The N-Terminus of the HIV-1 p6 Gag Protein Regulates Susceptibility to Degradation by IDE

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Transfection
2.2. HIV-1 Isolates and Expression Plasmids
2.3. Virus and VLP Isolation
2.4. Infection of Cells
2.5. SDS PAGE and Western Blotting
2.6. In Vitro Degradation Assay
2.7. Peptide Synthesis and Mass Spectrometry
2.8. In Silico Analysis
2.9. Ethical Statement
3. Results
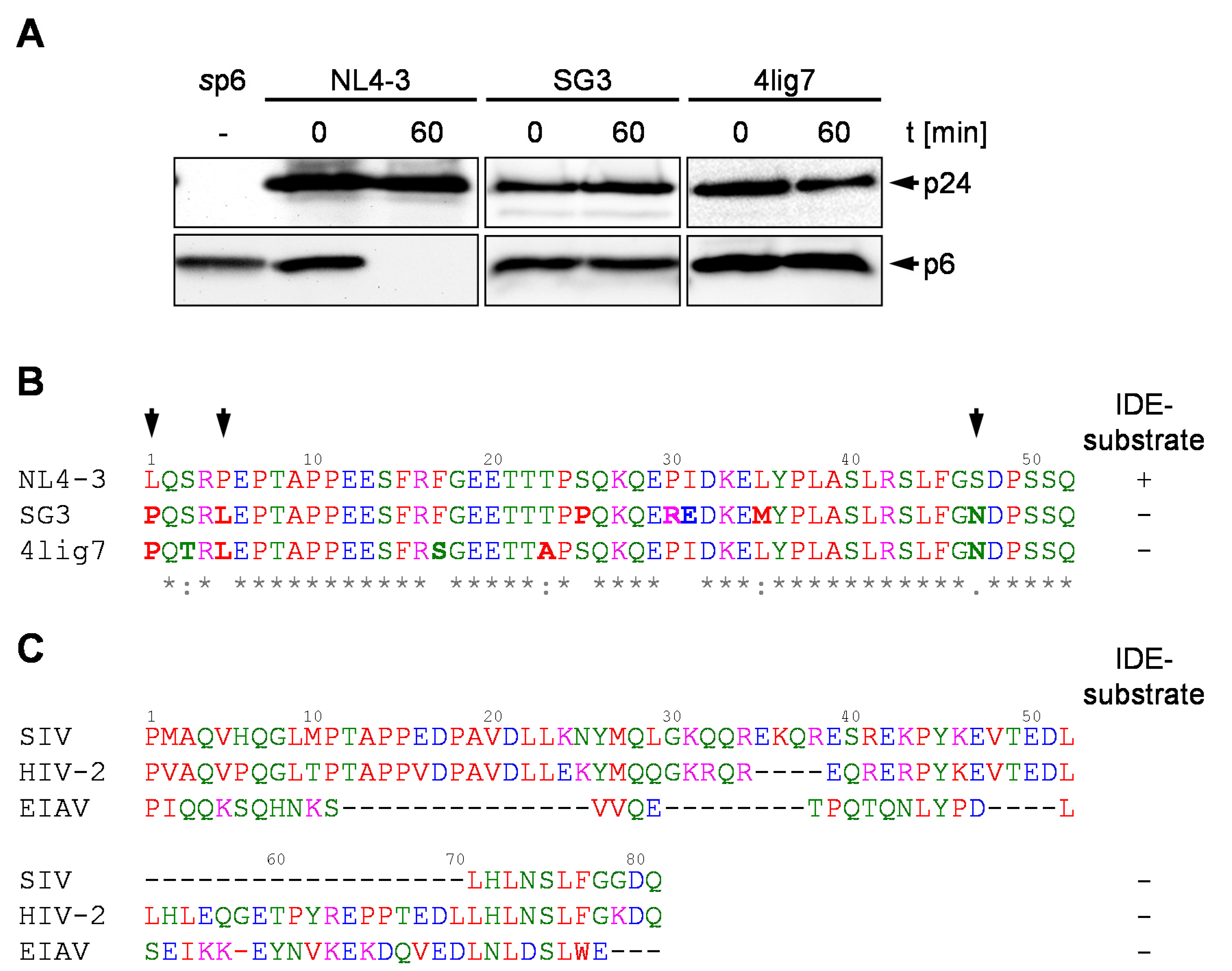
3.1. p6 Derived from the HIV-1 Isolates SG3 and 4lig7 is not Degraded by IDE
3.2. Proline at the N-Terminus of p6 Prevents IDE-Mediated Degradation
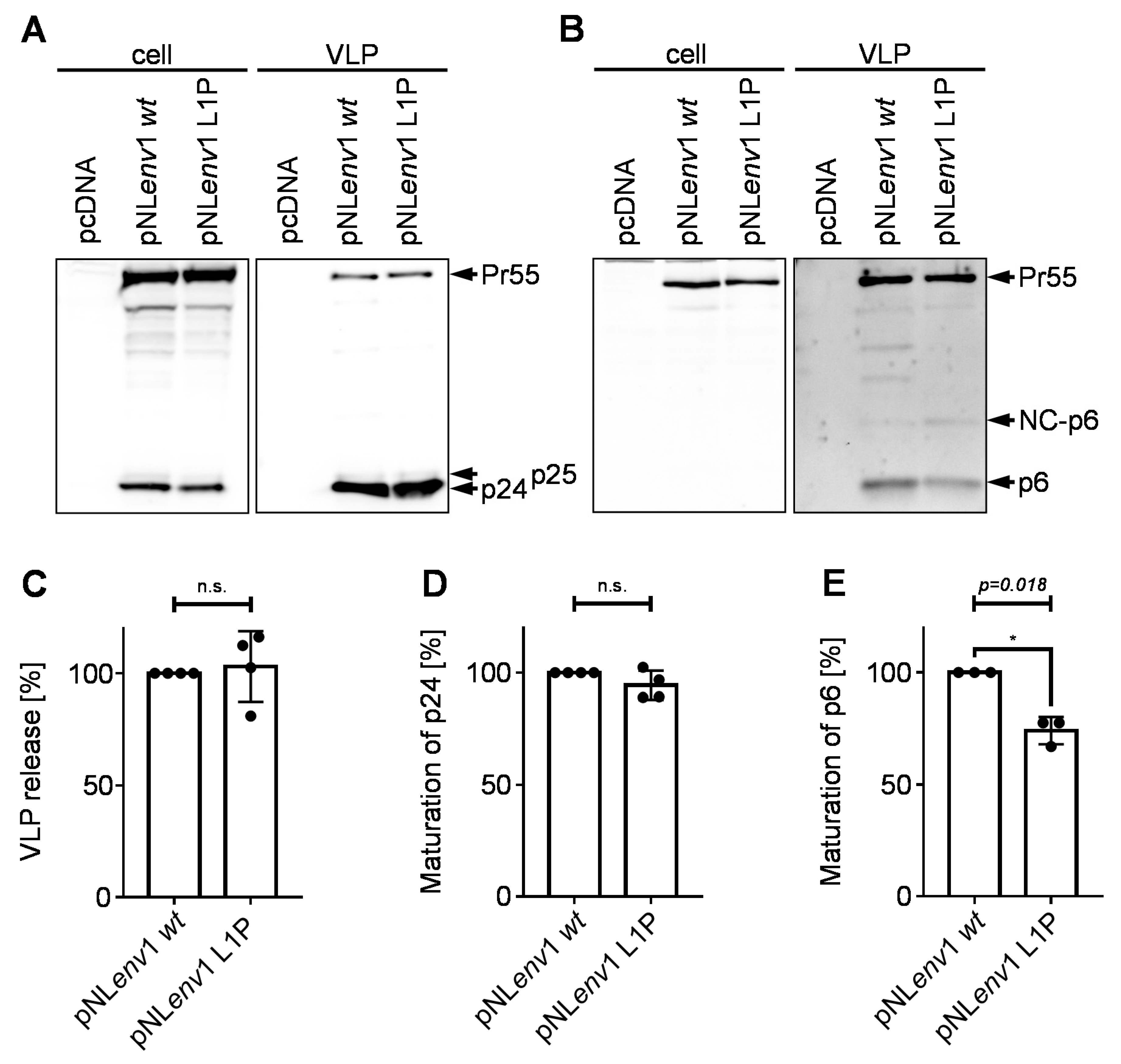
3.3. Effect of the pNLenv1 L1P Mutation on Late Steps of the HIV-1 Replication Cycle
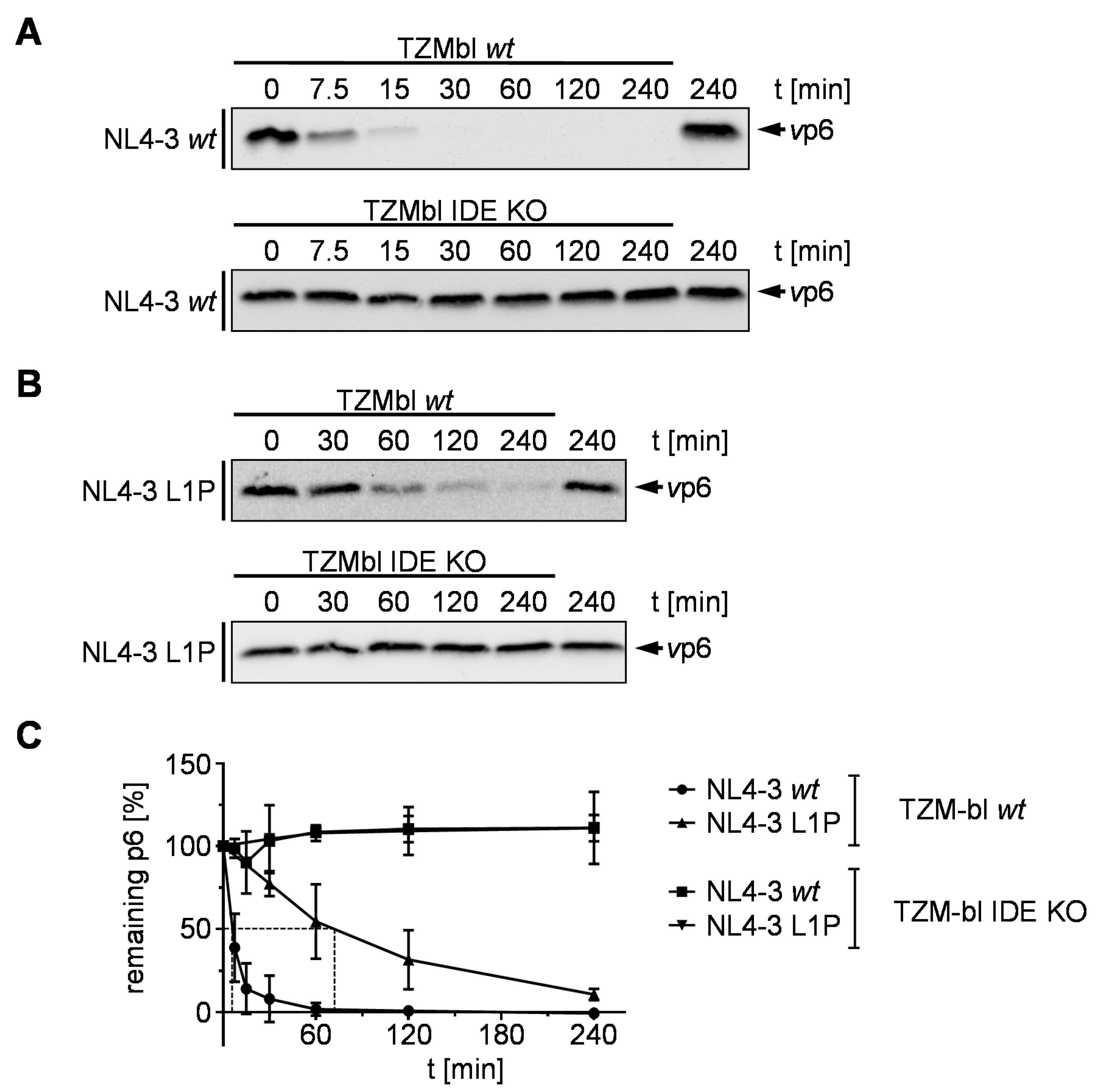
3.4. Stabilization by an N-Terminal Proline Renders HIV-1 Replication Resistant to Treatment with an IDE Inhibitor
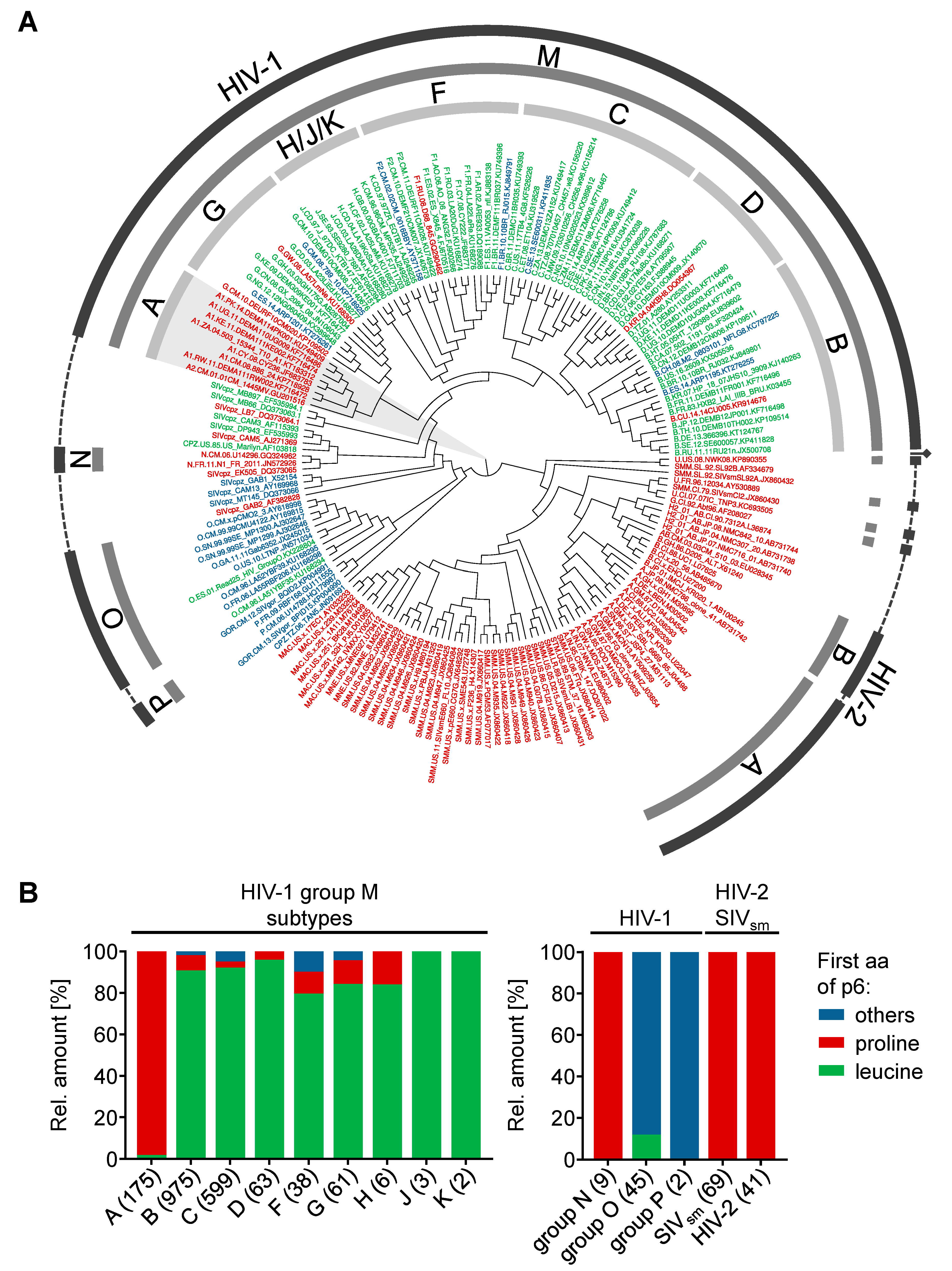
3.5. Phylogenetic Background of the N-Terminus of p6
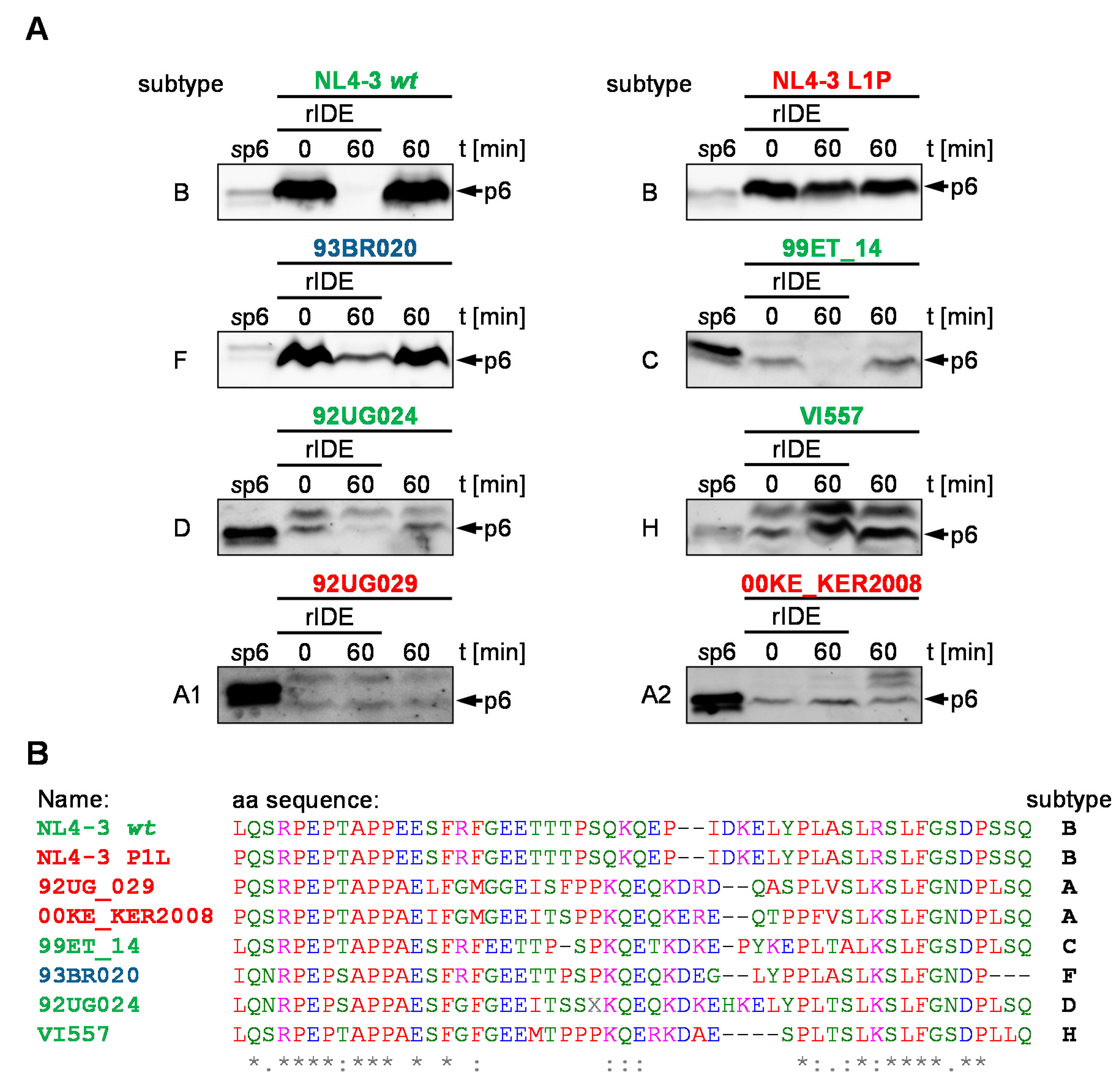
3.6. IDE-Susceptibility of p6 Derived from Various Subtypes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Veronese, F.D.; Rahman, R.; Copeland, T.D.; Oroszlan, S.; Gallo, R.C.; Sarngadharan, M.G. Immunological and chemical analysis of P6, the carboxyl-terminal fragment of HIV P15. AIDS Res. Hum. Retrovir. 1987, 3, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Darke, P.L.; Nutt, R.F.; Brady, S.F.; Garsky, V.M.; Ciccarone, T.M.; Leu, C.T.; Lumma, P.K.; Freidinger, R.M.; Veber, D.F.; Sigal, I.S. HIV-1 protease specificity of peptide cleavage is sufficient for processing of gag and pol polyproteins. Biochem. Biophys. Res. Commun. 1988, 156, 297–303. [Google Scholar] [CrossRef]
- Meek, T.D.; Dayton, B.D.; Metcalf, B.W.; Dreyer, G.B.; Strickler, J.E.; Gorniak, J.G.; Rosenberg, M.; Moore, M.L.; Magaard, V.W.; Debouck, C. Human immunodeficiency virus 1 protease expressed in Escherichia coli behaves as a dimeric aspartic protease. Proc. Natl. Acad. Sci. USA 1989, 86, 1841–1845. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, A.H.; Manchester, M.; Swanstrom, R. The activity of the protease of human immunodeficiency virus type 1 is initiated at the membrane of infected cells before the release of viral proteins and is required for release to occur with maximum efficiency. J. Virol. 1994, 68, 6782–6786. [Google Scholar] [PubMed]
- Göttlinger, H.G.; Dorfman, T.; Sodroski, J.G.; Haseltine, W.A. Effect of mutations affecting the p6 gag protein on human immunodeficiency virus particle release. Proc. Natl. Acad. Sci. USA 1991, 88, 3195–3199. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Orenstein, J.M.; Martin, M.A.; Freed, E.O. p6Gag is required for particle production from full-length human immunodeficiency virus type 1 molecular clones expressing protease. J. Virol. 1995, 69, 6810–6818. [Google Scholar] [PubMed]
- Strack, B.; Calistri, A.; Craig, S.; Popova, E.; Göttlinger, H.G. AIP1/ALIX is a binding partner for HIV-1 p6 and EIAV p9 functioning in virus budding. Cell 2003, 114, 689–699. [Google Scholar] [CrossRef]
- Fisher, R.D.; Chung, H.Y.; Zhai, Q.; Robinson, H.; Sundquist, W.I.; Hill, C.P. Structural and Biochemical Studies of ALIX/AIP1 and Its Role in Retrovirus Budding. Cell 2007, 128, 841–852. [Google Scholar] [CrossRef] [PubMed]
- Usami, Y.; Popov, S.; Gottlinger, H.G. Potent Rescue of Human Immunodeficiency Virus Type 1 Late Domain Mutants by ALIX/AIP1 Depends on Its CHMP4 Binding Site. J. Virol. 2007, 81, 6614–6622. [Google Scholar] [CrossRef]
- Carlton, J.G.; Martin-Serrano, J. Parallels between cytokinesis and retroviral budding: A role for the ESCRT machinery. Science 2007, 316, 1908–1912. [Google Scholar] [CrossRef]
- Morita, E.; Sandrin, V.; Chung, H.; Morham, S.G.; Gygi, S.P.; Rodesch, C.K.; Sundquist, W.I. Human ESCRT and ALIX proteins interact with proteins of the midbody and function in cytokinesis. EMBO J. 2007, 26, 4215–4227. [Google Scholar] [CrossRef] [PubMed]
- Wollert, T.; Hurley, J.H. Molecular mechanism of multivesicular body biogenesis by ESCRT complexes. Nature 2010, 464, 864–869. [Google Scholar] [CrossRef] [PubMed]
- Demirov, D.G.; Orenstein, J.M.; Freed, E.O. The Late Domain of Human Immunodeficiency Virus Type 1 p6 Promotes Virus Release in a Cell Type-Dependent Manner The Late Domain of Human Immunodeficiency Virus Type 1 p6 Promotes Virus Release in a Cell Type-Dependent Manner. J. Virol. 2002, 76, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Paxton, W.; Connor, R.I.; Landau, N.R. Incorporation of Vpr into human immunodeficiency virus type 1 virions: Requirement for the p6 region of gag and mutational analysis. J. Virol. 1993, 67, 7229–7237. [Google Scholar] [PubMed]
- Kondo, E.; Göttlinger, H.G. A conserved LXXLF sequence is the major determinant in p6gag required for the incorporation of human immunodeficiency virus type 1 Vpr. J. Virol. 1996, 70, 159–164. [Google Scholar] [PubMed]
- Hahn, F.; Schmalen, A.; Setz, C.; Friedrich, M.; Schlößer, S.; Kölle, J.; Spranger, R.; Rauch, P.; Fraedrich, K.; Reif, T.; et al. Proteolysis of mature HIV-1 p6 Gag protein by the insulin-degrading enzyme (IDE) regulates virus replication in an Env-dependent manner. PLoS ONE 2017, 12, e0174254. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Joachimiak, A.; Rich Rosner, M.; Tang, W.-J. Structures of human insulin-degrading enzyme reveal a new substrate recognition mechanism. Nature 2006, 443, 870–874. [Google Scholar] [CrossRef]
- Hulse, R.E.; Ralat, L.A.; Wei-Jen, T. Chapter 22 Structure, Function, and Regulation of Insulin-Degrading Enzyme. Vitam. Horm. 2009, 80, 635–648. [Google Scholar]
- Adames, N.; Blundell, K.; Ashby, M.N.; Boone, C. Role of yeast insulin-degrading enzyme homologs in propheromone processing and bud site selection. Science 1995, 270, 464–467. [Google Scholar] [CrossRef] [PubMed]
- VanderVere, P.S.; Bennett, T.M.; Oblong, J.E.; Lamppa, G.K. A chloroplast processing enzyme involved in precursor maturation shares a zinc-binding motif with a recently recognized family of metalloendopeptidases. Proc. Natl. Acad. Sci. USA 1995, 92, 7177–7181. [Google Scholar] [CrossRef] [PubMed]
- Finch, P.W.; Wilson, R.E.; Brown, K.; Hickson1, I.D.; Emmerson, P.T. Nucleic Acids Research Complete nucleotide sequence of the Escherichia coli ptr gene encoding protease III. Nucleic Acids Res. 1986, 14, 7695–7703. [Google Scholar] [CrossRef] [PubMed]
- Mirsky, I.A.; Broh-Kahn, R.H. The inactivation of insulin by tissue extracts; the distribution and properties of insulin inactivating extracts. Arch. Biochem. 1949, 20, 1–9. [Google Scholar] [PubMed]
- Bennett, R.G.; Duckworth, W.C.; Hamel, F.G. Degradation of Amylin by Insulin-degrading Enzyme. J. Biol. Chem. 2000, 275, 36621–36625. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Ali, M.A.; Cohen, J.I. Insulin Degrading Enzyme Is a Cellular Receptor Mediating Varicella-Zoster Virus Infection and Cell-to-Cell Spread. Cell 2006, 127, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Sarada, B.; Thiele, D.; Dang, T.; Lee, J.; Safavia, A.; Hersh, L.B.; Cottam, G.L. Anti-CD3 activation of human CD4+T cells increases expression of the intracellular β-endorphin endopeptidase (IDE/γ-EpGE). J. Neuroimmunol. 1998, 85, 59–68. [Google Scholar] [CrossRef]
- Hallé, M.; Tribout-Jover, P.; Lanteigne, A.M.; Boulais, J.; St-Jean, J.R.; Jodoin, R.; Girouard, M.P.; Constantin, F.; Migneault, A.; Renaud, F.; et al. Methods to monitor monocytes-mediated amyloid-beta uptake and phagocytosis in the context of adjuvanted immunotherapies. J. Immunol. Methods 2015, 424, 64–79. [Google Scholar] [CrossRef] [PubMed]
- Huet, T.; Cheynier, R.; Meyerhans, A.; Roelants, G.; Wain-Hobson, S. Genetic organization of a chimpanzee lentivirus related to HIV-1. Nature 1990, 345, 356–359. [Google Scholar] [CrossRef]
- Takehisa, J.; Kraus, M.H.; Ayouba, A.; Bailes, E.; Van Heuverswyn, F.; Decker, J.M.; Li, Y.; Rudicell, R.S.; Learn, G.H.; Neel, C.; et al. Origin and Biology of Simian Immunodeficiency Virus in Wild-Living Western Gorillas. J. Virol. 2009, 83, 1635–1648. [Google Scholar] [CrossRef]
- Vallari, A.; Holzmayer, V.; Harris, B.; Yamaguchi, J.; Ngansop, C.; Makamche, F.; Mbanya, D.; Kaptue, L.; Ndembi, N.; Gurtler, L.; et al. Confirmation of Putative HIV-1 Group P in Cameroon. J. Virol. 2011, 85, 1403–1407. [Google Scholar] [CrossRef]
- Simon, F.; Mauclère, P.; Roques, P.; Loussert-Ajaka, I.; Müller-Trutwin, M.C.; Saragosti, S.; Georges-Courbot, M.C.; Barré-Sinoussi, F.; Brun-VÉZINET, F. Identification of a new human immunodeficiency virus type 1 distinct from group M and group O. Nat. Med. 1998, 4, 1032–1037. [Google Scholar] [CrossRef]
- Keele, B.F.; Van Heuverswyn, F.; Li, Y.; Bailes, E.; Takehisa, J.; Santiago, M.L.; Bibollet-Ruche, F.; Chen, Y.; Wain, L.V.; Liegeois, F.; et al. Chimpanzee reservoirs of pandemic and nonpandemic HIV-1. Science 2006, 313, 523–526. [Google Scholar] [CrossRef] [PubMed]
- Peeters, M.; Gueye, A.; Mboup, S.; Bibollet-Ruche, F.; Ekaza, E.; Mulanga, C.; Ouedrago, R.; Gandji, R.; Mpele, P.; Dibanga, G.; et al. Geographical distribution of HIV-1 group O viruses in Africa. AIDS 1997, 11, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Korber, B.; Gaschen, B.; Yusim, K.; Thakallapally, R.; Kesmir, C.; Detours, V. Evolutionary and immunological implications of contemporary HIV-1 variation. Br. Med. Bull. 2001, 58, 19–42. [Google Scholar] [CrossRef] [PubMed]
- Sabino, E.C.; Shpaer, E.G.; Morgado, M.G.; Korber, B.T.; Diaz, R.S.; Bongertz, V.; Cavalcante, S.; Galvão-Castro, B.; Mullins, J.I.; Mayer, A. Identification of human immunodeficiency virus type 1 envelope genes recombinant between subtypes B and F in two epidemiologically linked individuals from Brazil. J. Virol. 1994, 68, 6340–6346. [Google Scholar] [PubMed]
- Robertson, D.L.; Hahn, B.H.; Sharp, P.M. Recombination in AIDS viruses. J. Mol. Evol. 1995, 40, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Robertson, D.L.; Sharp, P.M.; Mc Cutchan, F.E.; Hahn, B.H. Recombination in HIV-1. Nature 1995, 374, 124–126. [Google Scholar] [CrossRef] [PubMed]
- Verheyen, J.; Litau, E.; Sing, T.; Däumer, M.; Balduin, M.; Oette, M.; Fätkenheuer, G.; Rockstroh, J.K.; Schuldenzucker, U.; Hoffmann, D.; et al. Compensatory mutations at the HIV cleavage sites p7/p1 and p1/p6-gag in therapy-naive and therapy-experienced patients. Antivir. Ther. 2006, 11, 879–887. [Google Scholar] [PubMed]
- Adachi, A.; Gendelman, H.E.; Koenig, S.; Folks, T.; Willey, R.; Rabson, A.; Martin, M.A. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J. Virol. 1986, 59, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Aberham, C.; Kao, S.; Akari, H.; Gorelick, R.; Bour, S.; Strebel, K. Human immunodeficiency virus type 1 Vif protein is packaged into the nucleoprotein complex through an interaction with viral genomic RNA. J. Virol. 2001, 75, 7252–7265. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.K.; Fultz, P.N.; Keddie, E.; Saag, M.S.; Sharp, P.M.; Hahn, B.H.; Shaw, G.M. A Molecular Clone of HIV-1 Tropic and Cytopathic for Human and Chimpanzee Lymphocytes. Virology 1993, 194, 858–864. [Google Scholar] [CrossRef]
- Walter, H.; Schmidt, B.; Korn, K.; Vandamme, A.M.; Harrer, T.; Überla, K. Rapid, phenotypic HIV-1 drug sensitivity assay for protease and reverse transcriptase inhibitors. J. Clin. Virol. 1999, 13, 71–80. [Google Scholar] [CrossRef]
- Willey, R.L.; Smith, D.H.; Lasky, L.A.; Theodore, T.S.; Earl, P.L.; Moss, B.; Capon, D.J.; Martin, M.A. In vitro mutagenesis identifies a region within the envelope gene of the human immunodeficiency virus that is critical for infectivity. J. Virol. 1988, 62, 139–147. [Google Scholar] [PubMed]
- Laemmli, U.K. Nature Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Fossen, T.; Wray, V.; Bruns, K.; Rachmat, J.; Henklein, P.; Tessmer, U.; Maczurek, A.; Klinger, P.; Schubert, U. Solution structure of the human immunodeficiency virus type 1 p6 protein. J. Biol. Chem. 2005, 280, 42515–42527. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Cowley, A.; Uludag, M.; Gur, T.; McWilliam, H.; Squizzato, S.; Park, Y.M.; Buso, N.; Lopez, R. The EMBL-EBI bioinformatics web and programmatic tools framework. Nucleic Acids Res. 2015, 43, W580–W584. [Google Scholar] [CrossRef] [PubMed]
- McWilliam, H.; Li, W.; Uludag, M.; Squizzato, S.; Park, Y.M.; Buso, N.; Cowley, A.P.; Lopez, R. Analysis Tool Web Services from the EMBL-EBI. Nucleic Acids Res. 2013, 41, W597–W600. [Google Scholar] [CrossRef] [PubMed]
- Foley, B.; Leitner, T.; Apetrei, C.; Hahn, B.; Mizrachi, I.; Mullins, J.; Rambaut, A.; Wolinsky, S.; Korber, B. HIV Sequence Compendium 2016; LA-UR 16-25625; Theoretical Biology and Biophysics Group, Los Alamos National Laboratory: Los Alamos, NM, USA, 2016. [Google Scholar]
- Foley, B.; Leitner, T.; Apetrei, C.; Hahn, B.; Mizrachi, I.; Mullins, J.; Rambaut, A.; Wolinsky, S.; Korber, B. HIV Sequence Compendium 2015; LA-UR 15-27742; Theoretical Biology and Biophysics Group, Los Alamos National Laboratory: Los Alamos, NM, USA, 2015. [Google Scholar]
- Liu, K.; Raghavan, S.; Nelesen, S.; Linder, C.R.; Warnow, T. Rapid and accurate large-scale coestimation of sequence alignments and phylogenetic trees. Science 2009, 324, 1561–1564. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Warnow, T.J.; Holder, M.T.; Nelesen, S.M.; Yu, J.; Stamatakis, A.P.; Linder, C.R. SATé-II: Very Fast and Accurate Simultaneous Estimation of Multiple Sequence Alignments and Phylogenetic Trees. Syst. Biol. 2012, 61, 90–106. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Holder, M.T.; Sukumaran, J.; Mirarab, S.; Oaks, J. SATé Version 2.2.7. Available online: http://phylo.bio.ku.edu/software/sate/sate.html (accessed on 7 July 2017).
- Bogaardt, C.; Carvalho, L.; Hill, V.; O’Toole, A.; Rambaut, A.; Bedford, T.; Bollback, J.; Dudas, G.; Hall, M.; Hedge, J.; et al. FigTree Version 1.4.3. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 17 September 2017).
- Los Alamos National Laboratory HIV Sequence Database. Available online: http://www.hiv.lanl.gov/ (accessed on 9 October 2018).
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Schrodinger LLC the PyMOL Molecular Graphics System; Version 1.8; Schrödinger LLC: New York, NY, USA, 2015.
- Setz, C.; Friedrich, M.; Rauch, P.; Fraedrich, K.; Matthaei, A.; Traxdorf, M.; Schubert, U. Inhibitors of deubiquitinating enzymes block HIV-1 replication and augment the presentation of gag-derived MHC-I epitopes. Viruses 2017, 9, 222. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Bruns, K.; Roder, R.; Henklein, P.; Votteler, J.; Wray, V.; Schubert, U. Solution structure of the equine infectious anemia virus p9 protein: A rationalization of its different ALIX binding requirements compared to the analogous HIV-p6 protein. BMC Struct. Biol. 2009, 9, 74. [Google Scholar] [CrossRef] [PubMed]
- Solbak, S.M.Ø.; Reksten, T.R.; Hahn, F.; Wray, V.; Henklein, P.P.; Halskau, O.; Schubert, U.; Fossen, T.; Henklein, P.P.; Halskau, Ø.; Schubert, U.; et al. HIV-1 p6—A structured to flexible multifunctional membrane-interacting protein. Biochim. Biophys. Acta 2013, 1828, 816–823. [Google Scholar] [CrossRef] [PubMed]
- Maguire, M.F.; Guinea, R.; Griffin, P.; Macmanus, S.; Elston, R.C.; Wolfram, J.; Richards, N.; Hanlon, M.H.; Porter, D.J.T.; Wrin, T.; et al. Changes in Human Immunodeficiency Virus Type 1 Gag at Positions L449 and P453 Are Linked to I50V Protease Mutants In Vivo and Cause Reduction of Sensitivity to Amprenavir and Improved Viral Fitness In Vitro. J. Virol. 2002, 76, 7398–7406. [Google Scholar] [CrossRef] [PubMed]
- Dam, E.; Quercia, R.; Rbel Glass, B.; Descamps, D.; Launay, O.; Duval, X.; Krä Usslich, H.-G.; Hance, A.J.; Clavel, F.; Luban, J. Gag Mutations Strongly Contribute to HIV-1 Resistance to Protease Inhibitors in Highly Drug-Experienced Patients besides Compensating for Fitness Loss. PLoS Pathog. 2009, 5, e1000345. [Google Scholar] [CrossRef] [PubMed]
- Tözsér, J.; Shulenin, S.; Kádas, J.; Boross, P.; Bagossi, P.; Copeland, T.D.; Nair, B.C.; Sarngadharan, M.G.; Oroszlan, S. Human immunodeficiency virus type 1 capsid protein is a substrate of the retroviral proteinase while integrase is resistant toward proteolysis. Virology 2003, 310, 16–23. [Google Scholar] [CrossRef]
- Sauter, D.; Schindler, M.; Specht, A.; Landford, W.N.; Münch, J.; Kim, K.-A.; Votteler, J.; Schubert, U.; Bibollet-Ruche, F.; Keele, B.F.; et al. The evolution of pandemic and non-pandemic HIV-1 strains has been driven by Tetherin antagonism. Cell Host Microbe 2009, 19, 409–421. [Google Scholar] [CrossRef]
- Chen, Z.; Luckay, A.; Sodora, D.L.; Telfer, P.; Reed, P.; Gettie, A.; Kanu, J.M.; Sadek, R.F.; Yee, J.; Ho, D.D.; et al. Human immunodeficiency virus type 2 (HIV-2) seroprevalence and characterization of a distinct HIV-2 genetic subtype from the natural range of simian immunodeficiency virus-infected sooty mangabeys. J. Virol. 1997, 71, 3953–3960. [Google Scholar]
- Van Heuverswyn, F.; Li, Y.; Bailes, E.; Neel, C.; Lafay, B.; Keele, B.F.; Shaw, K.S.; Takehisa, J.; Kraus, M.H.; Loul, S.; et al. Genetic diversity and phylogeographic clustering of SIVcpzPtt in wild chimpanzees in Cameroon. Virology 2007, 368, 155–171. [Google Scholar] [CrossRef]
- Hemelaar, J.; Gouws, E.; Ghys, P.D.; Osmanov, S. Global and regional distribution of HIV-1 genetic subtypes and recombinants in 2004. AIDS 2006, 20, W13–W23. [Google Scholar] [CrossRef]
- Malet, I.; Roquebert, B.; Dalban, C.; Wirden, M.; Amellal, B.; Agher, R.; Simon, A.; Katlama, C.; Costagliola, D.; Calvez, V.; et al. Association of Gag cleavage sites to protease mutations and to virological response in HIV-1 treated patients. J. Infect. 2007, 54, 367–374. [Google Scholar] [CrossRef]
- Kletenkov, K.; Hoffmann, D.; Böni, J.; Yerly, S.; Aubert, V.; Schöni-Affolter, F.; Struck, D.; Verheyen, J.; Klimkait, T. Role of Gag mutations in PI resistance in the Swiss HIV cohort study: Bystanders or contributors? J. Antimicrob. Chemother. 2017, 72, 866–875. [Google Scholar] [CrossRef]
- Tamiya, S.; Mardy, S.; Kavlick, M.F.; Yoshimura, K.; Mistuya, H. Amino acid insertions near Gag cleavage sites restore the otherwise compromised replication of human immunodeficiency virus type 1 variants resistant to protease inhibitors. J. Virol. 2004, 78, 12030–12040. [Google Scholar] [CrossRef] [PubMed]
- Ibe, S.; Shibata, N.; Utsumi, M.; Kaneda, T. Selection of human immunodeficiency virus type 1 variants with an insertion mutation in the p6(gag) and p6(pol) genes under highly active antiretroviral therapy. Microbiol. Immunol. 2003, 47, 71–79. [Google Scholar] [CrossRef]
- Peters, S.; Munoz, M.; Yerly, S.; Sanchez-Merino, V.; Lopez-Galindez, C.; Perrin, L.; Larder, B.; Cmarko, D.; Fakan, S.; Meylan, P.; et al. Resistance to Nucleoside Analog Reverse Transcriptase Inhibitors Mediated by Human Immunodeficiency Virus Type 1 p6 Protein. J. Virol. 2001, 75, 9644–9653. [Google Scholar] [CrossRef] [PubMed]
- Abecasis, A.B.; Wensing, A.M.J.; Paraskevis, D.; Vercauteren, J.; Theys, K.; Van de Vijver, D.A.M.C.; Albert, J.; Asjö, B.; Balotta, C.; Beshkov, D.; et al. HIV-1 subtype distribution and its demographic determinants in newly diagnosed patients in Europe suggest highly compartmentalized epidemics. Retrovirology 2013, 10, 7. [Google Scholar] [CrossRef]
- Torrecilla, E.; Llaćer Delicado, T.; Holguín, Á. New findings in cleavage sites variability across groups, subtypes and recombinants of human immunodeficiency virus type 1. PLoS ONE 2014, 9, e88099. [Google Scholar] [CrossRef]
- HIV Sequence Database. Available online: http://www.hiv.lanl.gov/ (accessed on 6 July 2017).
- Patiño-Galindo, J.Á.; González-Candelas, F. The substitution rate of HIV-1 subtypes: A genomic approach. Virus Evol. 2017, 3. [Google Scholar] [CrossRef]
- Kurochkin, I.V. Amyloidogenic determinant as a substrate recognition motif of insulin-degrading enzyme. FEBS Lett. 1998, 427, 153–156. [Google Scholar] [CrossRef]
- Duckworth, W.C.; Kitabchi, A.E. Insulin and glucagon degradation by the same enzyme. Diabetes 1974, 23, 536–543. [Google Scholar] [CrossRef]
- Authier, F.; Bergeron, J.J.; Ou, W.J.; Rachubinski, R.A.; Posner, B.I.; Walton, P.A. Degradation of the cleaved leader peptide of thiolase by a peroxisomal proteinase. Proc. Natl. Acad. Sci. USA 1995, 92, 3859–3863. [Google Scholar] [CrossRef] [PubMed]
- Kaleebu, P.; Nankya, I.L.; Yirrell, D.L.; Shafer, L.A.; Kyosiimire-Lugemwa, J.; Lule, D.B.; Morgan, D.; Beddows, S.; Weber, J.; Whitworth, J.A.G. Relation between chemokine receptor use, disease stage, and HIV-1 subtypes A and D: Results from a rural Ugandan cohort. J. Acquir. Immune Defic. Syndr. 2007, 45, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Sacktor, N.; Nakasujja, N.; Skolasky, R.L.; Rezapour, M.; Robertson, K.; Musisi, S.; Katabira, E.; Ronald, A.; Clifford, D.B.; Laeyendecker, O.; et al. HIV Subtype D Is Associated with Dementia, Compared with Subtype A, in Immunosuppressed Individuals at Risk of Cognitive Impairment in Kampala, Uganda. Clin. Infect. Dis. 2009, 49, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Mirsky, I.A.; Perisutti, G. Effect of insulinase-inhibitor on hypoglycemic action of insulin. Science 1955, 122, 559–560. [Google Scholar] [CrossRef] [PubMed]
- Bennett, R.G.; Hamel, F.G.; Duckworth, W.C. An insulin-degrading enzyme inhibitor decreases amylin degradation, increases amylin-induced cytotoxicity, and increases amyloid formation in insulinoma cell cultures. Diabetes 2003, 52, 2315–2320. [Google Scholar] [CrossRef] [PubMed]
- Duckworth, W.C. Insulin and glucagon degradation by the kidney I. Subcellular distribution under different assay conditions. Biochim. Biophys. Acta Gen. Subj. 1976, 437, 518–530. [Google Scholar] [CrossRef]
- Iwata, N.; Tsubuki, S.; Takaki, Y.; Watanabe, K.; Sekiguchi, M.; Hosoki, E.; Kawashima-Morishima, M.; Lee, H.-J.; Hama, E.; Sekine-Aizawa, Y.; et al. Identification of the major Aβ1–42-degrading catabolic pathway in brain parenchyma: Suppression leads to biochemical and pathological deposition. Nat. Med. 2000, 6, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Bell, G.I.; Swain, W.F.; Pictet, R.; Cordell, B.; Goodman, H.M.; Rutter, W.J. Nucleotide sequence of a cDNA clone encoding human preproinsulin. Nature 1979, 282, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Lemaire, H.G.; Unterbeck, A.; Salbaum, J.M.; Masters, C.L.; Grzeschik, K.H.; Multhaup, G.; Beyreuther, K.; Müller-Hill, B. The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature 1987, 325, 733–736. [Google Scholar] [CrossRef] [PubMed]
- Laguette, N.; Benkirane, M. Frontiers in Microbiology; Frontiers Media SA: Lausanne, Switzerland, 2015; p. 142. [Google Scholar]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schmalen, A.; Karius-Fischer, J.; Rauch, P.; Setz, C.; Korn, K.; Henklein, P.; Fossen, T.; Schubert, U. The N-Terminus of the HIV-1 p6 Gag Protein Regulates Susceptibility to Degradation by IDE. Viruses 2018, 10, 710. https://doi.org/10.3390/v10120710
Schmalen A, Karius-Fischer J, Rauch P, Setz C, Korn K, Henklein P, Fossen T, Schubert U. The N-Terminus of the HIV-1 p6 Gag Protein Regulates Susceptibility to Degradation by IDE. Viruses. 2018; 10(12):710. https://doi.org/10.3390/v10120710
Chicago/Turabian StyleSchmalen, Adrian, Julia Karius-Fischer, Pia Rauch, Christian Setz, Klaus Korn, Petra Henklein, Torgils Fossen, and Ulrich Schubert. 2018. "The N-Terminus of the HIV-1 p6 Gag Protein Regulates Susceptibility to Degradation by IDE" Viruses 10, no. 12: 710. https://doi.org/10.3390/v10120710
APA StyleSchmalen, A., Karius-Fischer, J., Rauch, P., Setz, C., Korn, K., Henklein, P., Fossen, T., & Schubert, U. (2018). The N-Terminus of the HIV-1 p6 Gag Protein Regulates Susceptibility to Degradation by IDE. Viruses, 10(12), 710. https://doi.org/10.3390/v10120710