskakov, I.V.; Katorcha, E.; Makarava, N. Prion Strain-Specific Structure and Pathology: A View from the Perspective of Glycobiology. Viruses 2018, 10, 723. https://doi.org/10.3390/v10120723
Baskakov IV, Katorcha E, Makarava N. Prion Strain-Specific Structure and Pathology: A View from the Perspective of Glycobiology. Viruses. 2018; 10(12):723. https://doi.org/10.3390/v10120723
Chicago/Turabian StyleBaskakov, Ilia V., Elizaveta Katorcha, and Natallia Makarava. 2018. "Prion Strain-Specific Structure and Pathology: A View from the Perspective of Glycobiology" Viruses 10, no. 12: 723. https://doi.org/10.3390/v10120723
APA StyleBaskakov, I. V., Katorcha, E., & Makarava, N. (2018). Prion Strain-Specific Structure and Pathology: A View from the Perspective of Glycobiology. Viruses, 10(12), 723. https://doi.org/10.3390/v10120723