The Interplay between Human Cytomegalovirus and Pathogen Recognition Receptor Signaling


Abstract
:1. Introduction
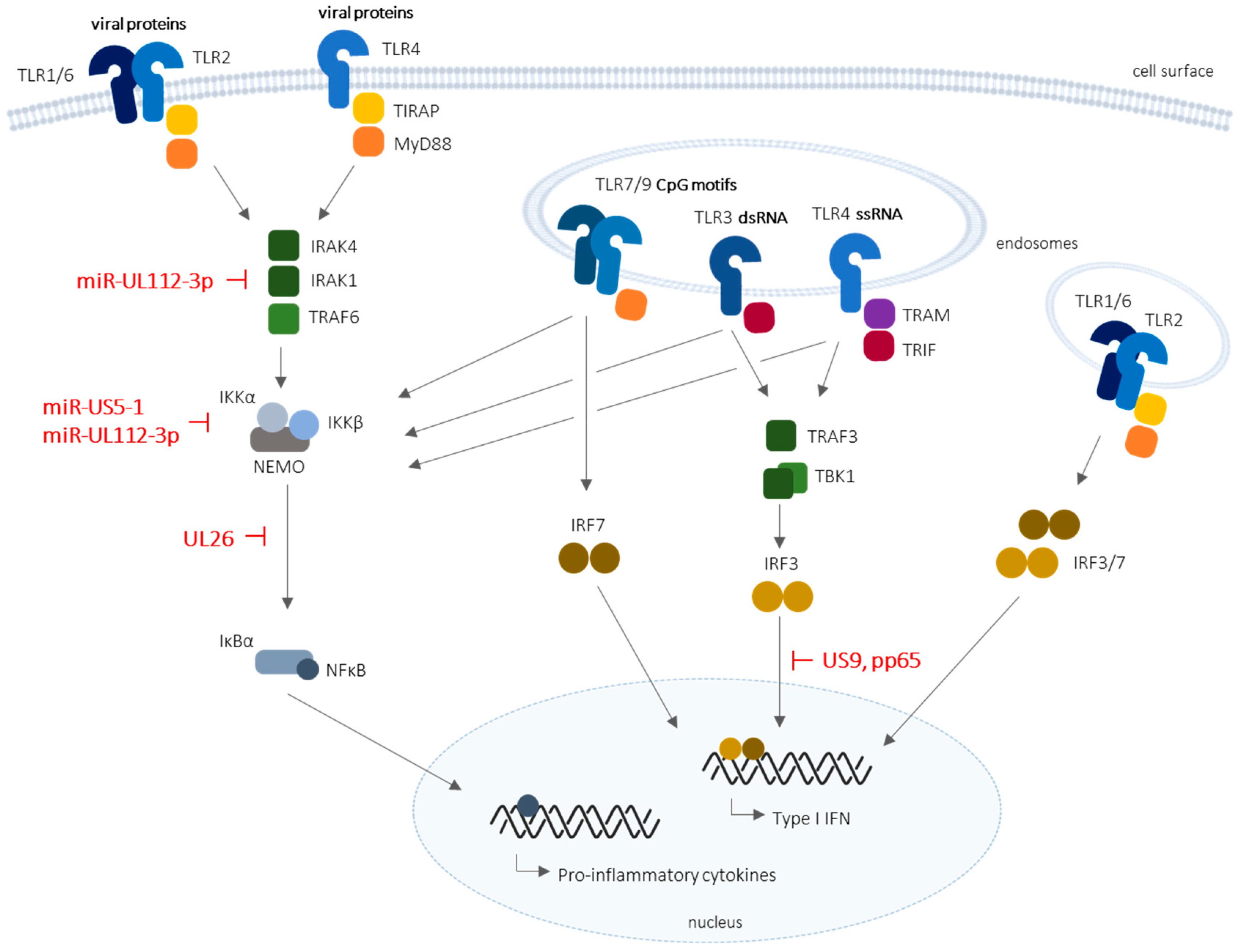
2. Toll-Like Receptors in HCMV Infection
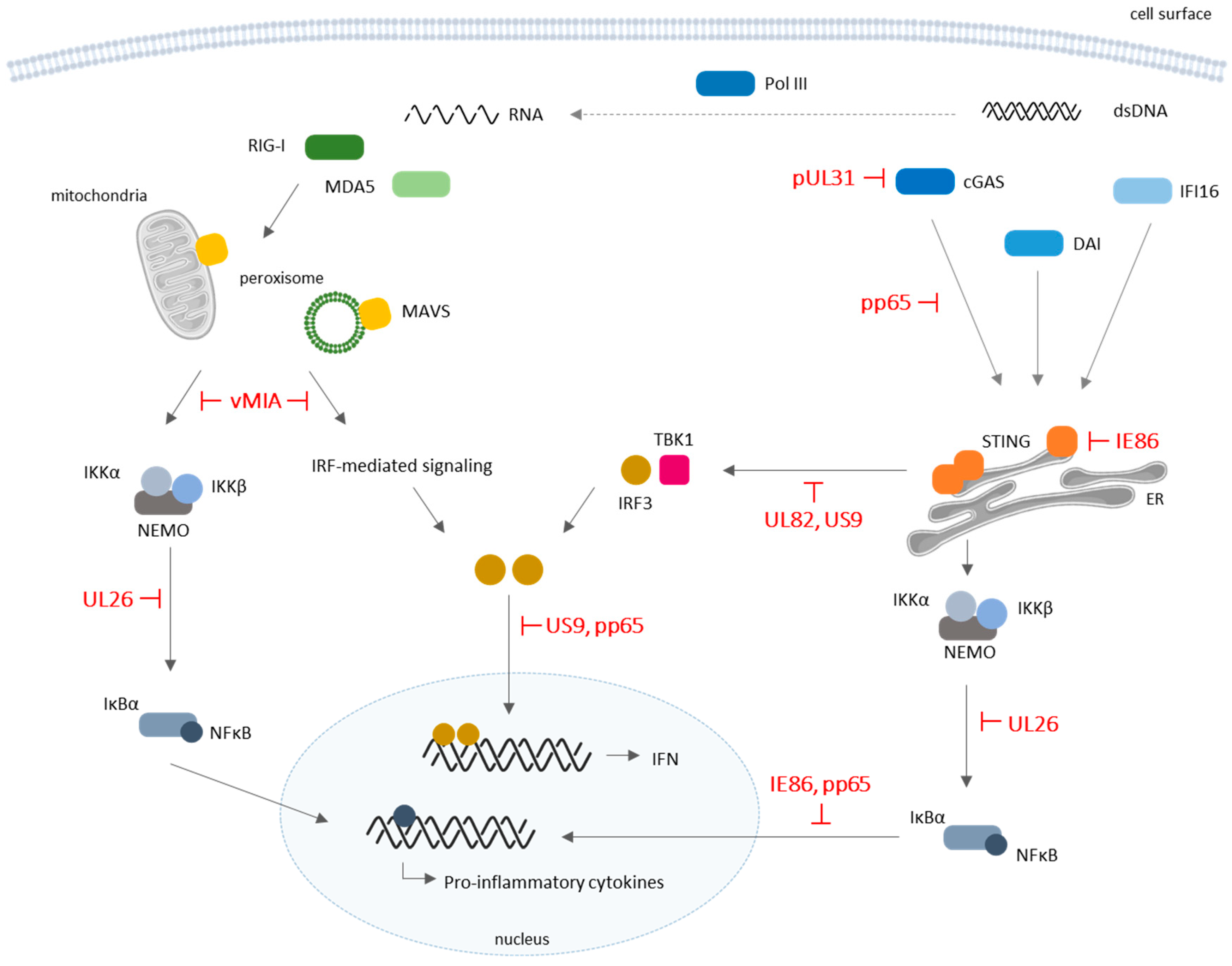
3. Cytosolic DNA Sensors in HCMV Infection
4. Is HCMV Antiviral Signaling Triggered by RNA Sensors?
5. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Cannon, M.J.; Schmid, D.S.; Hyde, T.B. Review of cytomegalovirus seroprevalence and demographic characteristics associated with infection. Rev. Med. Virol. 2010, 20, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Lancini, D.; Faddy, H.M.; Flower, R.; Hogan, C. Cytomegalovirus disease in immunocompetent adults. Med. J. Aust. 2014, 201, 578–580. [Google Scholar] [CrossRef] [PubMed]
- Dupont, L.; Reeves, M.B. Cytomegalovirus latency and reactivation: Recent insights into an age old problem. Rev. Med. Virol. 2016, 26, 75–89. [Google Scholar] [CrossRef] [PubMed]
- Limaye, A.; Kirby, K.; Rubenfeld, G.; Leisenring, W.; Bulger, E.; Neff, M.; Gibran, N.; Huand, M.; Santo, T.; Corey, L.; et al. Cytomegalovirus reactivation in critically ill immunocompetent patients. JAMA 2008, 300, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Pachnio, A.; Ciaurriz, M.; Begum, J.; Lal, N.; Zuo, J.; Beggs, A.; Moss, P. Cytomegalovirus Infection Leads to Development of High Frequencies of Cytotoxic Virus-Specific CD4+ T Cells Targeted to Vascular Endothelium. PLoS Pathog. 2016, 12, e1005832. [Google Scholar] [CrossRef] [PubMed]
- Kenneson, A.; Cannon, M.J. Review and meta-analysis of the epidemiology of congenital cytomegalovirus (CMV) infection. Rev. Med. Virol. 2007, 17, 253–276. [Google Scholar] [CrossRef] [PubMed]
- Grey, F.; Nelson, J. Identification and Function of Human Cytomegalovirus microRNAs. Gene Ther. 2009, 41, 186–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varnum, S.M.; Streblow, D.N.; Monroe, M.E.; Smith, P.; Auberry, K.J.; Paša-Tolić, L.; Wang, D.; Camp, D.G.; Rodland, K.; Wiley, S.; et al. Identification of Proteins in Human Cytomegalovirus (HCMV) Particles: The HCMV Proteome. J. Virol. 2004, 78, 10960–10966. [Google Scholar] [CrossRef] [PubMed]
- Kalejta, R.F. Tegument Proteins of Human Cytomegalovirus. Microbiol. Mol. Biol. Rev. 2008, 72, 249–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinzger, C.; Digel, M.; Jahn, G. Cytomegalovirus cell tropism. In Human Cytomegalovirus. Current Topics in Microbiology and Immunology; Shenk, T.E., Stinski, M.F., Eds.; Springer: Berlin/Heidelberg, Germany, 2008; Volume 325, pp. 63–83. [Google Scholar]
- Alwine, J.C. The Human Cytomegalovirus Assembly Compartment: A Masterpiece of Viral Manipulation of Cellular Processes That Facilitates Assembly and Egress. PLoS Pathog. 2012, 8, e1002878. [Google Scholar] [CrossRef] [PubMed]
- Cheung, A.K.L.; Abendroth, A.; Cunningham, A.L.; Slobedman, B. Viral gene expression during the establishment of human cytomegalovirus latent infection in myeloid progenitor cells. Blood 2006, 108, 3691–3699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isaacson, M.K.; Compton, T. Human Cytomegalovirus Glycoprotein B Is Required for Virus Entry and Cell-to-Cell Spread but Not for Virion Attachment, Assembly, or Egress. J. Virol. 2009, 83, 3891–3903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wille, P.T.; Wisner, T.W.; Ryckman, B.; Johnson, D.C. Human Cytomegalovirus (HCMV) Glycoprotein gB Promotes Virus Entry in Trans Acting as the Viral Fusion Protein Rather than as a Receptor-Binding Protein. MBio 2013, 4, e00332-13. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Huong, S.M.; Chiu, M.L.; Raab-Traub, N.; Huang, E.S. Epidermal growth factor receptor is a cellular receptor for human cytomegalovirus. Nature 2003, 424, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Huang, D.Y.; Huong, S.-M.; Huang, E.-S. Integrin αvβ3 is a coreceptor for human cytomegalovirus. Nat. Med. 2005, 11, 515–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogawa-Goto, K.; Tanaka, K.; Gibson, W.; Moriishi, E.; Miura, Y.; Kurata, T.; Irie, S.; Sata, T. Microtubule Network Facilitates Nuclear Targeting of Human Cytomegalovirus Capsid. J. Virol. 2003, 77, 8541–8547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bresnahan, W.; Shenk, T. A Subset of Viral Transcripts Packaged Within Human Cytomegalovirus Particles. Science 2000, 288, 2373–2376. [Google Scholar] [CrossRef] [PubMed]
- Melchjorsen, J. Learning from the messengers: Innate sensing of viruses and cytokine regulation of immunity-clues for treatments and vaccines. Viruses 2013, 5, 470–527. [Google Scholar] [CrossRef] [PubMed]
- Kumar, H.; Kawai, T.; Akira, S. Pathogen recognition by the innate immune system. Int. Rev. Immunol. 2011, 30, 16–34. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.R.; Kaminski, J.J.; Kurt-Jones, E.A.; Fitzgerald, K.A. Pattern recognition receptors and the innate immune response to viral infection. Viruses 2011, 3, 920–940. [Google Scholar] [CrossRef] [PubMed]
- Boehme, K.W.; Singh, J.; Perry, S.T.; Compton, T. Human Cytomegalovirus Elicits a Coordinated Cellular Antiviral Response via Envelope Glycoprotein B. J. Virol. 2004, 78, 1202–1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Netterwald, J.R.; Jones, T.R.; Britt, W.J.; Yang, S.; Mccrone, I.P.; Zhu, H. Postattachment Events Associated with Viral Entry Are Necessary for Induction of Interferon-Stimulated Genes by Human Cytomegalovirus. J. Virol. 2004, 78, 6688–6691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lester, S.N.; Li, K. Toll-like receptors in antiviral innate immunity. J. Mol. Biol. 2014, 426, 1246–1264. [Google Scholar] [CrossRef] [PubMed]
- Lund, J.; Sato, A.; Akira, S.; Medzhitov, R.; Iwasaki, A. Toll-like Receptor 9–mediated Recognition of Herpes Simplex Virus-2 by Plasmacytoid Dendritic Cells. J. Exp. Med. 2003, 198, 513–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boehme, K.W.; Guerrero, M.; Compton, T. Human Cytomegalovirus Envelope Glycoproteins B and H Are Necessary for TLR2 Activation in Permissive Cells. J. Immunol. 2006, 177, 7094–7102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexopoulou, L.; Czopik Holt, A.; Medzhitov, R.; Flavell, R.A. Recognition of double-stranded RNA and activation of NF-kappa B by Toll-like receptor 3. Nature 2001, 413, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Heil, F.; Hemmi, H.; Hochrein, H.; Ampenberger, F.; Kirschning, C.; Akira, S.; Lipford, G.; Wagner, H.; Bauer, S. Species-Specific Recognition of Single-Stranded RNA via Toll-like Receptor 7 and 8. Science 2004, 303, 1526–1529. [Google Scholar] [CrossRef] [PubMed]
- De Nardo, D. Toll-like receptors: Activation, signalling and transcriptional modulation. Cytokine 2015, 74, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Yew, K.H.; Carsten, B.; Harrison, C. Scavenger receptor A1 is required for sensing HCMV by endosomal TLR-3/-9 in monocytic THP-1 cells. Mol. Immunol. 2010, 47, 883–893. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, S.; Lowen, B.; Chan, G.; Davey, A.; Riddell, M.; Guilbert, L.J. Human Cytomegalovirus Interacts with Toll-like Receptor 2 and CD14 on Syncytiotrophoblasts to Stimulate Expression of TNFα mRNA and Apoptosis. Placenta 2009, 30, 994–1001. [Google Scholar] [CrossRef] [PubMed]
- Juckem, L.K.; Boehme, K.W.; Feire, A.L.; Compton, T. Differential Initiation of Innate Immune Responses Induced by Human Cytomegalovirus Entry into Fibroblast Cells. J. Immunol. 2008, 180, 4965–4977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mezger, M.; Bonin, M.; Kessler, T.; Gebhardt, F.; Einsele, H.; Loeffler, J. Toll-like receptor 3 has no critical role during early immune response of human monocyte-derived dendritic cells after infection with the human cytomegalovirus strain TB40E. Viral Immunol. 2009, 22, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Harwani, S.C.; Lurain, N.S.; Zariffard, M.R.; Spear, G.T. Differential inhibition of human cytomegalovirus (HCMV) by toll-like receptor ligands mediated by interferon-beta in human foreskin fibroblasts and cervical tissue. Virol. J. 2007, 4, 133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yew, K.H.; Carpenter, C.; Duncan, R.S.; Harrison, C.J. Human Cytomegalovirus Induces TLR4 Signaling Components in Monocytes Altering TIRAP, TRAM and Downstream Interferon-Beta and TNF-Alpha Expression. PLoS ONE 2012, 7, e44500. [Google Scholar] [CrossRef] [PubMed]
- Arcangeletti, M.C.; Germini, D.; Rodighiero, I.; Mirandola, P.; De Conto, F.; Medici, M.C.; Gatti, R.; Chezzi, C.; Calderaro, A. Toll-like receptor 4 is involved in the cell cycle modulation and required for effective human cytomegalovirus infection in THP-1 macrophages. Virology 2013, 440, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Iversen, A.-C.; Steinkjer, B.; Nilsen, N.; Bohnhorst, J.; Moen, S.H.; Vik, R.; Stephens, P.; Thomas, D.W.; Benedict, C.A.; Espevik, T. A proviral role for CpG in cytomegalovirus infection. J. Immunol. 2009, 182, 5672–5681. [Google Scholar] [CrossRef] [PubMed]
- Hancock, M.H.; Hook, L.M.; Mitchell, J.; Nelson, J.A. Human cytomegalovirus microRNAs miR-US5-1 and miR-UL112-3p block proinflammatory cytokine production in response to NF-κB-activating factors through direct downregulation of IKKα and IKKβ. MBio 2017, 8, e00109-17. [Google Scholar] [CrossRef] [PubMed]
- Landais, I.; Pelton, C.; Streblow, D.; DeFilippis, V.; McWeeney, S.; Nelson, J.A. Human Cytomegalovirus miR-UL112-3p Targets TLR2 and Modulates the TLR2/IRAK1/NFκB Signaling Pathway. PLoS Pathog. 2015, 11, e1004881. [Google Scholar] [CrossRef] [PubMed]
- Rathinam, V.A.K.; Fitzgerald, K.A. Cytosolic surveillance and antiviral immunity. Curr. Opin. Virol. 2011, 1, 455–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ori, D.; Murase, M.; Kawai, T. Cytosolic nucleic acid sensors and innate immune regulation. Int. Rev. Immunol. 2017, 36, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP Synthase is a Cytosolic DNA Sensor that Activates the Type-I Interferon Pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-D.; Wu, J.; Gao, D.; Wang, H.; Sun, L.; Chen, Z.J. Pivotal Roles of cGAS-cGAMP Signaling in Antiviral Defense and Immune Adjuvant Effects. Science 2013, 341, 1390–1394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gentili, M.; Kowal, J.; Tkach, M.; Satoh, T.; Lahaye, X.; Conrad, C.; Boyron, M.; Lombard, B.; Durand, S.; Kroemer, G.; et al. Transmisson of innate immune signaling by packaging of cGAMP in viral particles. Science 2015, 349, 1232–1236. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Ma, Z.; Barber, G.N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 2009, 461, 788–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, Y.; Chen, Z.J. STING Specifies IRF3 phosphorylation by TBK1 in the Cytosolic DNA Signaling Pathway. Sci. Signal 2013, 5, ra20. [Google Scholar] [CrossRef] [PubMed]
- Zhong, B.; Yang, Y.; Li, S.; Wang, Y.Y.; Li, Y.; Diao, F.; Lei, C.; He, X.; Zhang, L.; Tien, P.; et al. The Adaptor Protein MITA Links Virus-Sensing Receptors to IRF3 Transcription Factor Activation. Immunity 2008, 29, 538–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.G.; Wang, Y.Y.; Han, K.J.; Li, L.Y.; Zhai, Z.; Shu, H.B. VISA is an adapter protein required for virus-triggered IFN-β signaling. Mol. Cell 2005, 19, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Preston, C.M.; Harman, A.N.; Nicholl, M.J. Activation of Interferon Response Factor-3 in Human Cells Infected with Herpes Simplex Virus Type 1 or Human Cytomegalovirus. J. Virol. 2001, 75, 8909–8916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeFilippis, V.R.V.; Robinson, B.; Keck, T.M.; Hansen, S.G.; Nelson, J.A.; Früh, K.J.; Fru, K.J. Interferon regulatory factor 3 is necessary for induction of antiviral genes during human cytomegalovirus infection. J. Virol. 2006, 80, 1032–1037. [Google Scholar] [CrossRef] [PubMed]
- Lio, C.-W.J.; McDonald, B.; Takahashi, M.; Dhanwani, R.; Sharma, N.; Huang, J.; Pham, E.; Benedict, C.A.; Sharma, S. cGAS-STING Signaling Regulates Initial Innate Control of Cytomegalovirus Infection. J. Virol. 2016, 90, 7789–7797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paijo, J.; Döring, M.; Spanier, J.; Grabski, E.; Nooruzzaman, M.; Schmidt, T.; Witte, G.; Messerle, M.; Hornung, V.; Kaever, V.; et al. cGAS Senses Human Cytomegalovirus and Induces Type I Interferon Responses in Human Monocyte-Derived Cells. PLoS Pathog. 2016, 12, e1005546. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Liu, L.; Ma, D.; Liao, Y.; Lu, Y.; Huang, H.; Qin, W.; Liu, X.; Fang, F. Human cytomegalovirus triggers the assembly of AIM2 inflammasome in THP-1-derived macrophages. J. Med. Virol. 2017, 89, 2188–2195. [Google Scholar] [CrossRef] [PubMed]
- Gariano, G.R.; Dell’Oste, V.; Bronzini, M.; Gatti, D.; Luganini, A.; de Andrea, M.; Gribaudo, G.; Gariglio, M.; Landolfo, S. The intracellular DNA sensor IFI16 gene acts as restriction factor for human Cytomegalovirus replication. PLoS Pathog. 2012, 8. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Chen, J.; Cristea, I.M. Human cytomegalovirus tegument protein pUL83 inhibits IFI16- mediated DNA sensing for immune evasion. Cell Host Microbe 2013, 14. [Google Scholar] [CrossRef] [PubMed]
- DeFilippis, V.R.; Alvarado, D.; Sali, T.; Rothenburg, S.; Fruh, K. Human Cytomegalovirus Induces the Interferon Response via the DNA Sensor ZBP1. J. Virol. 2010, 84, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Fernandes-Alnemri, T.; Yu, J.-W.; Wu, J.; Datta, P.; Alnemri, E.S. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 2009, 458, 509–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unterholzner, L.; Keating, S.E.; Baran, M.; Horan, K.A.; Søren, B.; Sharma, S.; Sirois, C.M.; Jin, T.; Xiao, T.; Katherine, A.; et al. IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 2011, 11, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Ma, D.; Huang, H.; Lu, Y.; Liao, Y.; Liu, L.; Liu, X.; Fang, F. Interaction between HCMV pUL83 and human AIM2 disrupts the activation of the AIM2 inflammasome. Virol. J. 2017, 14, 34. [Google Scholar] [CrossRef] [PubMed]
- Biolatti, M.; Dell’Oste, V.; Pautasso, S.; von Einem, J.; Marschall, M.; Plachter, B.; Gariglio, M.; De Andrea, M.; Landolfo, S. Regulatory Interaction between the Cellular Restriction Factor IFI16 and Viral pp65 (pUL83) Modulates Viral Gene Expression and IFI16 Protein Stability. J. Virol. 2016, 90, 8238–8250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takaoka, A.; Wang, Z.; Choi, M.K.; Yanai, H.; Negishi, H.; Ban, T.; Lu, Y.; Miyagishi, M.; Kodama, T.; Honda, K.; et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 2007, 448, 501–505. [Google Scholar] [CrossRef] [PubMed]
- Browne, E.P.; Shenk, T. Human cytomegalovirus UL83-coded pp65 virion protein inhibits antiviral gene expression in infected cells. Proc. Natl. Acad. Sci. USA 2003, 100, 11439–11444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abate, D.A.; Watanabe, S.; Mocarski, E.S. Major human cytomegalovirus structural protein pp65 (ppUL83) prevents interferon response factor 3 activation in the interferon response. J. Virol. 2004, 78, 10995–11006. [Google Scholar] [CrossRef] [PubMed]
- Biolatti, M.; Dell’Oste, V.; Pautasso, S.; Gugliesi, F.; von Einem, J.; Krapp, C.; Jakobsen, M.R.; Borgogna, C.; Gariglio, M.; De Andrea, M.; et al. The Human Cytomegalovirus Tegument Protein pp65 (pUL83) Dampens Type I Interferon Production by Inactivating the DNA Sensor cGAS without Affecting STING. J. Virol. 2018, 92, e01774-17. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.-F.; Zou, H.-M.; Liao, B.-W.; Zhang, H.-Y.; Yang, Y.; Fu, Y.-Z.; Wang, S.-Y.; Luo, M.-H.; Wang, Y.-Y. Human Cytomegalovirus Protein UL31 Inhibits DNA Sensing of cGAS to Mediate Immune Evasion. Cell Host Microbe 2018, 24, 69–80.e4. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.Z.; Su, S.; Gao, Y.Q.; Wang, P.P.; Huang, Z.F.; Hu, M.M.; Luo, W.W.; Li, S.; Luo, M.H.; Wang, Y.Y.; et al. Human Cytomegalovirus Tegument Protein UL82 Inhibits STING-Mediated Signaling to Evade Antiviral Immunity. Cell Host Microbe 2017, 21, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Kim, Y.E.; Stinski, M.F.; Ahn, J.H.; Song, Y.J. Human cytomegalovirus IE2 86 kDa protein induces STING degradation and inhibits cGAMP-mediated IFN-β induction. Front. Microbiol. 2017, 8, 1854. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.T.; Bresnahan, W.A. Human Cytomegalovirus IE86 Attenuates Virus- and Tumor Necrosis Factor Alpha-Induced NFkB-Dependent Gene Expression. J. Virol. 2006, 80, 10763–10771. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.T.; Bresnahan, W.A. Human Cytomegalovirus Immediate-Early 2 Protein IE86 Blocks Virus-Induced Chemokine Expression. J. Virol. 2006, 80, 920–928. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Park, A.; Kang, S.; Lee, E.; Lee, T.A.; Ra, E.A.; Lee, J.; Lee, S.; Park, B. Human cytomegalovirus-encoded US9 targets MAVS and STING signaling to evade type i interferon immune responses. Nat. Commun. 2018, 9, 125. [Google Scholar] [CrossRef] [PubMed]
- Mathers, C.; Schafer, X.; Martinez-Sobrido, L.; Munger, J. The Human Cytomegalovirus UL26 Protein Antagonizes NF-κB Activation. J. Virol. 2014, 88, 14289–14300. [Google Scholar] [CrossRef] [PubMed]
- DeMeritt, I.B.; Podduturi, J.P.; Tilley, A.M.; Nogalski, M.T.; Yurochko, A.D. Prolonged activation of NF-κB by human cytomegalovirus promotes efficient viral replication and late gene expression. Virology 2006, 346, 15–31. [Google Scholar] [CrossRef] [PubMed]
- Poole, E.; King, C.A.; Sinclair, J.H.; Alcami, A. The UL144 gene product of human cytomegalovirus activates NFκB via a TRAF6-dependent mechanism. EMBO J. 2006, 25, 4390–4399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poole, E.; Groves, I.; Macdonald, A.; Pang, Y.; Alcami, A.; Sinclair, J. Identification of TRIM23 as a Cofactor Involved in the Regulation of NF-κB by Human Cytomegalovirus. J. Virol. 2009, 83, 3581–3590. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, M.; Kikuchi, M.; Matsumoto, K.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Foy, E.; Loo, Y.-M.; Gale, M.; Akira, S. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J. Immunol. 2005, 175, 2851–2858. [Google Scholar] [CrossRef] [PubMed]
- Belgnaoui, S.M.; Paz, S.; Hiscott, J. Orchestrating the interferon antiviral response through the mitochondrial antiviral signaling (MAVS) adapter. Curr. Opin. Immunol. 2011, 23, 564–572. [Google Scholar] [CrossRef] [PubMed]
- Biacchesi, S.; Merour, E.; Lamoureux, A.; Bernard, J.; Bremont, M. Both STING and MAVS Fish Orthologs Contribute to the Induction of Interferon Mediated by RIG-I. PLoS ONE 2012, 7, e47737. [Google Scholar] [CrossRef] [PubMed]
- Kowalinski, E.; Lunardi, T.; McCarthy, A.A.; Louber, J.; Brunel, J.; Grigorov, B.; Gerlier, D.; Cusack, S. Structural basis for the activation of innate immune pattern-recognition receptor RIG-I by viral RNA. Cell 2011, 147, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.; Ding, S.C.; Vela, A.; Kohlway, A.; Lindenbach, B.D.; Pyle, A.M. Structural insights into RNA recognition by RIG-I. Cell 2011, 147, 409–422. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Dixit, E.; Boulant, S.; Zhang, Y.; Lee, A.S.Y.; Odendall, C.; Shum, B.; Hacohen, N.; Chen, Z.J.; Whelan, S.P.; Fransen, M.; et al. Peroxisomes Are Signaling Platforms for Antiviral Innate Immunity. Cell 2010, 141, 668–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, D.C.; Gopalkrishnan, R.V.; Wu, Q.; Jankowsky, E.; Pyle, A.M.; Fisher, P.B. mda-5: An interferon-inducible putative RNA helicase with double-stranded RNA-dependent ATPase activity and melanoma growth-suppressive properties. Proc. Natl. Acad. Sci. USA 2002, 99, 637–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, F.; Sun, L.; Zheng, H.; Skaug, B.; Jiang, Q.-X.; Chen, Z.J. MAVS Forms Functional Prion-Like Aggregates to Activate and Propagate Antiviral Innate Immune Response. Cell 2011, 146, 448–461. [Google Scholar] [CrossRef] [PubMed]
- Bender, S.; Reuter, A.; Eberle, F.; Einhorn, E.; Binder, M.; Bartenschlager, R. Activation of Type I and III Interferon Response by Mitochondrial and Peroxisomal MAVS and Inhibition by Hepatitis C Virus. PLoS Pathog. 2016, 11, e1005264. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Cao, X.; Lu, J.; Huang, B.; Liu, Y.J.; Kato, N.; Shu, H.B.; Zhong, J. Hepatitis C virus NS4B blocks the interaction of STING and TBK1 to evade host innate immunity. J. Hepatol. 2013, 59, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.Y.; Chang, T.H.; Liang, J.J.; Chiang, R.L.; Lee, Y.L.; Liao, C.L.; Lin, Y.L. Dengue virus targets the adaptor protein MITA to subvert host innate immunity. PLoS Pathog. 2012, 8. [Google Scholar] [CrossRef] [PubMed]
- Holm, C.K.; Rahbek, S.H.; Gad, H.H.; Bak, R.O.; Jakobsen, M.R.; Jiang, Z.; Hansen, A.L.; Jensen, S.K.; Sun, C.; Thomsen, M.K.; et al. Influenza A virus targets a cGAS-independent STING pathway that controls enveloped RNA viruses. Nat. Commun. 2016, 7, 10680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguirre, S.; Maestre, A.M.; Pagni, S.; Patel, J.R.; Savage, T.; Gutman, D.; Maringer, K.; Bernal-Rubio, D.; Shabman, R.S.; Simon, V.; et al. DENV Inhibits Type I IFN Production in Infected Cells by Cleaving Human STING. PLoS Pathog. 2012, 8. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Goulet, M.-L.; Sze, A.; Hadj, S.B.; Belgnaoui, S.M.; Lababidi, R.R.; Zheng, C.; Fritz, J.H.; Olagnier, D.; Lin, R. RIG-I-Mediated STING Upregulation Restricts Herpes Simplex Virus 1 Infection. J. Virol. 2016, 90, 9406–9419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franz, K.M.; Neidermyer, W.J.; Tan, Y.-J.; Whelan, S.P.J.; Kagan, J.C. STING-dependent translation inhibition restricts RNA virus replication. Proc. Natl. Acad. Sci. USA 2018, 115, E2058–E2067. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.K.; Wang, Z.; Ban, T.; Yanai, H.; Lu, Y.; Koshiba, R.; Nakaima, Y.; Hangai, S.; Savitsky, D.; Nakasato, M.; et al. A selective contribution of the RIG-I-like receptor pathway to type I interferon responses activated by cytosolic DNA. Proc. Natl. Acad. Sci. USA 2009, 106, 17870–17875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melchjorsen, J.; Rintahaka, J.; Soby, S.; Horan, K.A.; Poltajainen, A.; Ostergaard, L.; Paludan, S.R.; Matikainen, S. Early Innate Recognition of Herpes Simplex Virus in Human Primary Macrophages Is Mediated via the MDA5/MAVS-Dependent and MDA5/MAVS/RNA Polymerase III-Independent Pathways. J. Virol. 2010, 84, 11350–11358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samanta, M.; Iwakiri, D.; Kanda, T.; Imaizumi, T.; Takada, K. EB virus-encoded RNAs are recognized by RIG-I and activate signaling to induce type I IFN. EMBO J. 2006, 25, 4207–4214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, Y.-H.; MacMillan, J.B.; Chen, Z.J. RNA Polymerase III Detects Cytosolic DNA and Induces Type-I Interferons Through the RIG-I Pathway. Cell 2009, 138, 576–591. [Google Scholar] [CrossRef] [PubMed]
- Ablasser, A.; Bauernfeind, F.; Hartmann, G.; Latz, E.; Fitzgerald, K.A.; Hornung, V. RIG-I dependent sensing of poly(dA-dT) via the induction of an RNA polymerase III transcribed RNA intermediate. Nat. Immunol. 2009, 10, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Marshall, E.E.; Bierle, C.J.; Brune, W.; Geballe, A.P. Essential Role for either TRS1 or IRS1 in Human Cytomegalovirus Replication. J. Virol. 2009, 83, 4112–4120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldmacher, V.S. vMIA, a viral inhibitor of apoptosis targeting mitochondria. Biochimie 2002, 84, 177–185. [Google Scholar] [CrossRef]
- McCormick, A.L.; Smith, V.L.; Chow, D.; Mocarski, E.S. Disruption of Mitochondrial Networks by the Human Cytomegalovirus UL37 Gene Product Viral Mitochondrion-Localized Inhibitor of Apoptosis. J. Virol. 2003, 77, 631–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castanier, C.; Garcin, D.; Vazquez, A.; Arnoult, D. Mitochondrial dynamics regulate the RIG-I-like receptor antiviral pathway. EMBO Rep. 2010, 11, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Magalhães, A.C.; Ferreira, A.R.; Gomes, S.; Vieira, M.; Gouveia, A.; Valença, I.; Islinger, M.; Nascimento, R.; Schrader, M.; Kagan, J.C.; et al. Peroxisomes are platforms for cytomegalovirus’ evasion from the cellular immune response. Sci. Rep. 2016, 6, 26028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, I. Degradation of RIG-I Following Cytomegalovirus Infection Is Independent of Apoptosis. Microbes Infect. 2009, 11, 973–979. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Viral Factor | Function | Cell Type (Strain) | Reference |
|---|---|---|---|
| miR-US5-1 | Targets IKKα and IKKβ to limit production of pro-inflammatory cytokines | NHDF cells (TB40/E) | [38] |
| miR-UL112-3p | Targets IKKα and IKKβ to limit production of pro-inflammatory cytokines | NHDF cells (TB40/E) | [38] |
| Targets and downregulates TLR2 and inhibits its dependent activation of IRAK1 and NF-κB signaling | NHDF cells (AD169) THP-1 (TB40E) | [39] | |
| pUL83 (pp65) | Inhibits IFN-α and antiviral gene expression by blocking IRF1 and NF-κB activity | HFF (AD169) | [62] |
| Modulates the rapid induction of an IFN-like response by inhibiting IRF3 activation | HFF (AD169) | [63] | |
| Dampens IFN-β production by selectively binding to cGAS, inactivating the cGAS/STING/IRF3 axis | HFF (TB40E) | [64] | |
| pUL31 | Downregulates antiviral gene expression by directly interacting with cGAS | HEK293T, HFF (AD169) | [65] |
| pUL82 | Prevents STING trafficking to the ER and impairs the formation of TBK1/IRF3/STING complexes | HEK293T, HFF, MLF (AD169) | [66] |
| pUL122 (IE86) | Mediates proteasome-dependent STING degradation and inhibits cellular transcription factors for IFN-β promoter activation | HFF (Towne) | [67] |
| NF-κB antagonist; suppresses NF-κB-dependent cytokine and chemokine gene expression | MRC5 fibroblasts (AD169) | [68] | |
| US9 | Inhibits IFN-β production and antiviral responses by targeting both MAVS- and STING-mediated signaling | HEK293T, HFF (AD169) | [70] |
| pUL26 | Antagonizes NF-κB activation by attenuating IKK phosphorylation | MRC5 (AD169) | [71] |
| pUL144 | Agonist of NF-κB-induced transcription via TRAF6 and TRIM23 | U373, HFF (AD169, TB40E) | [73,74] |
| vMIA (pUL37 × 1) | Inhibits mitochondrial MAVS-dependent antiviral signaling | HeLa (transfection) | [99] |
| Inhibits the peroxisomal MAVS-dependent antiviral signaling | HepG2, HFF, Mefs (transfection) | [100] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marques, M.; Ferreira, A.R.; Ribeiro, D. The Interplay between Human Cytomegalovirus and Pathogen Recognition Receptor Signaling. Viruses 2018, 10, 514. https://doi.org/10.3390/v10100514
Marques M, Ferreira AR, Ribeiro D. The Interplay between Human Cytomegalovirus and Pathogen Recognition Receptor Signaling. Viruses. 2018; 10(10):514. https://doi.org/10.3390/v10100514
Chicago/Turabian StyleMarques, Mariana, Ana Rita Ferreira, and Daniela Ribeiro. 2018. "The Interplay between Human Cytomegalovirus and Pathogen Recognition Receptor Signaling" Viruses 10, no. 10: 514. https://doi.org/10.3390/v10100514
APA StyleMarques, M., Ferreira, A. R., & Ribeiro, D. (2018). The Interplay between Human Cytomegalovirus and Pathogen Recognition Receptor Signaling. Viruses, 10(10), 514. https://doi.org/10.3390/v10100514