Characterizing the Different Effects of Zika Virus Infection in Placenta and Microglia Cells


Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Virus
2.2. Cell Infection
2.3. LIVE/DEAD Cellular Assay
2.4. RNA Extraction, cDNA, RT-qPCR, qPCR Arrays, and Standard Curve
2.5. Plaque Assays
2.6. siRNA Experiments
2.7. Signaling Pathway Analysis
2.8. Statistical Analysis
3. Results
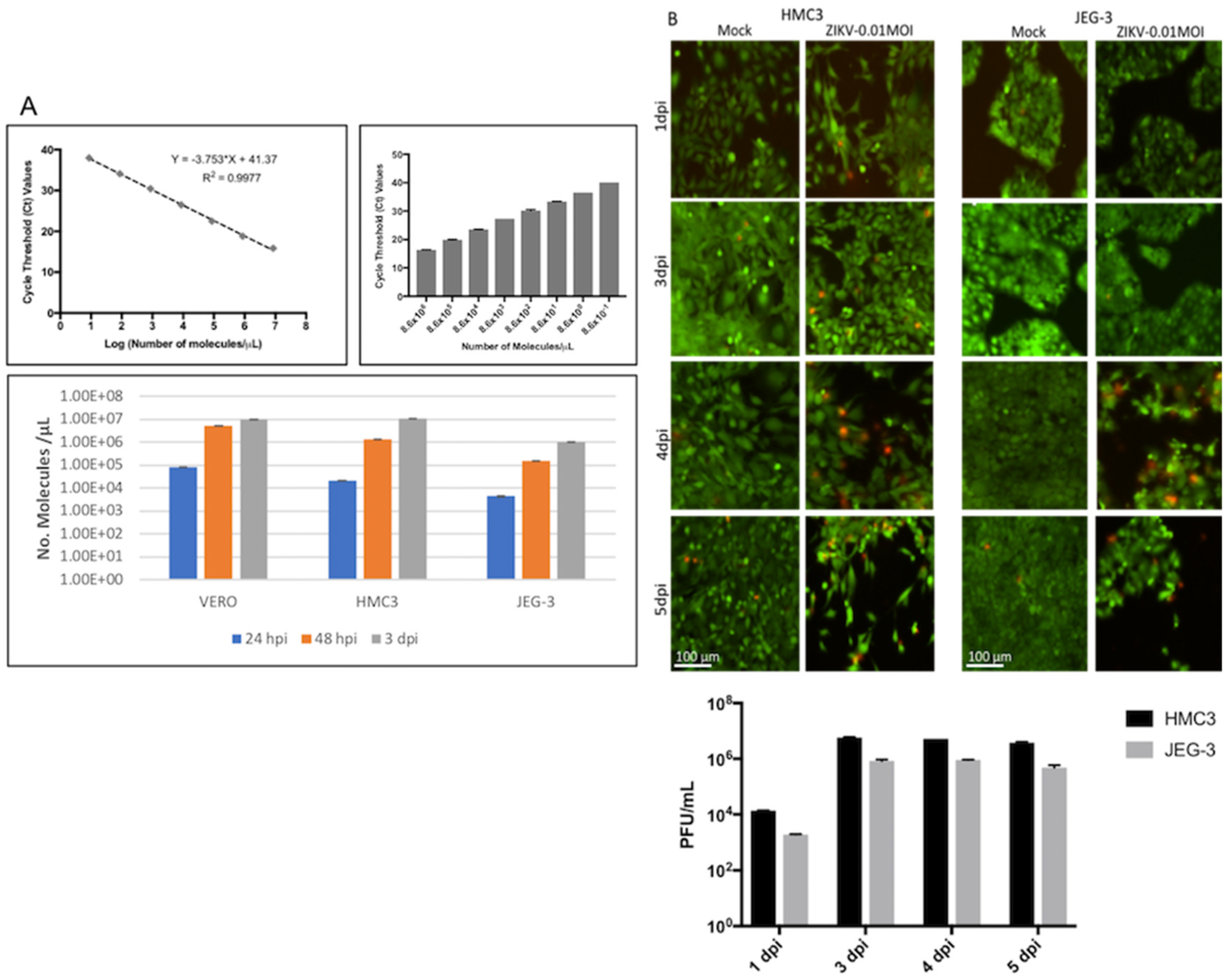
3.1. ZIKV Production, Titer, Cell Infection, and Cytopathic Effects (CPE)
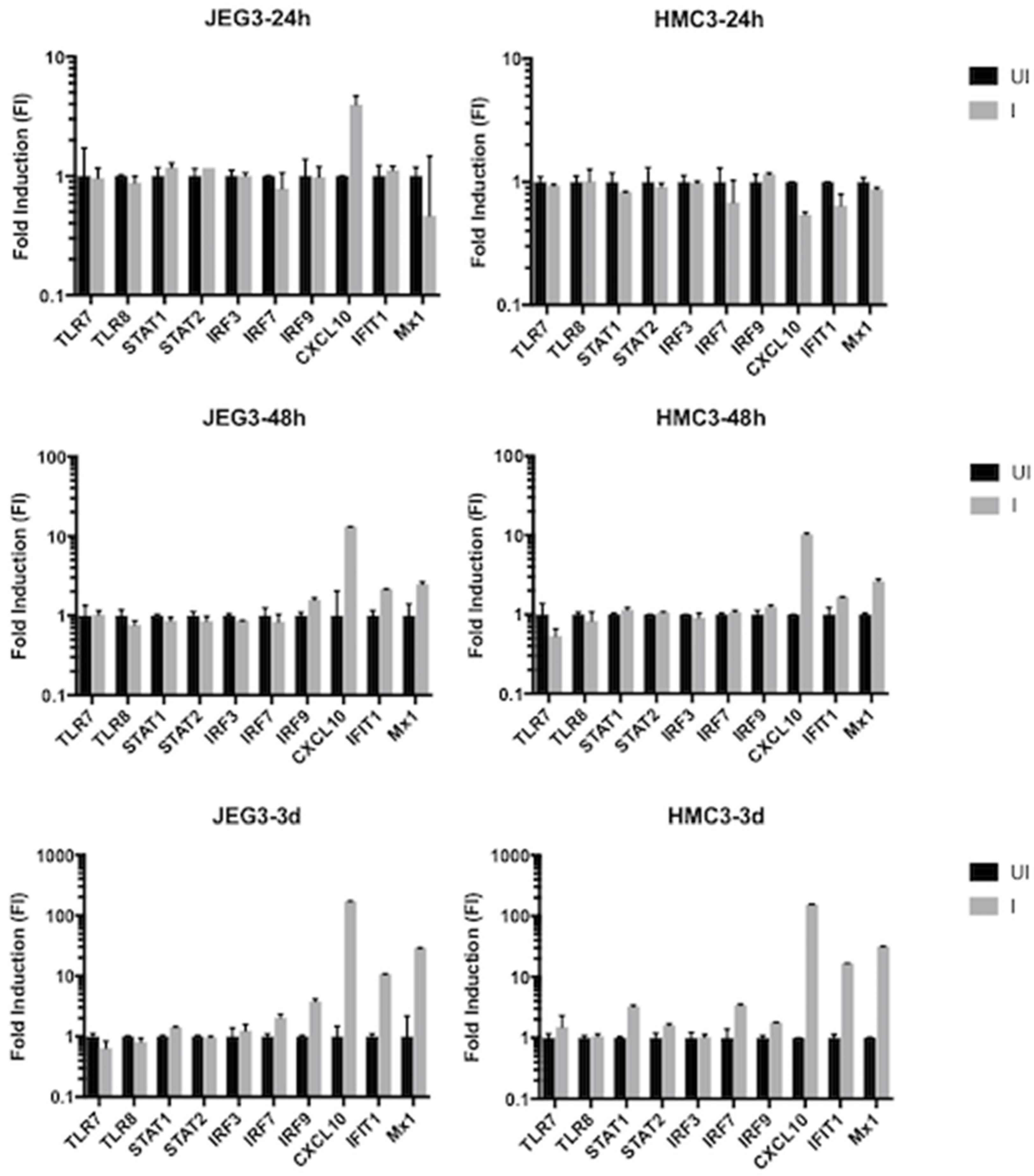
3.2. Host Intracellular Innate Immune Response and Differentially Affected Signaling Pathways
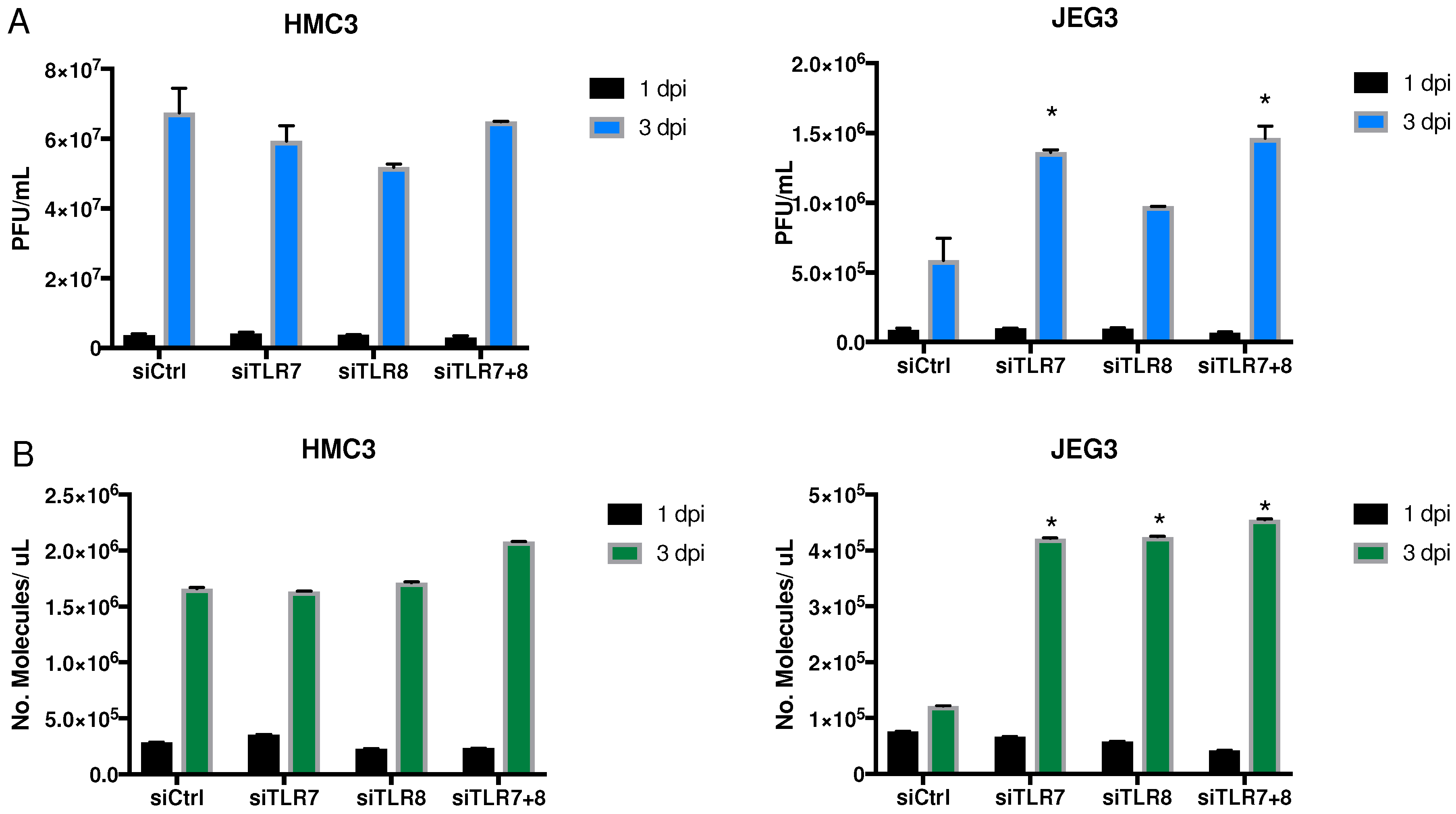
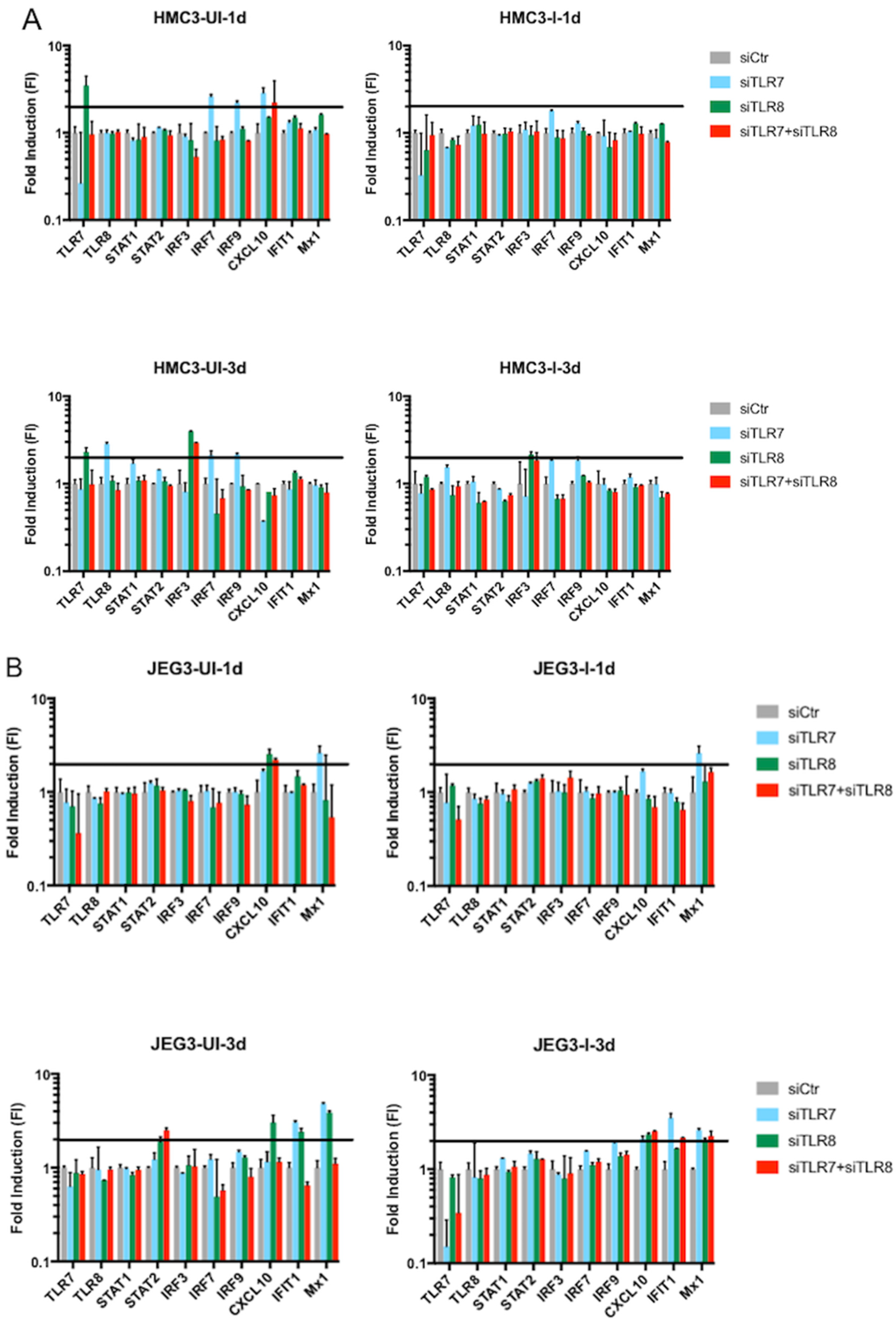
3.3. Effect of siRNA against TLR7, TLR8, or Both
3.3.1. Viral Replication
3.3.2. Innate Immune Response in Microglia and Placenta Cells
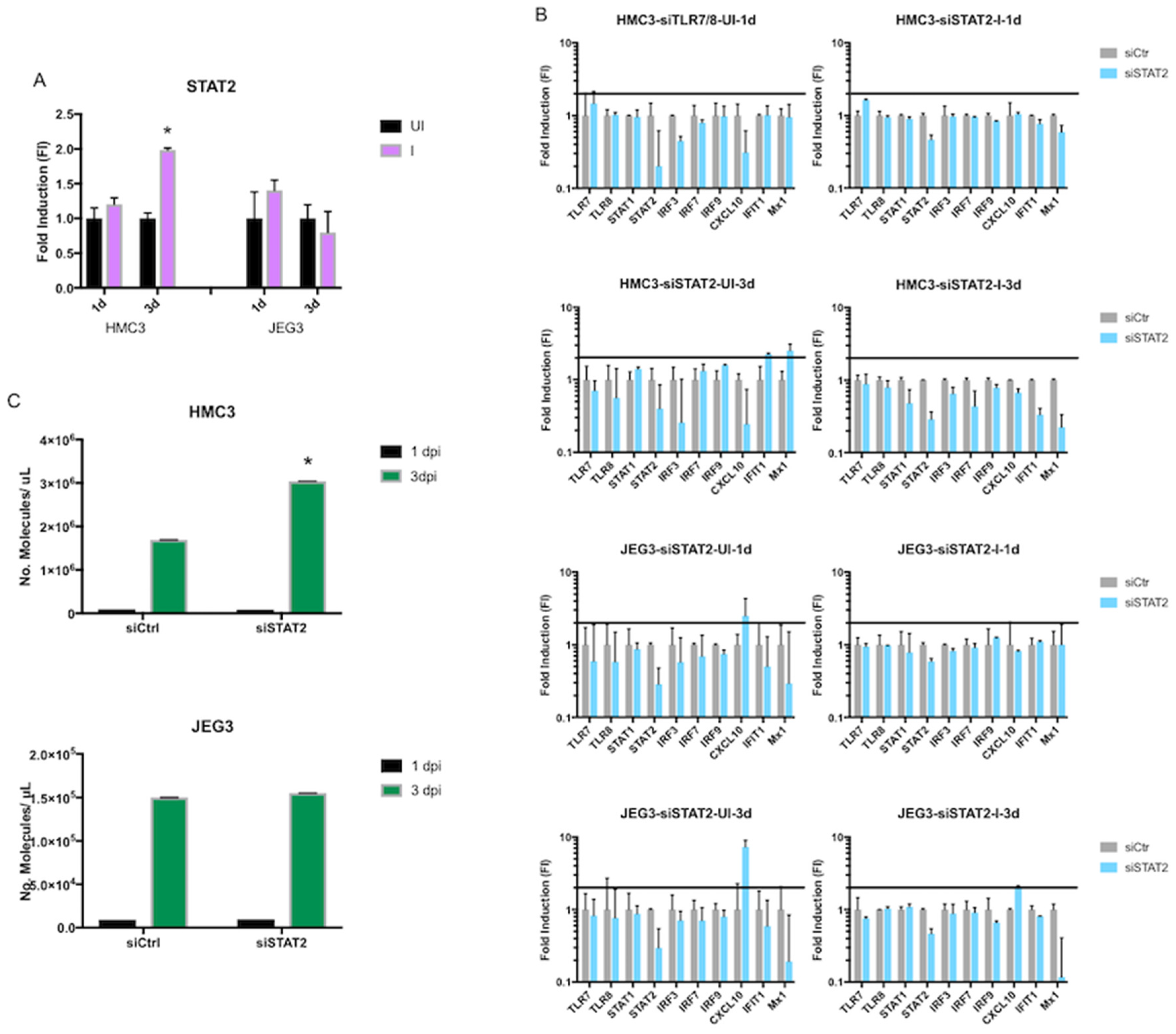
3.4. Role of STAT2 in Host Intracellular Response after ZIKV Infection
3.5. Role of Viral Receptor AXL in Microglia and Placenta Cells
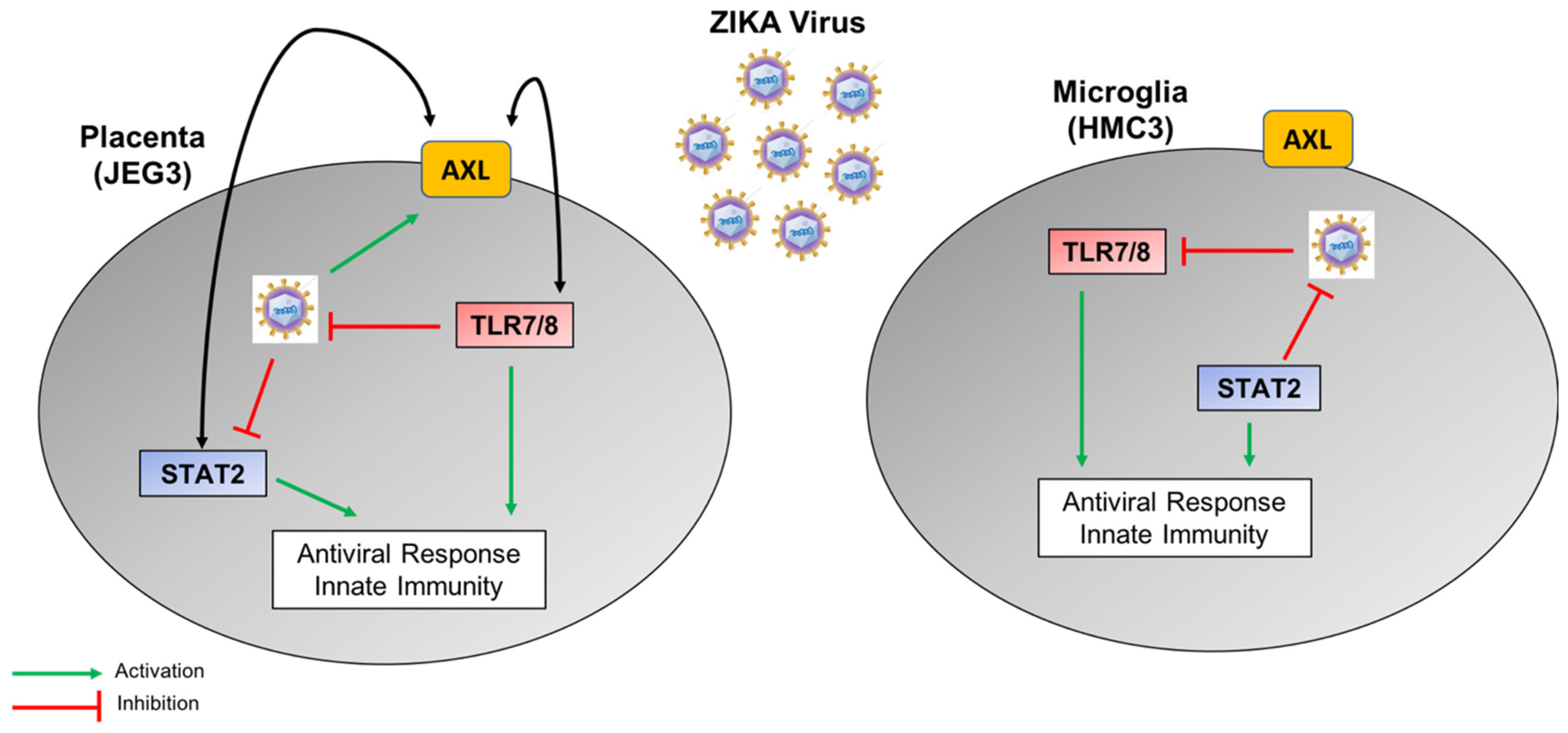
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dick, G.W.; Kitchen, S.F.; Haddow, A.J. Zika virus. I. Isolations and serological specificity. Trans. R. Soc. Trop. Med. Hyg. 1952, 46, 509–520. [Google Scholar] [CrossRef]
- White, M.K.; Wollebo, H.S.; David Beckham, J.; Tyler, K.L.; Khalili, K. Zika virus: An emergent neuropathological agent. Ann. Neurol. 2016, 80, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Russo, F.B.; Jungmann, P.; Beltrao-Braga, P.C.B. Zika infection and the development of neurological defects. Cell. Microbiol. 2017, 19. [Google Scholar] [CrossRef] [PubMed]
- Musso, D. Zika Virus Transmission from French Polynesia to Brazil. Emerg. Infect. Dis. 2015, 21, 1887. [Google Scholar] [CrossRef] [PubMed]
- D’Ortenzio, E.; Matheron, S.; Yazdanpanah, Y.; de Lamballerie, X.; Hubert, B.; Piorkowski, G.; Maquart, M.; Descamps, D.; Damond, F.; Leparc-Goffart, I. Evidence of Sexual Transmission of Zika Virus. N. Engl. J. Med. 2016, 374, 2193–2195. [Google Scholar] [CrossRef] [PubMed]
- Moreira, J.; Lamas, C.C.; Siqueira, A. Sexual Transmission of Zika Virus: Implications for Clinical Care and Public Health Policy. Clin. Infect. Dis. 2016, 63, 141–142. [Google Scholar] [CrossRef] [PubMed]
- McDonald, E.M.; Duggal, N.K.; Brault, A.C. Pathogenesis and sexual transmission of Spondweni and Zika viruses. PLoS Negl. Trop. Dis. 2017, 11, e0005990. [Google Scholar] [CrossRef] [PubMed]
- Musso, D.; Richard, V.; Teissier, A.; Stone, M.; Lanteri, M.C.; Latoni, G.; Alsina, J.; Reik, R.; Busch, M.P.; Recipient, E.; et al. Detection of Zika virus RNA in semen of asymptomatic blood donors. Clin. Microbiol. Infect. 2017, 23. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, D.A. The Origins and Emergence of Zika Virus, the Newest TORCH Infection: What’s Old Is New Again. Arch. Pathol. Lab. Med. 2017, 141, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Arora, N.; Sadovsky, Y.; Dermody, T.S.; Coyne, C.B. Microbial Vertical Transmission during Human Pregnancy. Cell Host Microbe 2017, 21, 561–567. [Google Scholar] [CrossRef] [PubMed]
- Jurado, K.A.; Simoni, M.K.; Tang, Z.; Uraki, R.; Hwang, J.; Householder, S.; Wu, M.; Lindenbach, B.D.; Abrahams, V.M.; Guller, S.; et al. Zika virus productively infects primary human placenta-specific macrophages. JCI Insight 2016, 1. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, A.Z.; Yu, W.; Hill, D.A.; Reyes, C.A.; Schwartz, D.A. Placental Pathology of Zika Virus: Viral Infection of the Placenta Induces Villous Stromal Macrophage (Hofbauer Cell) Proliferation and Hyperplasia. Arch. Pathol. Lab. Med. 2017, 141, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Adibi, J.J.; Marques, E.T., Jr.; Cartus, A.; Beigi, R.H. Teratogenic effects of the Zika virus and the role of the placenta. Lancet 2016, 387, 1587–1590. [Google Scholar] [CrossRef]
- Luo, H.; Winkelmann, E.R.; Fernandez-Salas, I.; Li, L.; Mayer, S.V.; Danis-Lozano, R.; Sanchez-Casas, R.M.; Vasilakis, N.; Tesh, R.; Barrett, A.D.; Weaver, S.C.; et al. Zika, dengue and yellow fever viruses induce differential anti-viral immune responses in human monocytic and first trimester trophoblast cells. Antiviral Res. 2018, 151, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Vanwalscappel, B.; Tada, T.; Landau, N.R. Toll-like receptor agonist R848 blocks Zika virus replication by inducing the antiviral protein viperin. Virology 2018, 522, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Grant, A.; Ponia, S.S.; Tripathi, S.; Balasubramaniam, V.; Miorin, L.; Sourisseau, M.; Schwarz, M.C.; Sanchez-Seco, M.P.; Evans, M.J.; Best, S.M.; et al. Zika Virus Targets Human STAT2 to Inhibit Type I Interferon Signaling. Cell Host Microbe 2016, 19, 882–890. [Google Scholar] [CrossRef] [PubMed]
- Hertzog, J.; Dias Junior, A.G.; Rigby, R.E.; Donald, C.L.; Mayer, A.; Sezgin, E.; Song, C.; Jin, B.; Hublitz, P.; Eggeling, C.; et al. Infection with a Brazilian isolate of Zika virus generates RIG-I stimulatory RNA and the viral NS5 protein blocks type I IFN induction and signaling. Eur. J. Immunol. 2018, 48, 1120–1136. [Google Scholar] [CrossRef] [PubMed]
- Pickett, B.E.; Sadat, E.L.; Zhang, Y.; Noronha, J.M.; Squires, R.B.; Hunt, V.; Liu, M.; Kumar, S.; Zaremba, S.; Gu, Z.; et al. ViPR: An open bioinformatics database and analysis resource for virology research. Nucleic Acids Res. 2012, 40, D593–D598. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Yamada, K.D.; Tomii, K.; Katoh, K. Parallelization of MAFFT for large-scale multiple sequence alignments. Bioinformatics 2018, 34, 2490–2492. [Google Scholar] [CrossRef] [PubMed]
- Pickett, B.E.; Liu, M.; Sadat, E.L.; Squires, R.B.; Noronha, J.M.; He, S.; Jen, W.; Zaremba, S.; Gu, Z.; Zhou, L.; et al. Metadata-driven comparative analysis tool for sequences (meta-CATS): An automated process for identifying significant sequence variations that correlate with virus attributes. Virology 2013, 447, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Tarca, A.L.; Draghici, S.; Khatri, P.; Hassan, S.S.; Mittal, P.; Kim, J.S.; Kim, C.J.; Kusanovic, J.P.; Romero, R. A novel signaling pathway impact analysis. Bioinformatics 2009, 25, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Nowakowski, T.J.; Pollen, A.A.; Di Lullo, E.; Sandoval-Espinosa, C.; Bershteyn, M.; Kriegstein, A.R. Expression Analysis Highlights AXL as a Candidate Zika Virus Entry Receptor in Neural Stem Cells. Cell Stem Cell 2016, 18, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Hamel, R.; Dejarnac, O.; Wichit, S.; Ekchariyawat, P.; Neyret, A.; Luplertlop, N.; Perera-Lecoin, M.; Surasombatpattana, P.; Talignani, L.; Thomas, F.; et al. Biology of Zika Virus Infection in Human Skin Cells. J. Virol. 2015, 89, 8880–8896. [Google Scholar] [CrossRef] [PubMed]
- Bayer, A.; Lennemann, N.J.; Ouyang, Y.; Bramley, J.C.; Morosky, S.; Marques, E.T., Jr.; Cherry, S.; Sadovsky, Y.; Coyne, C.B. Type III Interferons Produced by Human Placental Trophoblasts Confer Protection against Zika Virus Infection. Cell Host Microbe 2016, 19, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Quicke, K.M.; Bowen, J.R.; Johnson, E.L.; McDonald, C.E.; Ma, H.; O’Neal, J.T.; Rajakumar, A.; Wrammert, J.; Rimawi, B.H.; Pulendran, B.; et al. Zika Virus Infects Human Placental Macrophages. Cell Host Microbe 2016, 20, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, V.; Yuen, K.S.; Chan, J.F.; Chan, C.P.; Wang, P.H.; Cai, J.P.; Zhang, S.; Liang, M.; Kok, K.H.; Chan, C.P.; et al. Selective Activation of Type II Interferon Signaling by Zika Virus NS5 Protein. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Bowen, J.R.; Quicke, K.M.; Maddur, M.S.; O’Neal, J.T.; McDonald, C.E.; Fedorova, N.B.; Puri, V.; Shabman, R.S.; Pulendran, B.; Suthar, M.S. Zika Virus Antagonizes Type I Interferon Responses during Infection of Human Dendritic Cells. PLoS Pathog. 2017, 13, e1006164. [Google Scholar] [CrossRef] [PubMed]
- Meertens, L.; Labeau, A.; Dejarnac, O.; Cipriani, S.; Sinigaglia, L.; Bonnet-Madin, L.; Le Charpentier, T.; Hafirassou, M.L.; Zamborlini, A.; Cao-Lormeau, V.M.; et al. Axl Mediates ZIKA Virus Entry in Human Glial Cells and Modulates Innate Immune Responses. Cell Rep. 2017, 18, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Wells, M.F.; Salick, M.R.; Wiskow, O.; Ho, D.J.; Worringer, K.A.; Ihry, R.J.; Kommineni, S.; Bilican, B.; Klim, J.R.; Hill, E.J.; et al. Genetic Ablation of AXL Does Not Protect Human Neural Progenitor Cells and Cerebral Organoids from Zika Virus Infection. Cell Stem Cell 2016, 19, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Yang, Y.F.; Yang, Y.; Zou, P.; Chen, J.; He, Y.; Shui, S.L.; Cui, Y.R.; Bai, R.; Liang, Y.J.; et al. AXL promotes Zika virus infection in astrocytes by antagonizing type I interferon signalling. Nat. Microbiol. 2018, 3, 302–309. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HMC3 (Human Microglia) | JEG3 (Human Placenta) | |||||||
|---|---|---|---|---|---|---|---|---|
| 1 dpi (I vs. UI) | 3 dpi (I vs. UI) | Infected (3 dpi vs. 1 dpi) | Uninfected (3 dpi vs. 1 dpi) | 1 dpi (I vs. UI) | 3 dpi (I vs, UI) | Infected (3 dpi vs. 1 dpi) | Uninfected (3 dpi vs. 1 dpi) | |
| Jak-STAT signaling pathway | 7.88 × 10−6 | 6.85 × 10−8 | 2.77 × 10−8 | 6.58 × 10−8 | 7.90 × 10−4 | 1.26 × 10−7 | 1.06 × 10−13 | 2.05 × 10−7 |
| NF-kappa B signaling pathway | 2.05 × 10−3 | 2.13 × 10−11 | 6.23 × 10−10 | 5.13 × 10−10 | 8.00 × 10−8 | 2.12 × 10−9 | 2.16 × 10−9 | 1.02 × 10−11 |
| Toll-like receptor signaling pathway | 9.21 × 10−12 | 1.44 × 10−29 | 5.48 × 10−28 | 2.87 × 10−20 | 1.25 × 10−23 | 1.02 × 10−31 | 3.44 × 10−33 | 1.22 × 10−27 |
| Toll-like receptor TLR1:TLR2 cascade | N.S. | 1.55 × 10−5 | 1.43 × 10−7 | 1.04 × 10−5 | 4.89 × 10−3 | 9.80 × 10−11 | 1.20 × 10−8 | 7.44 × 10−4 |
| Toll-like receptor TLR6:TLR2 cascade | N.S. | 1.36 × 10−5 | 1.54 × 10−7 | 1.17 × 10−5 | 4.92 × 10−3 | 9.00 × 10−11 | 1.24 × 10−8 | 7.25 × 10−4 |
| TRIF-mediated TLR3/TLR4 signaling | N.S. | 7.07 × 10−7 | 3.30 × 10−7 | 4.47 × 10−7 | 5.53 × 10−3 | 1.11 × 10−8 | 2.12 × 10−3 | 4.70 × 10−7 |
| TRAF6-mediated IRF7 activation in TLR7/8 or 9 signaling | 2.23 × 10−2 | 2.20 × 10−3 | 6.63 × 10−9 | 9.52 × 10−6 | N.S. | N.S. | 1.98 × 10−3 | 1.69 × 10−8 |
| Caspase activation via extrinsic apoptotic signaling pathway | N.S. | N.S. | N.S. | 2.53 × 10−2 | 8.02 × 10−3 | 2.24 × 10−8 | 4.23 × 10−4 | 2.34 × 10−4 |
| IFN-alpha signaling pathway | N.S. | 3.34 × 10−3 | 2.63 × 10−3 | 7.99 × 10−3 | N.S. | 1.07 × 10−5 | 2.40 × 10−7 | N.S. |
| PI3K-Akt signaling pathway | N.S. | 2.34 × 10−3 | 1.50 × 10−3 | N.S. | 2.86 × 10−2 | 4.47 × 10−5 | 4.66 × 10−6 | N.S. |
| Toll-like receptor 7/8 (TLR7/8) cascade | N.S. | 7.57 × 10−6 | 5.30 × 10−10 | 8.80 × 10−6 | N.S. | N.S. | 3.18 × 10−2 | 1.00 × 10−5 |
| Toll-like receptor 9 (TLR9) cascade | N.S. | 9.55 × 10−6 | 6.03 × 10−10 | 1.17 × 10−5 | N.S. | N.S. | 3.77 × 10−2 | 1.28 × 10−5 |
| TRAF6-mediated induction of NFkB and MAP kinases upon TLR7/8 or 9 activation | N.S. | 5.13 × 10−4 | 5.82 × 10−8 | 1.97 × 10−4 | N.S. | N.S. | 2.74 × 10−2 | 5.23 × 10−4 |
| TRAF6-mediated IRF7 activation | N.S. | 4.14 × 10−4 | 2.92 × 10−4 | 3.78 × 10−6 | N.S. | N.S. | 5.97 × 10−4 | 9.19 × 10−3 |
| Regulation of nuclear SMAD2/3 signaling | N.S. | 7.12 × 10−5 | 1.93 × 10−3 | N.S. | N.S. | N.S. | N.S. | 4.02 × 10−2 |
| TRAF3-dependent IRF activation pathway | N.S. | 2.58 × 10−3 | 2.02 × 10−3 | 3.55 × 10−3 | N.S. | N.S. | N.S. | N.S. |
| TRIF-mediated programmed cell death | N.S. | N.S. | N.S. | N.S. | N.S. | 1.59 × 10−8 | 1.94 × 10−3 | 8.05 × 10−4 |
| Apoptosis | N.S. | 2.53 × 10−2 | N.S. | 3.22 × 10−2 | N.S. | N.S. | N.S. | N.S. |
| Regulation of IFNA signaling | N.S. | N.S. | N.S. | N.S. | N.S. | 1.85 × 10−2 | 2.23 × 10−4 | N.S. |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martinez Viedma, M.d.P.; Pickett, B.E. Characterizing the Different Effects of Zika Virus Infection in Placenta and Microglia Cells. Viruses 2018, 10, 649. https://doi.org/10.3390/v10110649
Martinez Viedma MdP, Pickett BE. Characterizing the Different Effects of Zika Virus Infection in Placenta and Microglia Cells. Viruses. 2018; 10(11):649. https://doi.org/10.3390/v10110649
Chicago/Turabian StyleMartinez Viedma, Maria del Pilar, and Brett E. Pickett. 2018. "Characterizing the Different Effects of Zika Virus Infection in Placenta and Microglia Cells" Viruses 10, no. 11: 649. https://doi.org/10.3390/v10110649
APA StyleMartinez Viedma, M. d. P., & Pickett, B. E. (2018). Characterizing the Different Effects of Zika Virus Infection in Placenta and Microglia Cells. Viruses, 10(11), 649. https://doi.org/10.3390/v10110649