Metagenomic Next-Generation Sequencing Reveals Individual Composition and Dynamics of Anelloviruses during Autologous Stem Cell Transplant Recipient Management

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Metagenomic Workflow and Bioinformatic Analysis
2.3. TTV Quantification
2.4. Statistical Analysis
2.5. Ethics Statement
3. Results
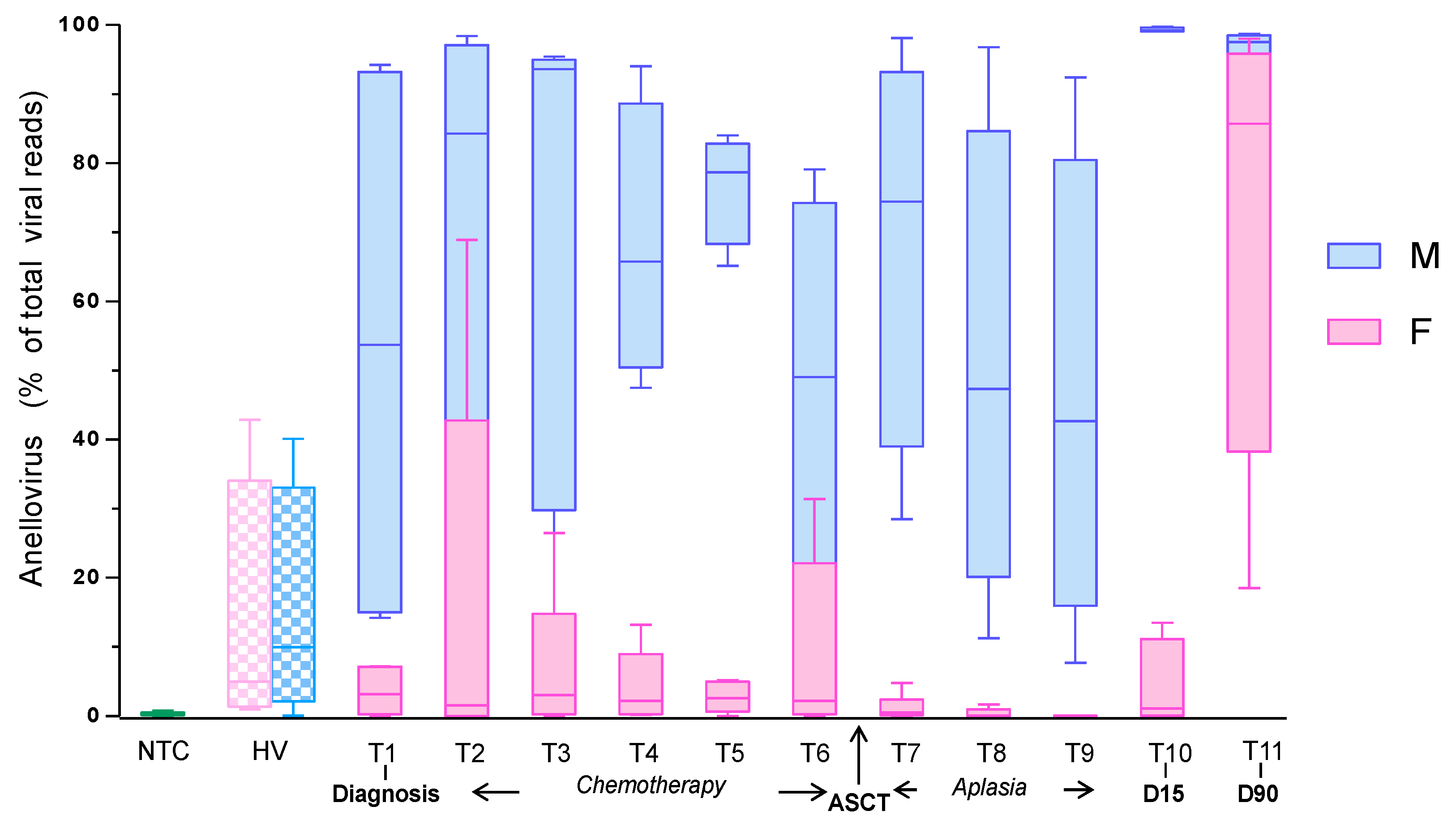
3.1. Anellovirus Dynamics within the Plasma Virome
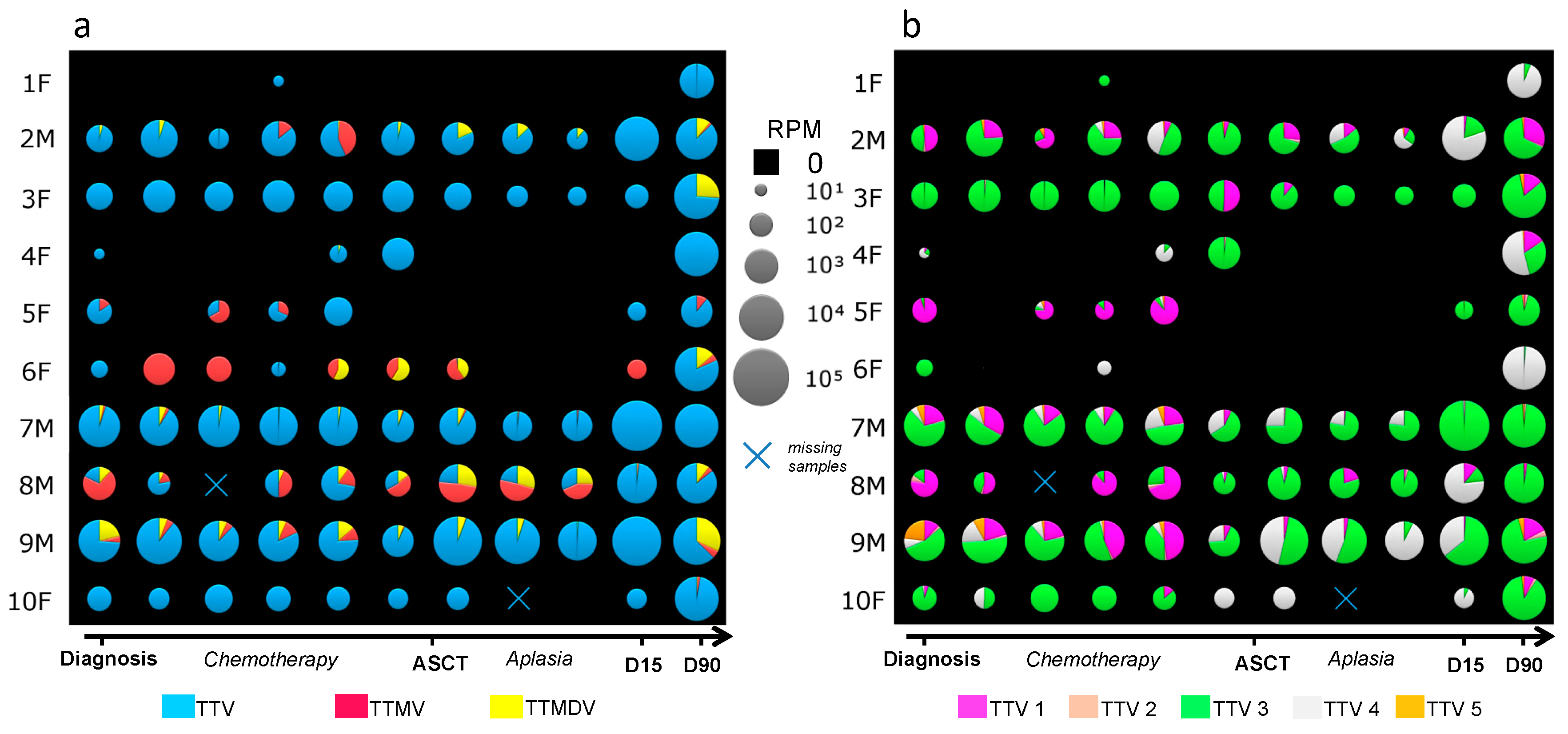
3.2. Anellovirus Abundance and Composition among Individuals
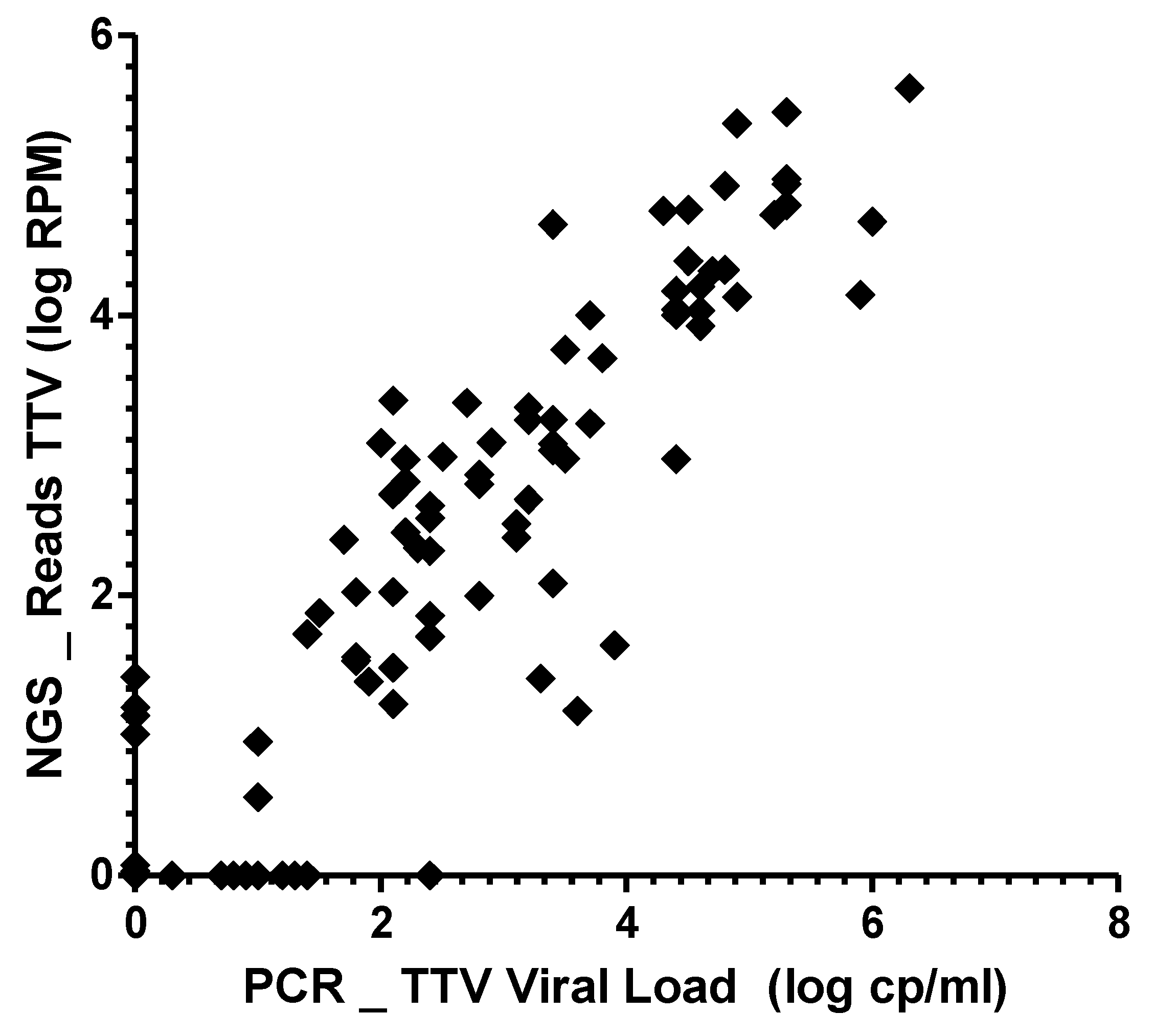
3.3. Correlation between mNGS and qPCR Assays for TTV
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Moreto, A.; Fariñas-Alvarez, C.; Puente, M.; Ocejo-Vinyals, J.G.; Sánchez-Velasco, P.; Horcajada, J.P.; Batlle, A.; Montes, C.; Santos, F.; Conde, E.; et al. Mannose-binding lectin gene variants and infections in patients receiving autologous stem cell transplantation. BMC Immunol. 2014, 15, 17. [Google Scholar] [CrossRef] [PubMed]
- De Vlaminck, I.; Khush, K.K.; Strehl, C.; Kohli, B.; Luikart, H.; Neff, N.F.; Okamoto, J.; Snyder, T.M.; Cornfield, D.N.; Nicolls, M.R.; et al. Temporal response of the human virome to immunosuppression and antiviral therapy. Cell 2013, 155, 1178–1187. [Google Scholar] [CrossRef] [PubMed]
- Strassl, R.; Schiemann, M.; Doberer, K.; Görzer, I.; Puchhammer-Stöckl, E.; Eskandary, F.; Kikic, Ž.; Gualdoni, G.A.; Vossen, M.G.; Rasoul-Rockenschaub, S.; et al. Quantification of Torque Teno Virus Viremia as a Prospective Biomarker for Infectious Disease in Kidney Allograft Recipients. J. Infect. Dis. 2018, 218, 1191–1199. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.K.; Relman, D.A.; Pinsky, B.A. The Human Virome: Implications for Clinical Practice in Transplantation Medicine. J. Clin. Microbiol. 2017, 55, 2884–2893. [Google Scholar] [CrossRef] [PubMed]
- Focosi, D.; Antonelli, G.; Pistello, M.; Maggi, F. Torquetenovirus: The human virome from bench to bedside. Clin. Microbiol. Infect. 2016, 22, 589–593. [Google Scholar] [CrossRef] [PubMed]
- Maggi, F.; Andreoli, E.; Lanini, L.; Fornai, C.; Vatteroni, M.; Pistello, M.; Presciuttini, S.; Bendinelli, M. Relationships between Total Plasma Load of Torquetenovirus (TTV) and TTV Genogroups Carried. J. Clin. Microbiol. 2005, 43, 4807–4810. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, F.; Shan, T.-L.; Deng, X.; Delwart, E.; Feng, X.-P. A novel species of torque teno mini virus (TTMV) in gingival tissue from chronic periodontitis patients. Sci. Rep. 2016, 6, 26739. [Google Scholar] [CrossRef] [PubMed]
- Rocchi, J.; Ricci, V.; Albani, M.; Lanini, L.; Andreoli, E.; Macera, L.; Pistello, M.; Ceccherini-Nelli, L.; Bendinelli, M.; Maggi, F. Torquetenovirus DNA drives proinflammatory cytokines production and secretion by immune cells via toll-like receptor 9. Virology 2009, 394, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Vu, D.-L.; Cordey, S.; Simonetta, F.; Brito, F.; Docquier, M.; Turin, L.; van Delden, C.; Boely, E.; Dantin, C.; Pradier, A.; et al. Human pegivirus persistence in the human blood virome after allogeneic haematopoietic stem cell transplantation. Clin. Microbiol. Infect. 2018. [Google Scholar] [CrossRef] [PubMed]
- Bal, A.; Pichon, M.; Picard, C.; Casalegno, J.S.; Valette, M.; Schuffenecker, I.; Billard, L.; Vallet, S.; Vilchez, G.; Cheynet, V.; et al. Quality control implementation for universal characterization of DNA and RNA viruses in clinical respiratory samples using single metagenomic next-generation sequencing workflow. BMC Infect. Dis. 2018, 18, 537. [Google Scholar] [CrossRef] [PubMed]
- Bazinet, A.L.; Ondov, B.D.; Sommer, D.D.; Ratnayake, S. BLAST-based validation of metagenomic sequence assignments. PeerJ 2018, 6, e4892. [Google Scholar] [CrossRef] [PubMed]
- Biagini, P. Classification of TTV and related viruses (anelloviruses). Curr. Top. Microbiol. Immunol. 2009, 331, 21–33. [Google Scholar] [PubMed]
- Kulifaj, D.; Durgueil-Lariviere, B.; Meynier, F.; Munteanu, E.; Pichon, N.; Dubé, M.; Joannes, M.; Essig, M.; Hantz, S.; Barranger, C.; et al. Development of a standardized real time PCR for Torque teno viruses (TTV) viral load detection and quantification: A new tool for immune monitoring. J. Clin. Virol. 2018, 105, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Moustafa, A.; Xie, C.; Kirkness, E.; Biggs, W.; Wong, E.; Turpaz, Y.; Bloom, K.; Delwart, E.; Nelson, K.E.; Venter, J.C.; et al. The blood DNA virome in 8000 humans. PLoS Pathog. 2017, 13, e1006292. [Google Scholar] [CrossRef]
- Blatter, J.A.; Sweet, S.C.; Conrad, C.; Danziger-Isakov, L.A.; Faro, A.; Goldfarb, S.B.; Hayes, D.; Melicoff, E.; Schecter, M.; Storch, G.; et al. Anellovirus loads are associated with outcomes in pediatric lung transplantation. Pediatr. Transplant. 2018, 22, e13069. [Google Scholar] [CrossRef] [PubMed]
- Segura-Wang, M.; Görzer, I.; Jaksch, P.; Puchhammer-Stöckl, E. Temporal dynamics of the lung and plasma viromes in lung transplant recipients. PLoS ONE 2018, 13, e0200428. [Google Scholar] [CrossRef] [PubMed]
- Haloschan, M.; Bettesch, R.; Görzer, I.; Weseslindtner, L.; Kundi, M.; Puchhammer-Stöckl, E. TTV DNA plasma load and its association with age, gender, and HCMV IgG serostatus in healthy adults. Age 2014, 36, 9716. [Google Scholar] [CrossRef] [PubMed]
- Hirokawa, K.; Utsuyama, M.; Hayashi, Y.; Kitagawa, M.; Makinodan, T.; Fulop, T. Slower immune system aging in women versus men in the Japanese population. Immun. Ageing A 2013, 10, 19. [Google Scholar] [CrossRef] [PubMed]
- Béland, K.; Dore-Nguyen, M.; Gagné, M.-J.; Patey, N.; Brassard, J.; Alvarez, F.; Halac, U. Torque Teno virus in children who underwent orthotopic liver transplantation: New insights about a common pathogen. J. Infect. Dis. 2014, 209, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Focosi, D.; Maggi, F.; Albani, M.; Macera, L.; Ricci, V.; Gragnani, S.; Di Beo, S.; Ghimenti, M.; Antonelli, G.; Bendinelli, M.; et al. Torquetenovirus viremia kinetics after autologous stem cell transplantation are predictable and may serve as a surrogate marker of functional immune reconstitution. J. Clin. Virol. 2010, 47, 189–192. [Google Scholar] [CrossRef] [PubMed]
- Albert, E.; Solano, C.; Giménez, E.; Focosi, D.; Pérez, A.; Macera, L.; Piñana, J.L.; Boluda, J.C.H.; Maggi, F.; Navarro, D. The kinetics of torque teno virus plasma DNA load shortly after engraftment predicts the risk of high-level CMV DNAemia in allogeneic hematopoietic stem cell transplant recipients. Bone Marrow Transplant. 2018, 53, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Wohlfarth, P.; Leiner, M.; Schoergenhofer, C.; Hopfinger, G.; Goerzer, I.; Puchhammer-Stoeckl, E.; Rabitsch, W. Torquetenovirus Dynamics and Immune Marker Properties in Patients Following Allogeneic Hematopoietic Stem Cell Transplantation: A Prospective Longitudinal Study. Biol. Blood Marrow Transplant. 2018, 24, 194–199. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bal, A.; Sarkozy, C.; Josset, L.; Cheynet, V.; Oriol, G.; Becker, J.; Vilchez, G.; Sesques, P.; Mallet, F.; Pachot, A.; et al. Metagenomic Next-Generation Sequencing Reveals Individual Composition and Dynamics of Anelloviruses during Autologous Stem Cell Transplant Recipient Management. Viruses 2018, 10, 633. https://doi.org/10.3390/v10110633
Bal A, Sarkozy C, Josset L, Cheynet V, Oriol G, Becker J, Vilchez G, Sesques P, Mallet F, Pachot A, et al. Metagenomic Next-Generation Sequencing Reveals Individual Composition and Dynamics of Anelloviruses during Autologous Stem Cell Transplant Recipient Management. Viruses. 2018; 10(11):633. https://doi.org/10.3390/v10110633
Chicago/Turabian StyleBal, Antonin, Clémentine Sarkozy, Laurence Josset, Valérie Cheynet, Guy Oriol, Jérémie Becker, Gaëlle Vilchez, Pierre Sesques, François Mallet, Alexandre Pachot, and et al. 2018. "Metagenomic Next-Generation Sequencing Reveals Individual Composition and Dynamics of Anelloviruses during Autologous Stem Cell Transplant Recipient Management" Viruses 10, no. 11: 633. https://doi.org/10.3390/v10110633
APA StyleBal, A., Sarkozy, C., Josset, L., Cheynet, V., Oriol, G., Becker, J., Vilchez, G., Sesques, P., Mallet, F., Pachot, A., Morfin, F., Lina, B., Salles, G., Reynier, F., Trouillet-Assant, S., & Brengel-Pesce, K. (2018). Metagenomic Next-Generation Sequencing Reveals Individual Composition and Dynamics of Anelloviruses during Autologous Stem Cell Transplant Recipient Management. Viruses, 10(11), 633. https://doi.org/10.3390/v10110633