Communities of Phytoplankton Viruses across the Transition Zone of the St. Lawrence Estuary

 , and
, and 
Abstract
1. Introduction
2. Materials and Methods
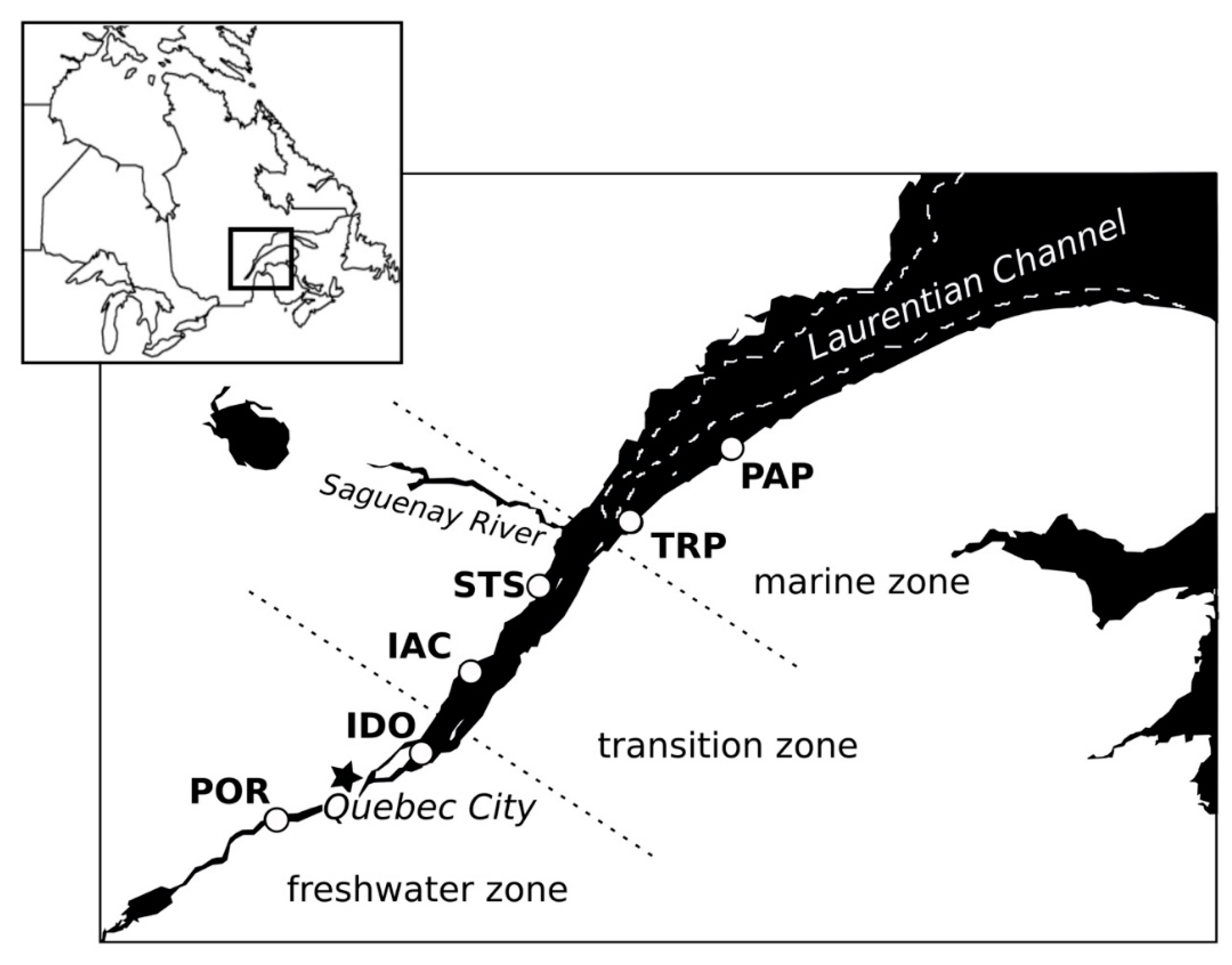
2.1. Sampling Locations
2.2. Sample Collection and Filtration
2.3. Nucleic-Acid Extraction
2.4. High-Throughput Sequencing of Viral Nucleic Acids
2.5. 18S rRNA and 18S rDNA High-Throughput Sequencing
2.6. Analysis of Reads
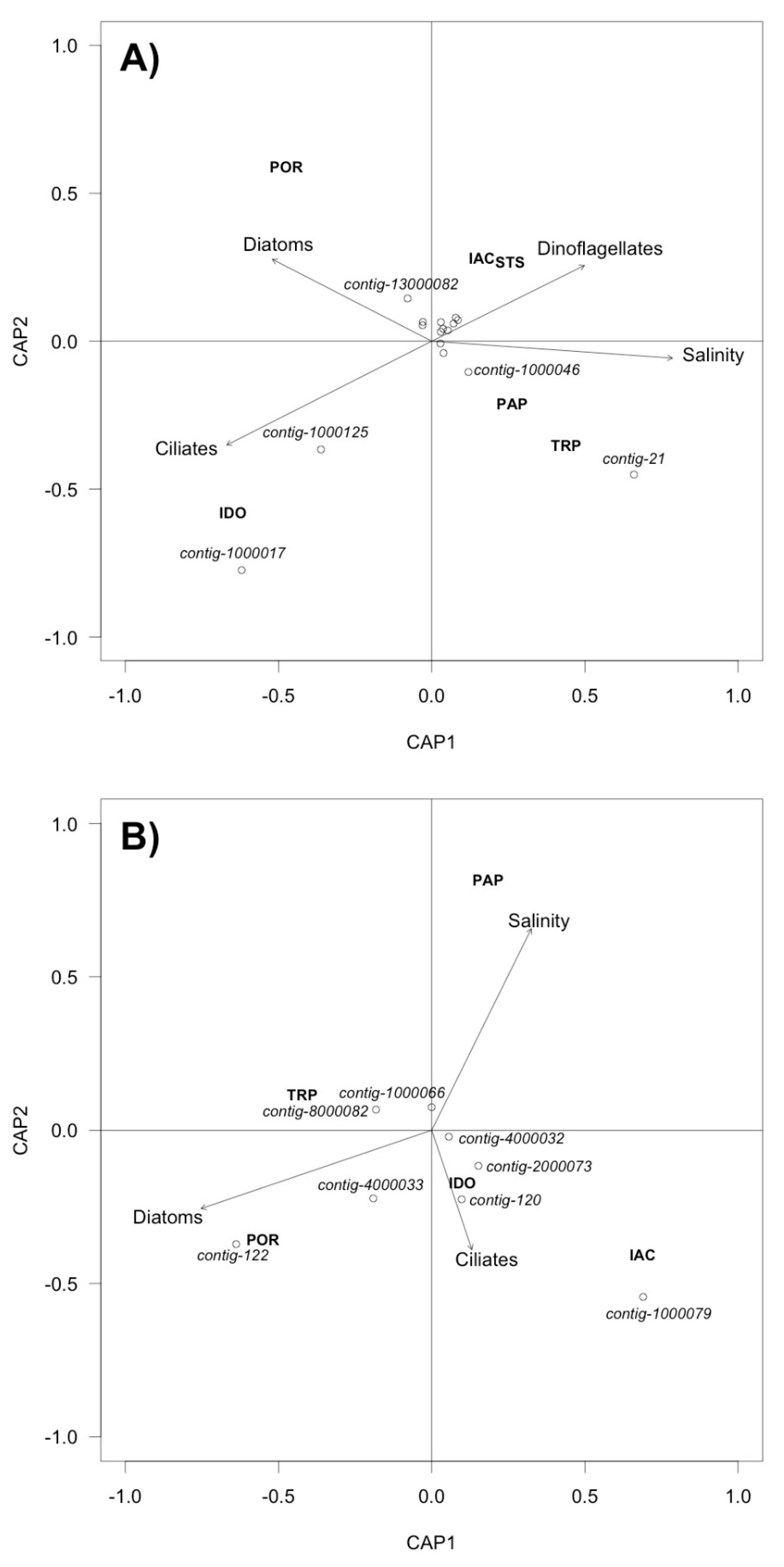
2.7. Statistical Analysis
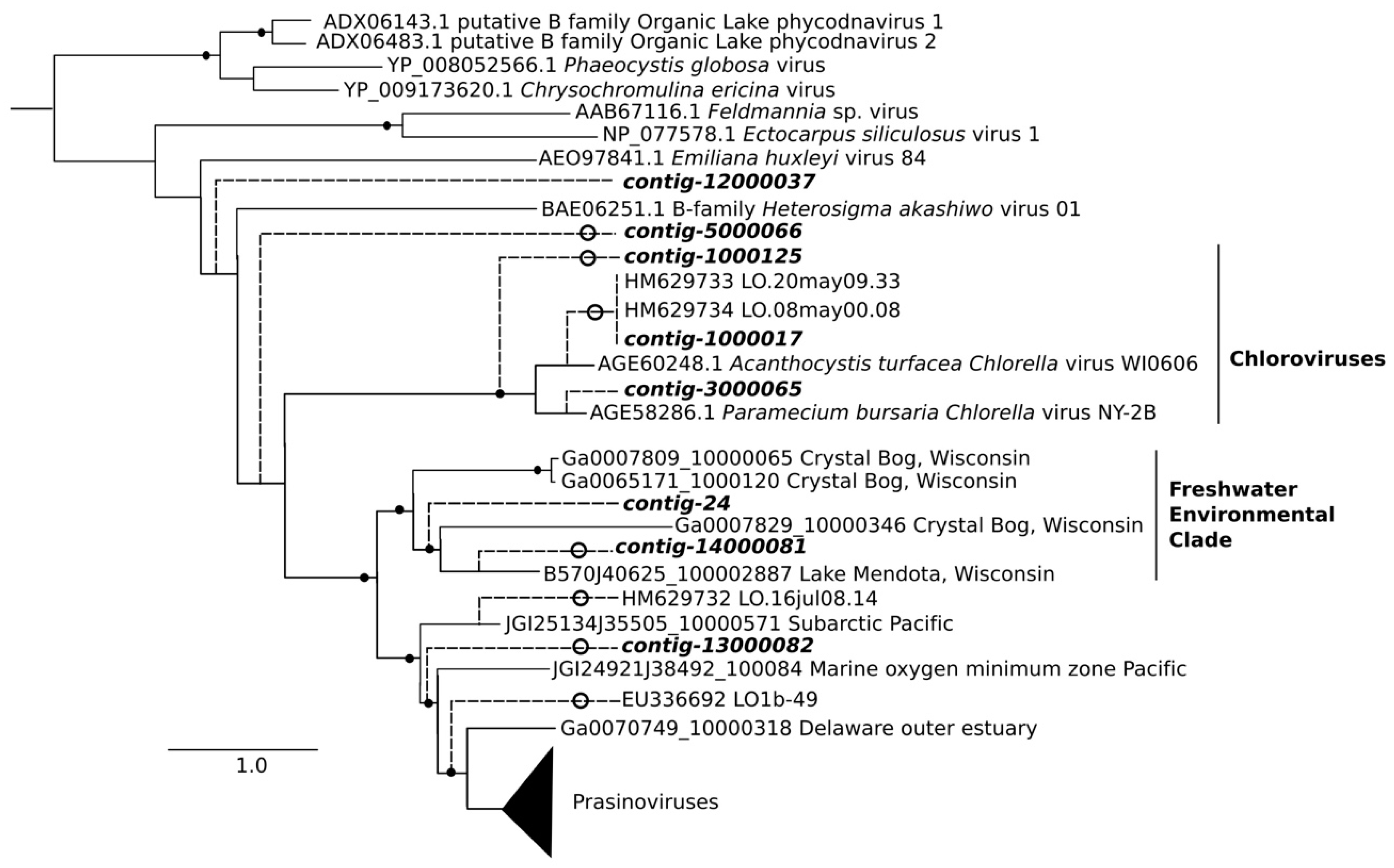
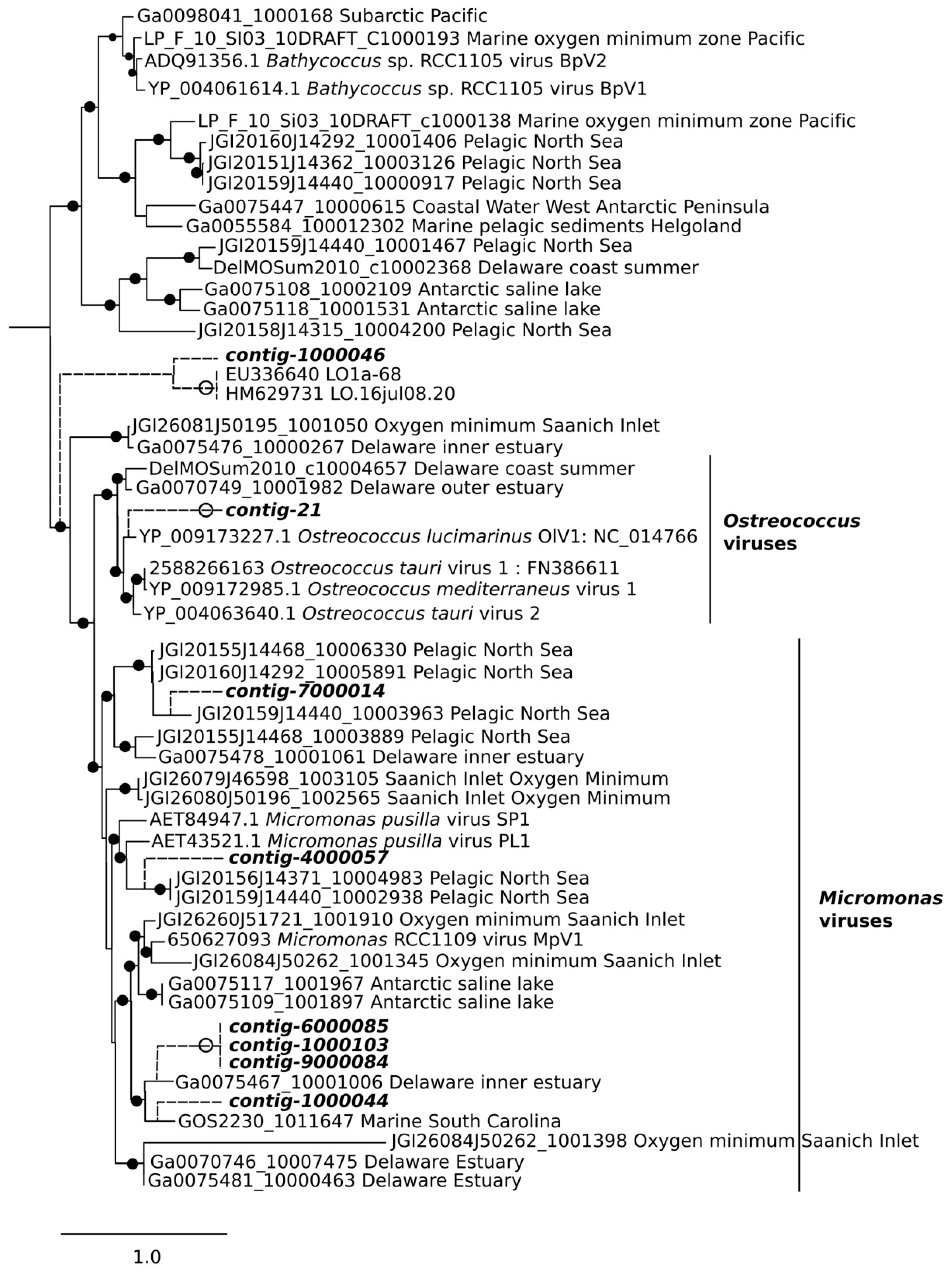
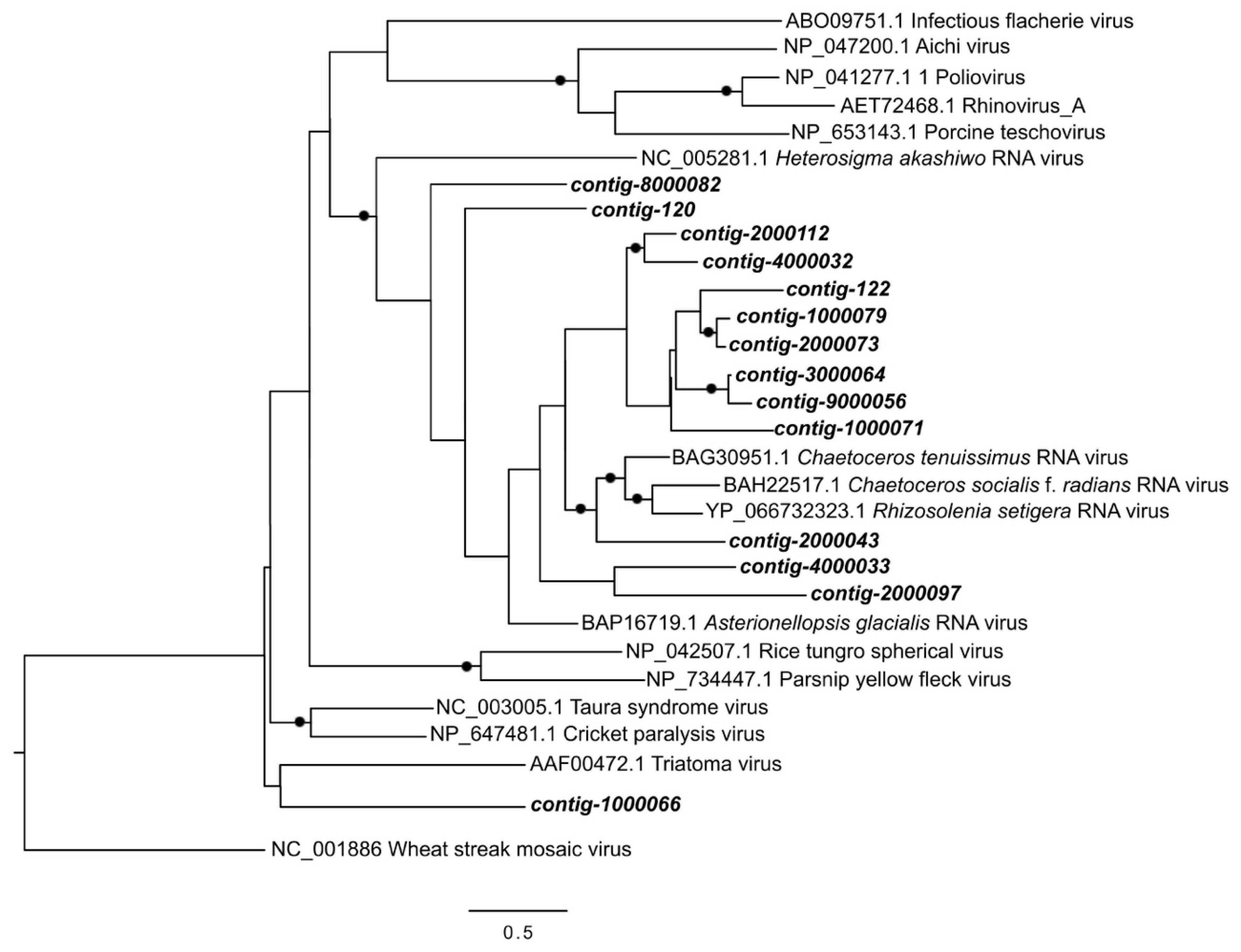
2.8. Phylogenetic Analysis
3. Results
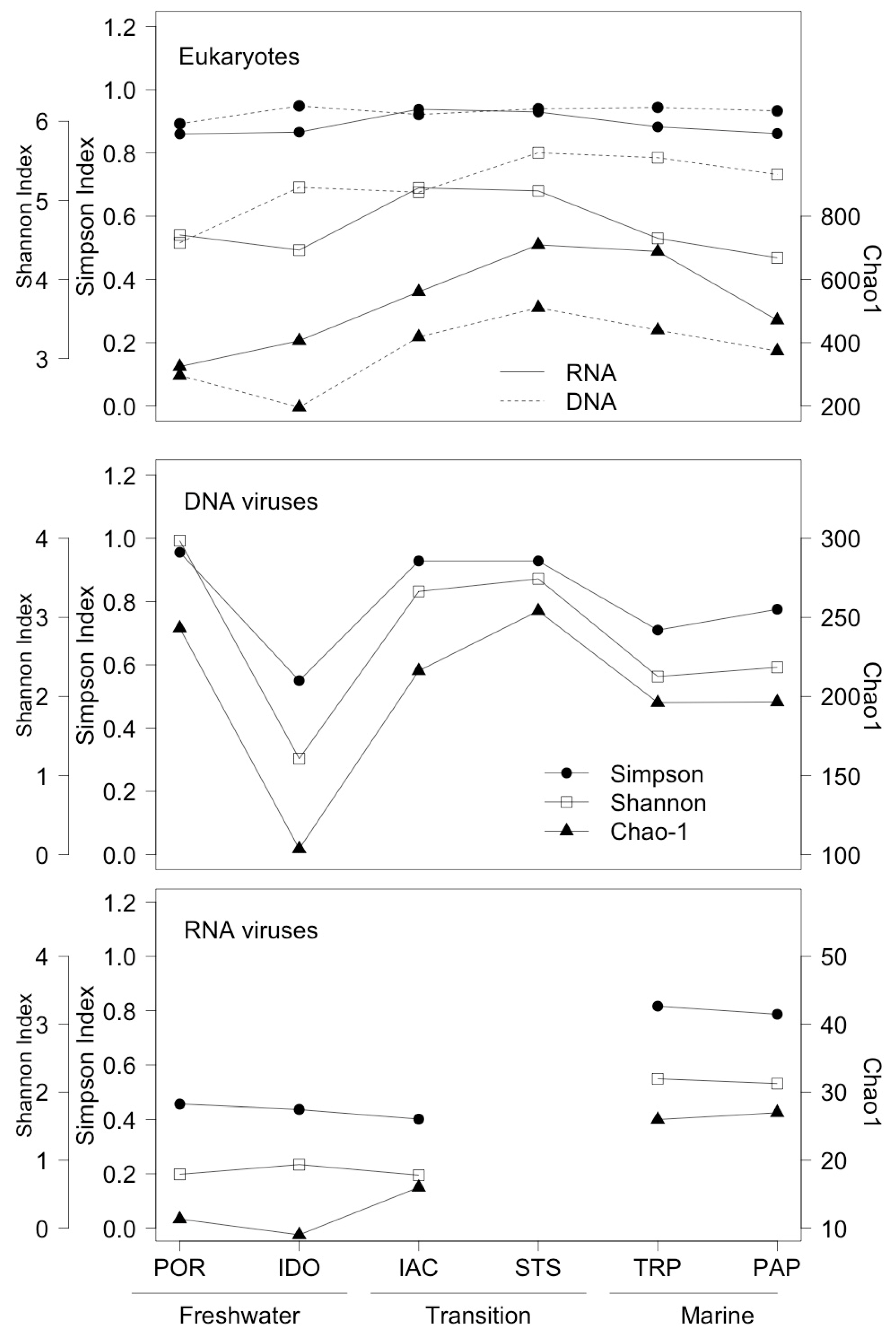
3.1. Environmental Parameters
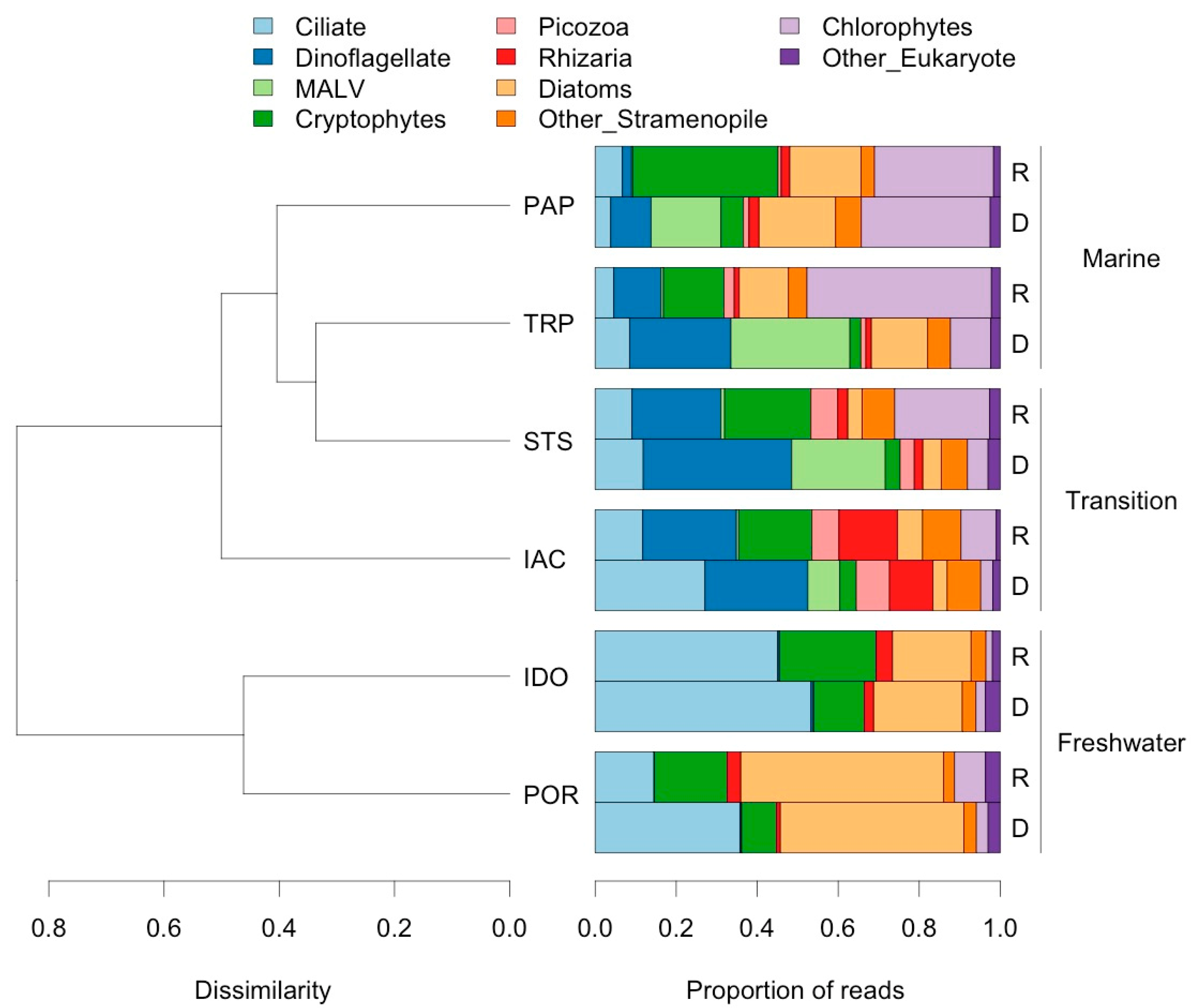
3.2. Analysis of Eukaryotic Communities
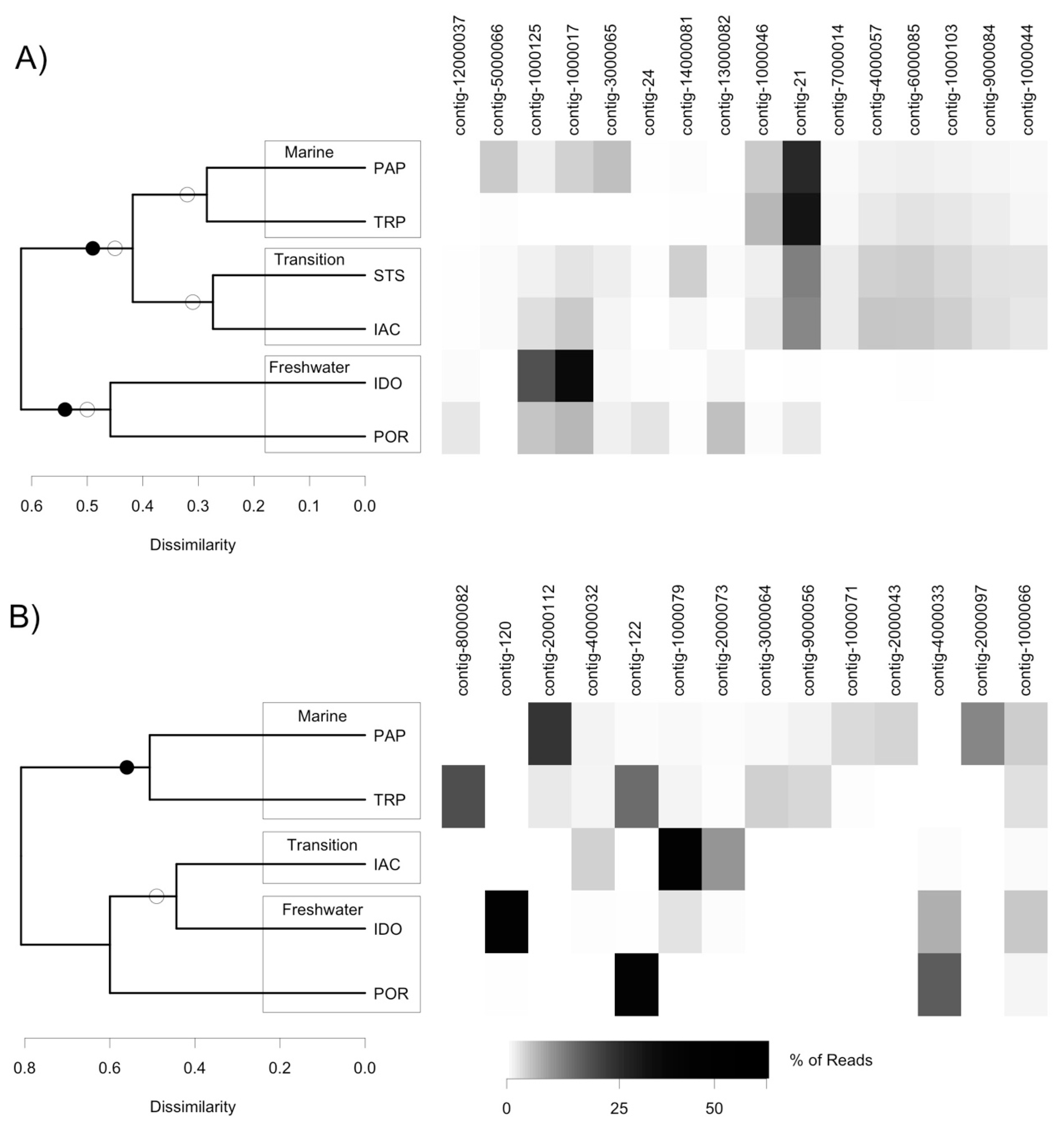
3.3. Analysis of DNA Pol and RdRP Sequences
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vincent, W.F.; Dodson, J.J. The St. Lawrence River, Canada-USA: The need for an ecosystem-level understanding of large rivers. Japan J. Limnol. 1999, 60, 29–50. [Google Scholar] [CrossRef]
- Lovejoy, C.; Vincent, W.F.; Frenette, J.J.; Dodson, J.J. Microbial gradients in a turbid estuary: Application of a new method for protozoan community analysis. Limnol. Oceanogr. 1993, 38, 1295–1303. [Google Scholar] [CrossRef]
- Dodson, J.J.; Dauvin, J.-C.; Ingram, R.G.; d’Anglejan, B. Abundance of larval rainbow smelt (Osmerus mordax) in relation to the maximum turbidity zone and associated macroplanktonic fauna of the middle St. Lawrence estuary. Estuaries 1989, 12, 66–81. [Google Scholar] [CrossRef]
- Lapierre, J.-F.; Frenette, J.-J. Advection of freshwater phytoplankton in the St. Lawrence River estuarine turbidity maximum as revealed by sulfur-stable isotopes. Mar. Ecol. Prog. Ser. 2008, 372, 19–29. [Google Scholar] [CrossRef]
- Thérriault, J.-C.; Legendre, L.; Demers, S. Oceanography and ecology of phytoplankton in the St. Lawrence Estuary. In Oceanography of a Large-Scale Estuarine System; El-Sabh, M.I., Silverberg, N., Eds.; Springer: New York, NY, USA, 1990; Volume 39, pp. 269–295. [Google Scholar]
- Vincent, W.F.; Dodson, J.J.; Bertrand, N.; Frenette, J.-J. Photosynthetic and bacterial production gradients in a larval fish nursery: The St. Lawrence River transition zone. Mar. Ecol. Prog. Ser. 1996, 139, 227–238. [Google Scholar] [CrossRef]
- Findlay, S.; Pace, M.L.; Lints, D.; Cole, J.J.; Caraco, N.F.; Peierls, B. Weak coupling of bacterial and algal production in a heterotrophic ecosystem: The Hudson River estuary. Limnol. Oceanogr. 1991, 36, 268–278. [Google Scholar] [CrossRef]
- Martineau, C.; Vincent, W.F.; Frenette, J.-J.; Dodson, J.J. Primary consumers and particulate organic matter: Isotopic evidence of strong selectivity in the estuarine transition zone. Limnol. Oceanogr. 2004, 49, 1679–1686. [Google Scholar] [CrossRef]
- Brussaard, C.P.D. Viral control of phytoplankton populations—A review. J. Eukaryot. Microbiol. 2004, 51, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Sime-Ngando, T.; Gosselin, M.; Roy, S.; Chanut, J.-P. Significance of planktonic ciliated protozoa in the lower St. Lawrence Estuary: Comparison with bacterial, phytoplankton, and particulate organic carbon. Aquat. Microb. Ecol. 1995, 9, 243–258. [Google Scholar] [CrossRef]
- Winkler, G.; Dodson, J.J.; Bertrand, N.; Thivierge, D.; Vincent, W.F. Trophic coupling across the St. Lawrence River estuarine transition zone. Mar. Ecol. Prog. Ser. 2003, 251, 59–73. [Google Scholar] [CrossRef]
- Peduzzi, P. Virus ecology of fluvial systems: A blank spot on the map? Biol. Rev. 2015, 91, 937–949. [Google Scholar] [CrossRef] [PubMed]
- Short, S.M. The ecology of viruses that infect eukaryotic algae. Environ. Microbiol. 2012, 14, 2253–2271. [Google Scholar] [CrossRef] [PubMed]
- Nagasaki, K.; Kim, J.-J.; Tomaru, Y.; Takao, Y.; Nagai, S. Isolation and characterization of a novel virus infecting Teleaulax amphioxeia (Cryptophyceae). Plankton Benthos Res. 2009, 4, 122–124. [Google Scholar] [CrossRef]
- Culley, A.; Lang, A.S.; Suttle, C.A. Metagenomic analysis of coastal RNA virus communities. Science 2006, 312, 1795–1798. [Google Scholar] [CrossRef] [PubMed]
- Tomaru, Y.; Mizumoto, H.; Nagasaki, K. Virus resistance in the toxic bloom-forming dinoflagellate Heterocapsa circularisquama to single-stranded RNA virus infection. Environ. Microbiol. 2009, 11, 2915–2923. [Google Scholar] [CrossRef] [PubMed]
- Nagasaki, K. Dinoflagellates, diatoms and their viruses. J. Microbiol. 2008, 46, 235–243. [Google Scholar] [CrossRef] [PubMed]
- King, A.M.; Lefkowitz, E.; Adams, M.J.; Carstens, E.B. (Eds.) Virus Taxonomy: Ninth Report of the International Committee on Taxonomy of Viruses; Elsevier: Amsterdam, Netherlands, 2011. [Google Scholar]
- Steward, G.F.; Culley, A.I.; Mueller, J.A.; Wood-Charlson, E.M.; Belcaid, M.; Poisson, G. Are we missing half of the viruses in the ocean? ISME J. 2013, 7, 672–679. [Google Scholar] [CrossRef] [PubMed]
- Mueller, J.A.; Culley, A.I.; Steward, G.F. Variables influencing extraction of nucleic acids from microbial plankton (viruses, bacteria, and protists) collected on nanoporous aluminum oxide filters. Appl. Environ. Microbiol. 2014, 80, 3930–3942. [Google Scholar] [CrossRef] [PubMed]
- Culley, A.; Suttle, C.A.; Steward, G.F. Characterization of the diversity of marine RNA viruses. In Manual of Aquatic Viral Ecology; Wilhelm, S.W., Weinbauer, M.G., Suttle, C.A., Eds.; Association for the Sciences of Limnology and Oceanography: Waco, TX, USA, 2010; pp. 193–201. [Google Scholar]
- Chen, F.; Suttle, C.A. Amplification of DNA Polymerase gene fragments from viruses infecting microalgae. Appl. Environ. Microbiol. 1995, 61, 1274–1278. [Google Scholar] [CrossRef] [PubMed]
- Short, S.M.; Rusanova, O.; Staniewski, M.A. Novel phycodnavirus genes amplified from Canadian freshwater environments. Aquat. Microb. Ecol. 2011, 63, 61–67. [Google Scholar] [CrossRef]
- Hopkins, M.; Kailasan, S.; Cohen, A.; Roux, S.; Tucker, K.P.; Shevenell, A.; Agbandje-McKenna, M.; Breitbart, M. Diversity of environmental single-stranded DNA phages revealed by PCR amplification of the partial major capsid protein. ISME J. 2014, 8, 2093–2103. [Google Scholar] [CrossRef] [PubMed]
- Comeau, A.M.; Li, W.K.W.; Tremblay, J.-É.; Carmack, E.C.; Lovejoy, C. Arctic Ocean Microbial Community Structure before and after the 2007 Record Sea Ice Minimum. PLoS ONE 2011, 6, e27492. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Boisvert, S.; Raymond, F.; Godzarisid, É.; Laviolette, F.; Corbeil, J. Ray Meta: Scalable de novo metagenome assembly and profiling. Genome Biol. 2012, 13, R122. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Schaeffer, L.; Pimentel, H.; Bray, N.; Melsted, P.; Pachter, L. Pseudoalignment for metagenomic read assignment. Bioinformatics 2017, 33, 2082–2088. [Google Scholar] [CrossRef] [PubMed]
- Raoult, D.; Audic, S.; Robert, C.; Abergel, C.; Renesto, P.; Ogata, H.; La Scola, B.; Suzan, M.; Claverie, J.-M. The 1.2-megabase genome sequence of mimivirus. Science 2004, 306, 1344–1350. [Google Scholar] [CrossRef] [PubMed]
- Nagasaki, K.; Shirai, Y.; Tomaru, Y.; Nishida, K.; Pietrokovski, S. Algal viruses with distinct intraspecies host specificities include identical intein elements. Appl. Environ. Microbiol. 2005, 71, 3599–3607. [Google Scholar] [CrossRef] [PubMed]
- Culley, A.; Asuncion, B.F.; Steward, G.F. Detection of inteins among diverse DNA polymerase genes of uncultivated members of the Phycodnaviridae. ISME J. 2009, 3, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Lovejoy, C.; Comeau, A.M.; Thaler, M. Curated reference database of SSU rRNA for northern marine and freshwater communities of Archaea, Bacteria and microbial eukaryotes. Nordicana D 2015. [Google Scholar] [CrossRef]
- Hammer, Ø.; Harper, D.A.T. Paleontological Data Analysis, version 2.17; Wiley-Blackwell: Hoboken, NJ, USA, 2005; p. 229. ISBN 978-1405115445. [Google Scholar]
- Maillet, N.; Collet, G.; Vannier, T.; Lavenier, D.; Peterlongo, P. COMMET: Comparing and combining multiple metagenomic datasets. In Proceedings of the IEEE International Conference on Bioinformatics and Biomedicine, Belfast, UK, 2–5 November 2014. [Google Scholar]
- Lozupone, C.; Hamady, M.; Knight, R. UniFrac—An online tool for comparing microbial community diversity in a phylogenetic context. BMC Bioinform. 2006, 7, 371. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Kuma, K.; Toh, H.; Miyata, T. MAFFT version 5: Improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 2005, 33, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Legendre, P.; Andersson, M.J. Distance-based redundancy analysis: Testing multispecies responses in multifactorial ecological experiments. Ecol. Monogr. 1999, 69, 1–24. [Google Scholar] [CrossRef]
- Shankar, V.; Agans, R.; Paliy, O. Advantages of phylogenetic distance based constrained ordination analyses for the examination of microbial communities. Nat. Sci. Rep. 2017, 7, 6481. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J.; Blanchet, F.G.; Kindet, R.; Legendre, P.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; Stevens, M.H.; Wagner, H. vegan: Community Ecology Package. R Package Version 2.4-4. Available online: http://CRAN.R-project.org/package=vegan (accessed on 24 August 2017).
- Paez-Espino, D.; Chen, I.M.; Palaniappan, K.; Ratner, A.; Chu, K.; Szeto, E.; Pillay, M.; Huang, J.; Markowitz, V.M.; Nielsen, T.; et al. IMG/VR: A database of cultured and uncultured DNA viruses and retroviruses. Nucleic Acids Res. 2017, 45, D457–D465. [Google Scholar] [CrossRef] [PubMed]
- Braithwaite, D.K.; Ito, J. Compilation, alignment, and phylogenetic relationships of DNA polymerases. Nucleic Acids Res. 1993, 21, 787–807. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V. The phylogeny of RNA-dependent RNA polymerases of positive-strand RNA viruses. J. Gen. Virol. 1991, 72, 2197–2206. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RaxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of tax and mixed models. Bioinformatics 2006, 22, 2688–2690. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.A.; Krompass, D.; Stamatakis, A. Performance, accuracy, and web server for evolutionary placement of short sequence reads under maximum likelihood. Syst. Biol. 2011, 60, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Ingram, R.G.; El-Sabh, M.I. Fronts and mesoscale features in the St. Lawrence Estuary. Coast. Estuar. Stud. 1990, 39, 71–93. [Google Scholar] [CrossRef]
- Jane, S.F.; Wilcox, T.M.; McKelvey, K.S.; Young, M.K.; Schwartz, M.K.; Lowe, W.H.; Letcher, B.H.; Whiteley, A.R. Distance, flow and PCR inhibition: eDNA dynamics in two headwater streams. Mol. Ecol. Resour. 2015, 15, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Balzano, S.; Sarno, D.; Kooistra, W.H.C. Effects of salinity on the growth rate and morphology of ten Skeletonema strains. J. Plankt. Res. 2011, 33, 937–945. [Google Scholar] [CrossRef]
- Charvet, S.; Vincent, W.F.; Comeau, A.; Lovejoy, C. Pyrosequencing analysis of the protist communities in a High Arctic meromictic lake: DNA preservation and change. Front. Microbiol. 2012, 3, 422. [Google Scholar] [CrossRef] [PubMed]
- Gustavsen, J.A.; Winget, D.M.; Tian, X.; Suttle, C.A. High temporal and spatial diversity in marine RNA viruses implies that they have an important role in mortality and structuring plankton communities. Front. Microbiol. 2014, 5, 703. [Google Scholar] [CrossRef] [PubMed]
- Suttle, C.A. Marine viruses—Major players in the global ecosystem. Nat. Rev. Microbiol. 2007, 5, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Jacquet, S. Contrasting diversity of phycodnavirus signature genes in two large and deep western European lakes. Environ. Microbiol. 2014, 16, 759–773. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primers targeting RNA-dependent RNA-polymerase (Picornavirales): | |||
| Primer | Sequence | Hybridization Temperature (°C) | Reference |
| Mpl.sc2F | ITWGCIGGIGATTWCA | 43.3 | [21] |
| Mpl.sc2R | CKYTTCARRAAWTCAGCATC | 43.3 | [21] |
| RdRp1 | GGRGAYTACASCIRWTTTGAT | 50 | [21] |
| RdRp2 | MACCCAACKMCKCTTSARRAA | 50 | [21] |
| Primers targeting DNA-dependent DNA polymerase (Phycodnaviridae): | |||
| Primer | Sequence | Hybridization Temperature (°C) | Reference |
| AVS1 | GARGGIGCIACIGTIYTIGAYGC | 44.9 | [22] |
| AVS2 | GCIGCRTAICKYTTYTTISWRTA | 44.9 | [22] |
| ChlvdF | CCWATCGCAGCWCTMGATTTTG | 52 | [23] |
| ChlvdR | ATCTCVCCBGCVARCCACTT | 52 | [23] |
| DNA Viruses | RNA Viruses | ||||||
|---|---|---|---|---|---|---|---|
| Station | Salinity | Seqs. | Contigs | Unique Contigs (%) | Seqs. | Contigs | Unique Contigs (%) |
| Portneuf (POR) | 0.11 | 369,201 | 316 | 4.7 | 70,103 | 29 | 34 |
| Île d’Orléans (IDO) | 0.13 | 12,264 | 99 | 0 | 28,605 | 17 | 18 |
| Isle-aux-Coudres (IAC) | 23.17 | 86,707 | 252 | 0.4 | 705,577 | 35 | 26 |
| St-Siméon (STS) | 28.77 | 1,371,765 | 387 | 3.3 | NA | NA | NA |
| Trois-Pistoles (TRP) | 29.37 | 2,397,455 | 381 | 3.8 | 4368 | 34 | 26 |
| Pointe-au-Père (PAP) | 29.57 | 3,494,306 | 392 | 2.8 | 15,526 | 37 | 36 |
| Contribution | Contribution | ||||
|---|---|---|---|---|---|
| DNA | % | Cum. | RNA | % | Cum. |
| Ciliate—Choreotrichida | 11.3 | 11.3 | Diatom—Skeletonema | 12.9 | 12.9 |
| Diatom—Skeletonema | 10.4 | 21.6 | Chlorophyte—Ostreococcus | 8.3 | 21.2 |
| Diatom—Thalassiosirales | 7.3 | 28.9 | Cryptophyte—Cryptomonas | 8.3 | 29.4 |
| Dinoflagellate—Heterocapsa rotundata | 6.2 | 35.2 | Ciliate—Stokesia | 6.4 | 35.8 |
| Marine Alveolate—Unclassified | 5.8 | 40.1 | Cryptophyte—Plagioselmis/Teleaulax | 6.0 | 41.8 |
| Dinoflagellate—Gymnodiniales | 5.2 | 46.1 | Dinoflagellate—Heterocapsa rotundata | 5.5 | 47.3 |
| Marine Alveolate—Guillou II.1 | 4.6 | 50.7 | Cryptophyte—Teleaulax gracilis | 5.4 | 52.7 |
| Chlorophyte—Ostreococcus | 4.5 | 55.2 | Chlorophyte—Mamiellophyceae | 3.9 | 60.5 |
| Cryptophyte—Cryptomonas | 2.8 | 61.7 | Diatom—Thalassiosirales | 2.4 | 63.0 |
| Picozoa—NW617.02 | 2.2 | 63.9 | Diatom—Thalassiosira | 2.4 | 65.3 |
| Ciliate—Oligotrichida | 2.2 | 66.0 | Picozoa—NW617.02 | 2.4 | 68 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Labbé, M.; Raymond, F.; Lévesque, A.; Thaler, M.; Mohit, V.; Audet, M.; Corbeil, J.; Culley, A. Communities of Phytoplankton Viruses across the Transition Zone of the St. Lawrence Estuary. Viruses 2018, 10, 672. https://doi.org/10.3390/v10120672
Labbé M, Raymond F, Lévesque A, Thaler M, Mohit V, Audet M, Corbeil J, Culley A. Communities of Phytoplankton Viruses across the Transition Zone of the St. Lawrence Estuary. Viruses. 2018; 10(12):672. https://doi.org/10.3390/v10120672
Chicago/Turabian StyleLabbé, Myriam, Frédéric Raymond, Alice Lévesque, Mary Thaler, Vani Mohit, Martyne Audet, Jacques Corbeil, and Alexander Culley. 2018. "Communities of Phytoplankton Viruses across the Transition Zone of the St. Lawrence Estuary" Viruses 10, no. 12: 672. https://doi.org/10.3390/v10120672
APA StyleLabbé, M., Raymond, F., Lévesque, A., Thaler, M., Mohit, V., Audet, M., Corbeil, J., & Culley, A. (2018). Communities of Phytoplankton Viruses across the Transition Zone of the St. Lawrence Estuary. Viruses, 10(12), 672. https://doi.org/10.3390/v10120672