Effect of Type-I Interferon on Retroviruses

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Retroviruses
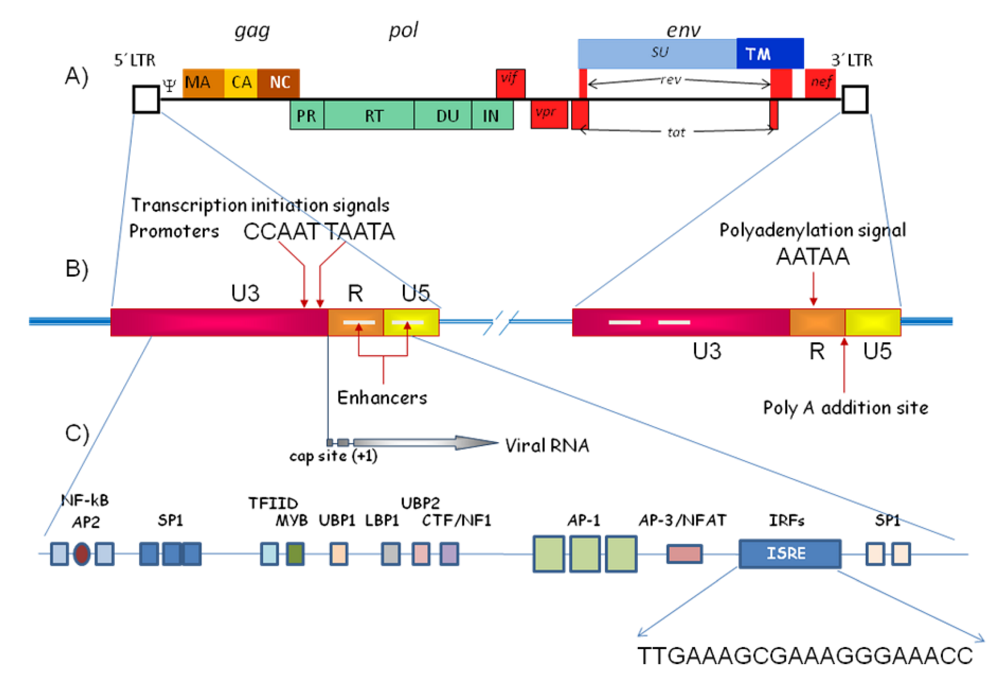
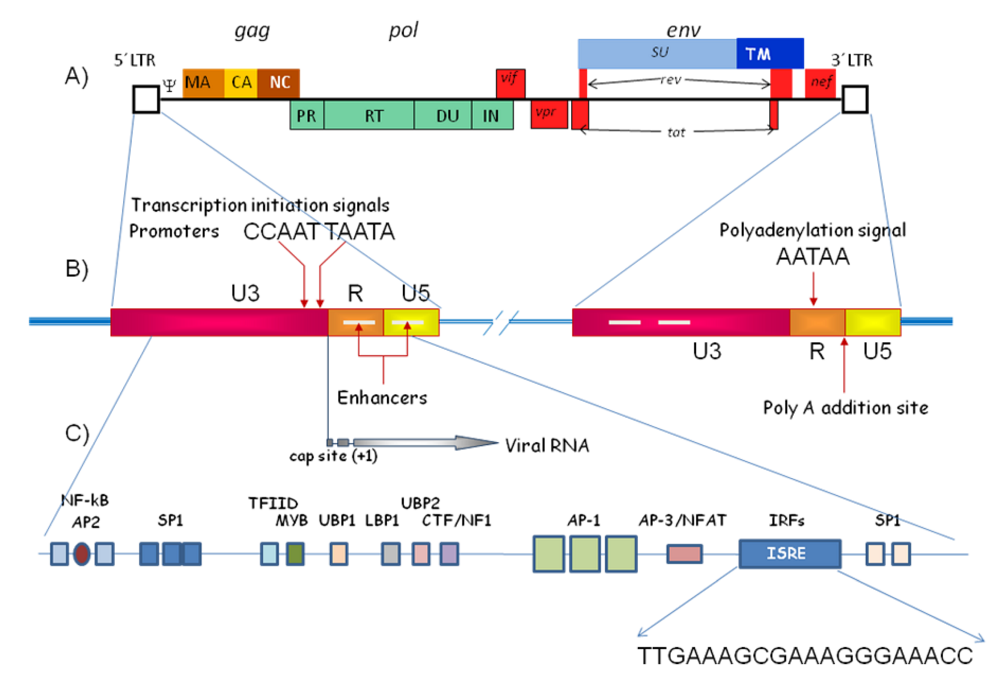
1.1. Long Terminal Repeats (LTR)
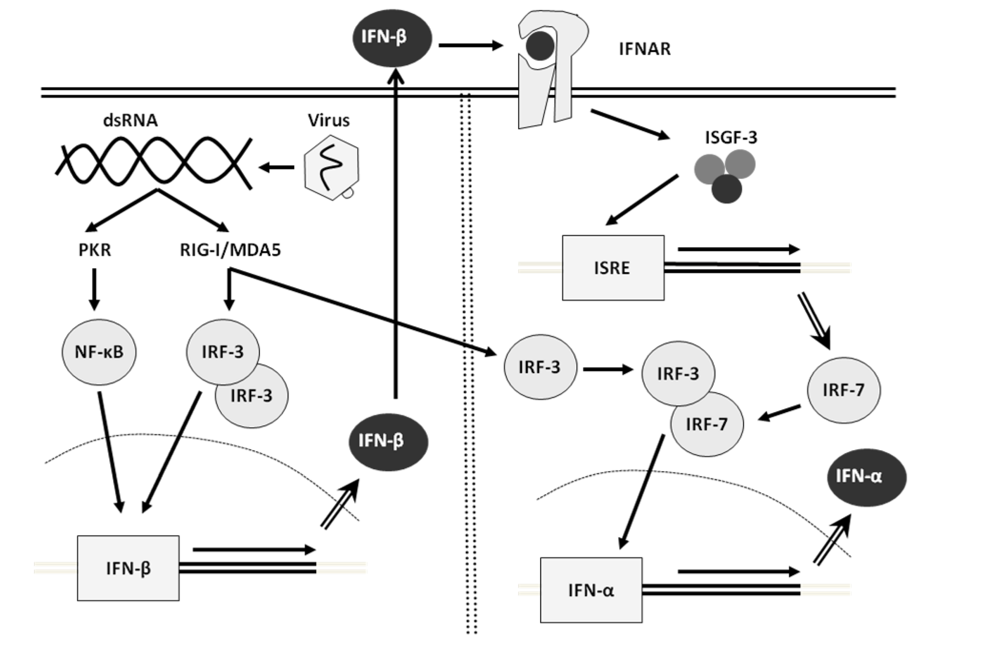
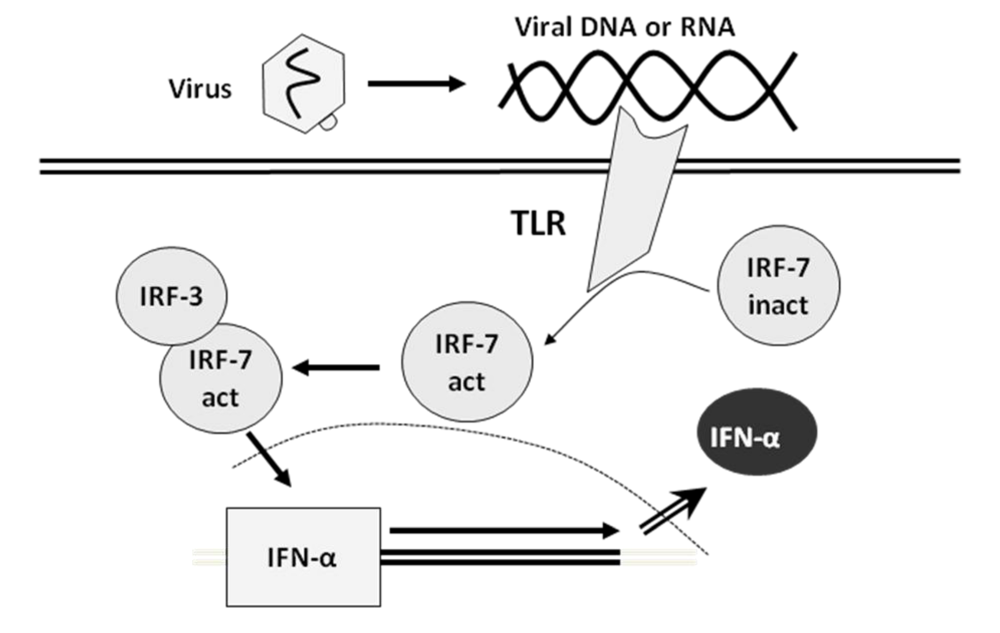
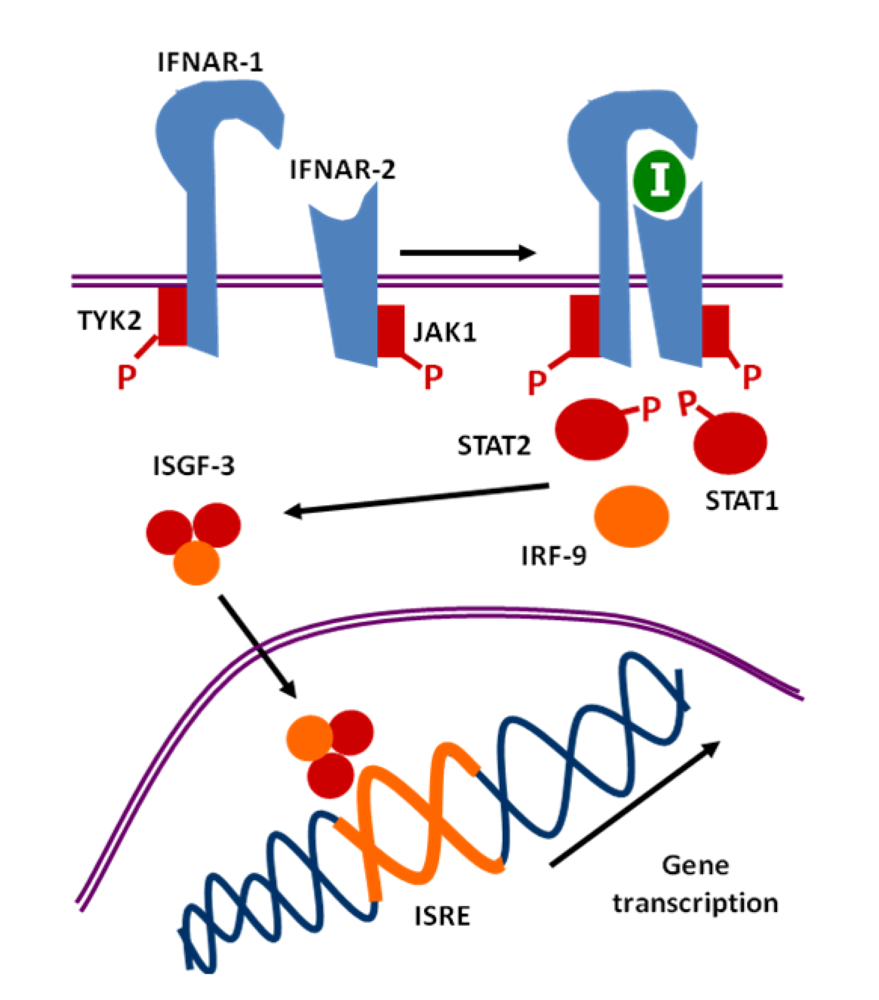
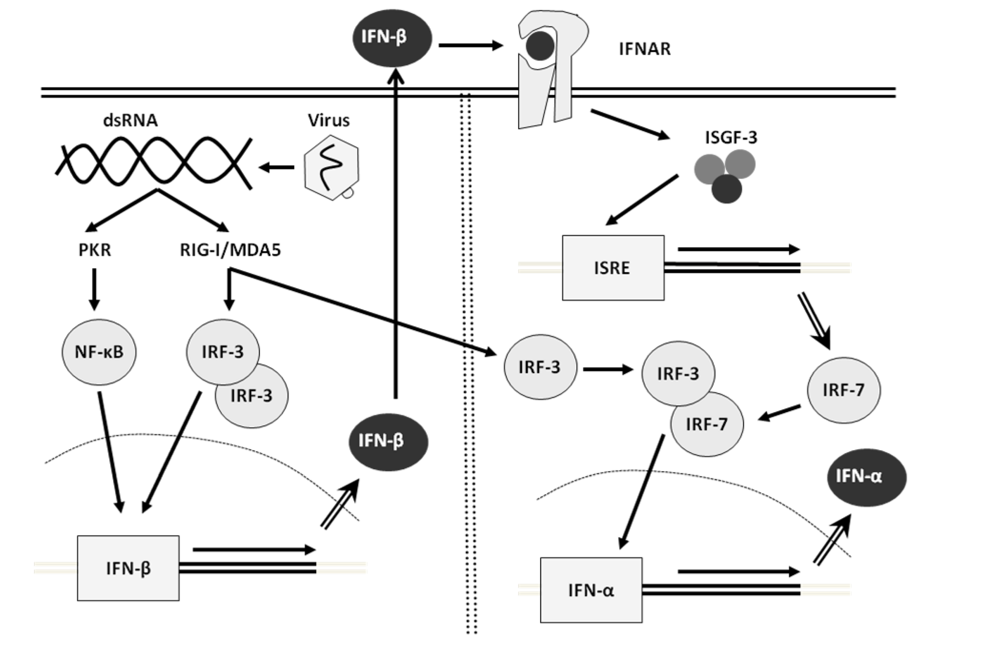
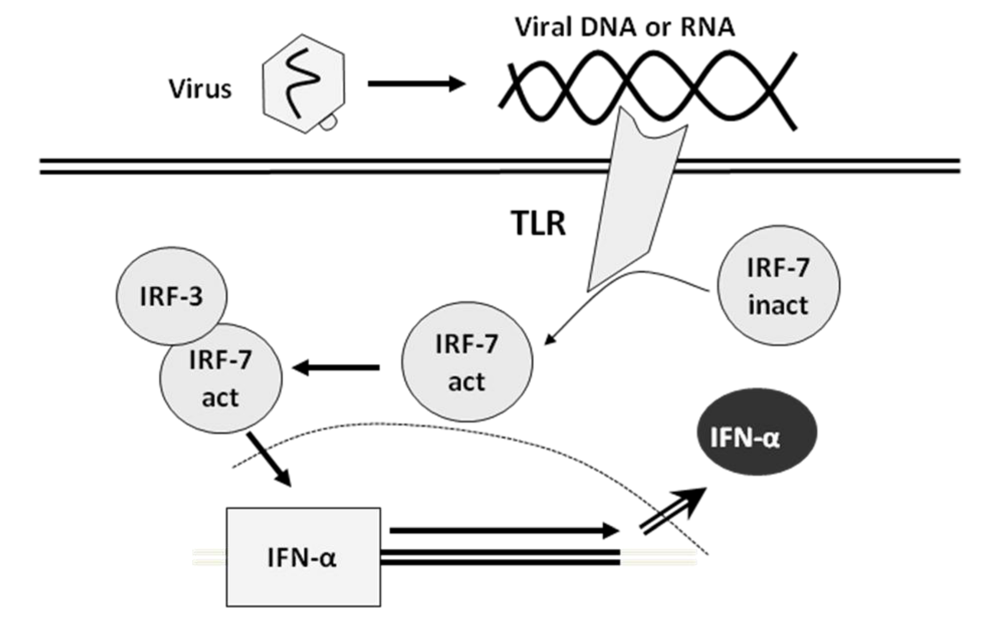
2. Interferon
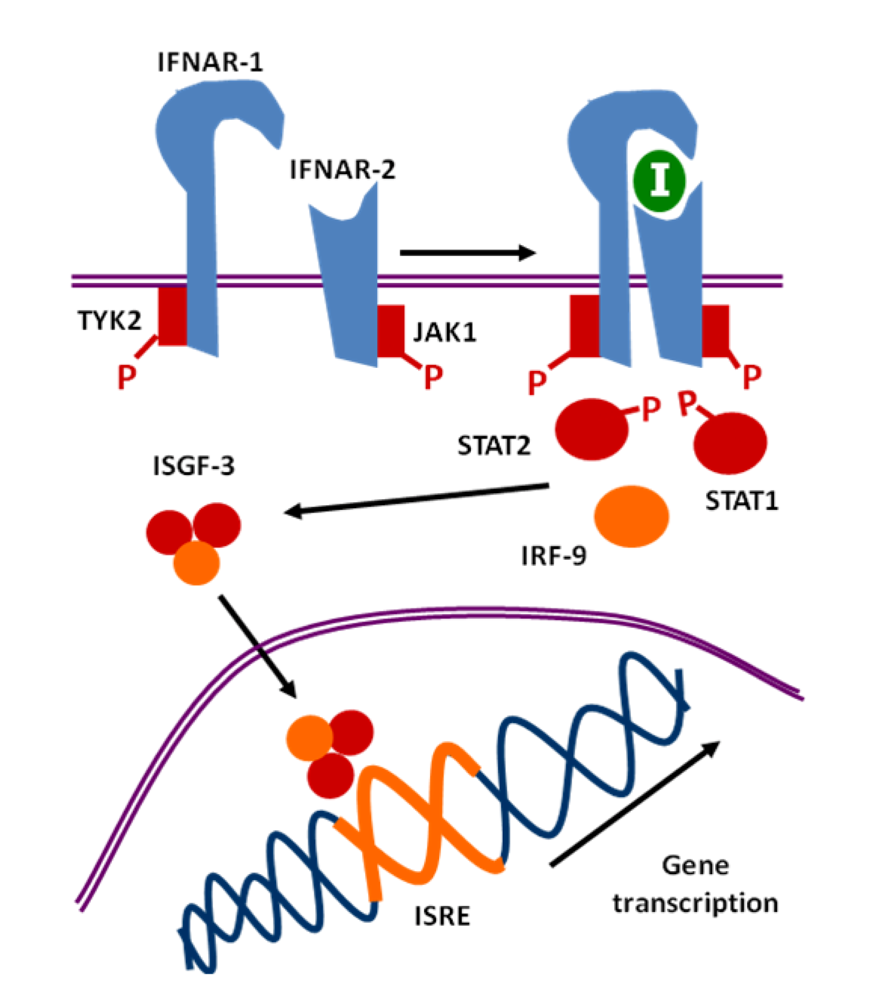
- IFN-I levels in natural and experimental retroviral infection, and whether they are protective or deleterious, and association to specific syndromes, such as dementia
- Possible impairment of pDCs as the cause of altered IFN levels
- Effects of IFN on virogenesis
- Effects of IFN at the cellular level: induction of apoptosis in infected and non infected cells
- Effects of IFN at the molecular level: presence of ISREs in retroviral genome (LTRs)
- Mechanisms by which retroviruses may evade the effect of IFN-I
5. Effects of IFN at the Cellular Level: Apoptosis
6. Direct Effects of Type I Interferons at the Molecular Level: ISREs
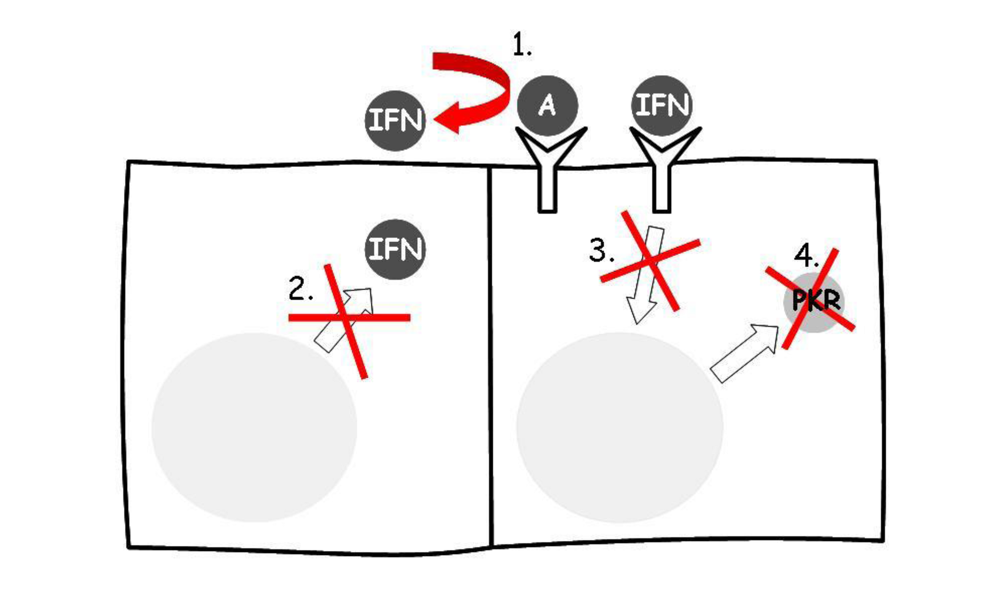
7. Mechanisms by which Retroviruses May Evade the Effects of Type I Inteferons
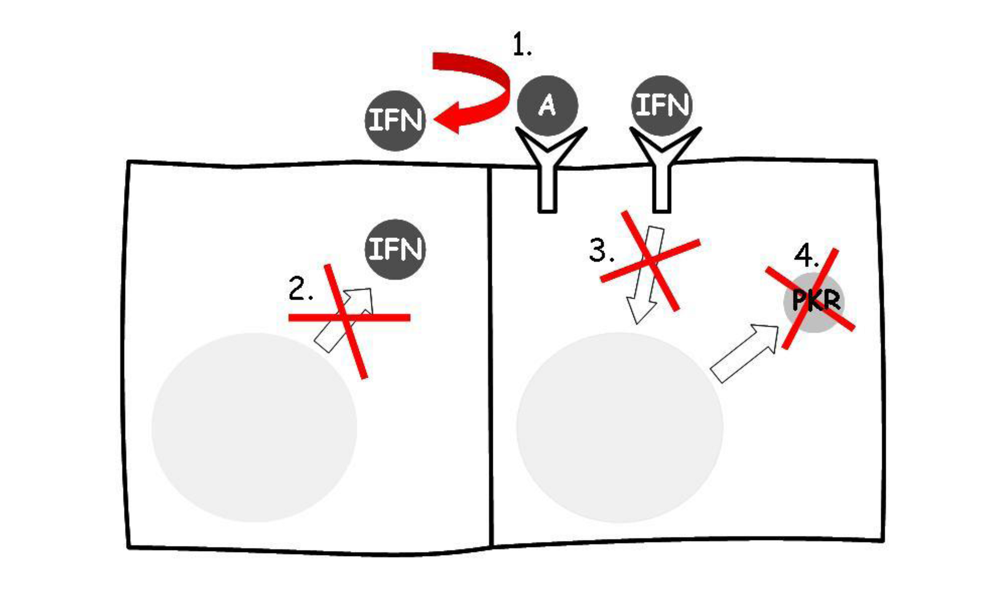
8. Other Type I Interferons
8.1. Interferon-tau (IFN-τ)
8.2. Interferon-omega (IFN-ω)
References and Notes
- Abbate, I.; Dianzani, F.; Capobianchi, M. R. Activation of signal transduction and apoptosis in healthy lymphomonocytes exposed to bystander HIV-1-infected cells. Clin. Exp. Immunol. 2000, 122, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Aboud, M.; Kimchi, R.; Bakhanashvili, M.; Salzberg, S. Intracellular production of virus particles and viral components in NIH/3T3 cells chronically infected with Moloney murine leukemia virus: effect of interferon. J. Virol. 1981, 40, 830–838. [Google Scholar] [PubMed]
- Aboud, M.; Malik, Z.; Bari, S.; Kimchi, R.; Hassan, Y.; Salzberg, S. Effect of interferon on the formation and release of intracellular virions in NIH/3T3 cells chronically infected with Moloney murine leukemia virus. J. Interferon Res. 1983, 3, 33–44. [Google Scholar] [PubMed]
- Agy, M.B.; Acker, R.L.; Sherbert, C.H.; Katze, M.G. Interferon treatment inhibits virus replication in HIV-1- and SIV-infected CD4+ T-cell lines by distinct mechanisms: evidence for decreased stability and aberrant processing of HIV-1 proteins. Virology 1995, 214, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M.; Cordero, M.; Almeida, J.; Orfao, A. Different subsets of peripheral blood dendritic cells show distinct phenotypic and functional abnormalities in HIV-1 infection. AIDS 2005, 19, 261–271. [Google Scholar] [PubMed]
- Alsharifi, M.; Müllbacher, A.; Regner, M. Interferon type I responses in primary and secondary infections. Immunol. Cell. Biol. 2008, 86, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Andrieu, M.; Chassin, D.; Desoutter, J.F.; Bouchaert, I.; Baillet, M.; Hanau, D.; Guillet, J.G.; Hosmalin, A. Downregulation of major histocompatibility class I on human dendritic cells by HIV Nef impairs antigen presentation to HIV-specific CD8+ T lymphocytes. AIDS Res. Hum. Retroviruses 2001, 17, 1365–1370. [Google Scholar] [PubMed]
- Babé, L.M.; Unal, A.; Craik, C.S. Obstruction of HIV-1 particle release by interferon-α occurs before viral protease processing and is independent of envelope glycoprotein. J. Interferon Cytokine Res. 1997, 17, 287–93. [Google Scholar] [PubMed]
- Baca-Regen, L.; Heinzinger , N.; Stevenson , M.; Gendelman, HE. A interferon-induced antiretroviral activities: restriction of viral nucleic acid synthesis and progeny virion production in human immunodeficiency virus type 1-infected monocytes . J. Virol. 1994, 68, 7559–7565. [Google Scholar] [PubMed]
- Balachandran, S.; Kim, C.N.; Yeh, W.C.; Mak, T.W.; Bhalla, K.; Barber, G.N. Activation of the dsRNA-dependent protein kinase, PKR, induces apoptosis through FADD-mediated death signaling. EMBO J. 1998, 17, 6888–6902. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, S.L.; Powell, T.D.; Sellins, K.S.; Radecki, S.V.; Cohen, J.J.; Milhausen, M.J. The biological effects of five feline IFN-α subtypes. Vet. Immunol. Immunopathol. 2004, 99, 153–167. [Google Scholar] [PubMed]
- Bandyopadhyay, SK.; Leonard Jr., G.T. ; Bandyopadhyay, T.; Stark, G.R.; Sen, G.C. Transcriptional induction by double-stranded RNA is mediated by interferon-stimulated response elements without activation of interferon-stimulated gene factor 3. J. Biol. Chem. 1995, 270, 19624–19629. [Google Scholar] [CrossRef] [PubMed]
- Barber, G.N. The interferons and cell death: guardians of the cell or accomplices of apoptosis? Semin. Cancer Biol.. 2000, 10, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Barron, M.A.; Blyveis., N.; Palmer, B.E.; MaWhinney, S.; Wilson, C.C. Influence of plasma viremia on defects in number and immunophenotype of blood dendritic cell subsets in human immunodeficiency virus 1-infected individuals . J. Infect. Dis. 2003, 187, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Battistini, A.; Marsili, G.; Sgarbanti, M.; Ensoli, B.; Hiscott, J. IRF regulation of HIV-1 long terminal repeat activity. J. Interferon Cytokine Res. 2002, 22, 27–37. [Google Scholar] [PubMed]
- Bekisz, J.; Schmeisser, H.; Hernandez, J.; Goldman, ND.; Zoon, KC. Human interferons α, beta and omega. Growth Factors 2004, 22, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Belardelli, F.; Ferrantini, M.; Proietti, E.; Kirkwood, J.M. Interferon-α in tumor immunity and immunotherapy. Cytokine Growth Factor Rev. 2002, 13, 119–134. [Google Scholar] [CrossRef] [PubMed]
- Boasso, A.; Shearer, G.M. Chronic innate immune activation as a cause of HIV-1 immunopathogenesis. Clin. Immunol. 2008, 126, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Bolinger, C.; Boris-Lawrie, K. Mechanisms employed by retroviruses to exploit host factors for translational control of a complicated proteome . Retrovirology 2009, 6:8, 20. [Google Scholar]
- Boxus, M.; Twizere, J.C.; Legros, S.; Dewulf, J.F.; Kettmann, R.; Willems, L. The HTLV-1 Tax interactome. Retrovirology 2008, 5, 76. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.A.; Rose, J.K. Sorting of GPI-anchored proteins to glycolipid-enriched membrane subdomains during transport to the apical cell surface. Cell 1992, 68, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Bushman, F.D. Targeting survival: integration site selection by retroviruses and LTR-retrotransposons. Cell 2003, 115, 135–138. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, A.; Yang, B.; Gendelman, H.E.; Persidsky, Y.; Kanmogne, G.D. STAT1 signaling modulates HIV-1-induced inflammatory responses and leukocyte transmigration across the blood-brain barrier. Blood 2008, 111, 2062–2072. [Google Scholar] [CrossRef] [PubMed]
- Chawla-Sarkar, M.; Lindner, DJ.; Liu, Y.F.; Williams, B.R.; Sen, G.C.; Silverman, R.H.; Borden, E.C. Apoptosis and interferons: role of interferon-stimulated genes as mediators of apoptosis. Apoptosis 2003, 8, 237–249. [Google Scholar] [CrossRef]
- Chehimi, J.; Campbell, D.E.; Azzoni, L.; Bacheller, D.; Papasavvas, E.; Jerandi, G.; Mounzer, K.; Kostman, J.; Trinchieri, G.; Montaner, L.J. Persistent decreases in blood plasmacytoid dendritic cell number and function despite effective highly active antiretroviral therapy and increased blood myeloid dendritic cells in HIV-infected individuals . J. Immunol. 2002, 168, 4796–4801. [Google Scholar] [PubMed]
- Clayette, P.; Martin, M.; Dereuddre-Bosquet, N.; Tournay, V.; Gras, G.; Martal, J.; Dormont, D. IFN-tau, a new interferon type I with antiretroviral activity. Pathol. Biol. (Paris) 1999, 47, 553–559. [Google Scholar] [PubMed]
- Coccia, E.M.; Krust, B.; Hovanessian, A.G. Specific inhibition of viral protein synthesis in HIV-infected cells in response to interferon treatment. J. Biol. Chem. 1994, 269, 23087–23094. [Google Scholar] [PubMed]
- Collado, V.M.; Gómez-Lucía, E.; Tejerizo, G.; Miró, G.; Escolar, E.; Martín, S.; Doménech, A. Effect of type I interferons on the expression of feline leukaemia virus. Vet. Microbiol. 2007, 123, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Collado, V.M.; Doménech, A.; Ballesteros, N.A.; Ramos, P.; Miró, G.; Gómez-Lucía, E. Efecto in vitro del interferón de tipo I en la expresión del virus de la inmunodeficiencia felina (FIV) . Salamanca, Spain, 2009; p. 274. [Google Scholar]
- Collado, V.M. unpublished results . [CrossRef] [PubMed]
- Colonna, M.; Krug, A.; Cella, M. Interferon-producing cells: on the front line in immune responses against pathogens. Curr. Opin. Immunol. 2002, 14, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Czarniecki, C.W.; Sreevalsan, T.; Friedman, R.M.; Panet, A. Dissociation of interferon effects on murine leukemia virus and encephalomyocarditis virus replication in mouse cells . J. Virol. 1981, 37, 827–831. [Google Scholar] [PubMed]
- de Mari, K.; Maynard, L.; Sanquer, A.; Lebreux, B.; Eun, H.M. Therapeutic effects of recombinant feline interferon-omega on feline leukemia virus (FeLV)-infected and FeLV/feline immunodeficiency virus (FIV)-coinfected symptomatic cats. J. Vet. Intern. Med. 2004, 18, 477–478. [Google Scholar] [CrossRef] [PubMed]
- De Parseval, A; Elder, J.H. Demonstration that orf2 encodes the feline immunodeficiency virus transactivating (Tat) protein and characterization of a unique gene product with partial rev activity . J. Virol. 1999, 73, 608–617. [Google Scholar] [PubMed]
- Dereuddre-Bosquet, N.; Clayette, P.; Martin, M.; Mabondzo, A.; Frétier, P.; Gras, G.; Martal, J.; Dormont, D. Anti-HIV potential of a new interferon, interferon-tau (trophoblastin). J. Acquir. ImmuneDefic. Syndr. Hum. Retrovirol. 1996, 11, 241–246. [Google Scholar] [CrossRef]
- Dianzani, F.; Castilletti, C.; Gentile, M.; Gelderblom, H.R.; Frezza, F.; Capobianchi, M.R. Effects of IFN α on late stages of HIV-1 replication cycle. Biochimie 1998, 80, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Diebold, S.S.; Montoya, M.; Unger, H.; Alexopoulou, L.; Roy, P.; Haswell, L.E.; Al-Shamkhani, A.; Flavell, R.; Borrow, P.; Reis e Sousa, C. Viral infection switches non-plasmacytoid dendritic cells into high interferon producers. Nature 2003, 424, 324–328. [Google Scholar] [CrossRef] [PubMed]
- Diop, O.M.; Ploquin, M.J.; Mortara, L.; Faye, A.; Jacquelin, B.; Kunkel, D.; Lebon, P.; Butor, C.; Hosmalin, A.; Barré-Sinoussi, F.; Müller-Trutwin, M.C. Plasmacytoid dendritic cell dynamics and α interferon production during Simian immunodeficiency virus infection with a nonpathogenic outcome. J. Virol. 2008, 82, 5145–5152. [Google Scholar] [CrossRef] [PubMed]
- Donaghy, H.; Pozniak, A.; Gazzard, B.; Qazi, N.; Gilmour, J.; Gotch, F.; Patterson, S. Loss of blood CD11c(+) myeloid and CD11c(-) plasmacytoid dendritic cells in patients with HIV-1 infection correlates with HIV-1 RNA virus load. Blood 2001, 98, 2574–2576. [Google Scholar] [CrossRef] [PubMed]
- Donaghy, H.; Gazzard, B.; Gotch, F.; Patterson, S. Dysfunction and infection of freshly isolated blood myeloid and plasmacytoid dendritic cells in patients infected with HIV-1. Blood 2003, 101, 4505–4511. [Google Scholar] [CrossRef] [PubMed]
- Endo-Munoz, L.; Warby, T.; Harrich, D.; McMillan, NA. Phosphorylation of HIV Tat by PKR increases interaction with TAR RNA and enhances transcription . Virol. J. 2005, 2:17, 13. [Google Scholar]
- Estaquier, J.; Idziorek, T.; de Bels, F.; Barré-Sinoussi, F.; Hurtrel, B.; Aubertin, A.M.; Venet, A.; Mehtali, M.; Muchmore, E.; Michel, P.; Mouton, Y.; Girard, M.; Ameisen, J.C. Programmed cell death and AIDS: significance of T-cell apoptosis in pathogenic and nonpathogenic primate lentiviral infections. Proc. Natl. Acad. Sci. USA 1994, 91, 9431–9435. [Google Scholar] [CrossRef]
- Eyster, M.E.; Goedert, J.J.; Poon, M.C.; Preble, O.T. Acid-labile α interferon. A possible preclinical marker for the acquired immunodeficiency syndrome in hemophilia. N. Engl. J. Med. 1983, 309, 583–586. [Google Scholar] [PubMed]
- Feldman, S.; Stein, D.; Amrute, S.; Denny, T.; Garcia, Z.; Kloser, P.; Sun, Y.; Megjugorac, N.; Fitzgerald-Bocarsly, P. Decreased interferon-α production in HIV-infected patients correlates with numerical and functional deficiencies in circulating type 2 dendritic cell precursors. Clin. Immunol. 2001, 101, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Ratner, L. Human T-cell leukemia virus type 1 blunts signaling by interferon α. Virology 2008, 374, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Heyden, N.V.; Ratner, L. Α interferon inhibits human T-cell leukemia virus type 1 assembly by preventing Gag interaction with rafts. J. Virol. 2003, 77, 13389–13395. [Google Scholar] [CrossRef] [PubMed]
- Finke, J. S.; Shodell, M.; Shah, K.; Siegal, F. P.; Steinman, R.M. Dendritic cell numbers in the blood of HIV-1 infected patients before and after changes in antiretroviral therapy. J. Clin. Immunol. 2004, 24, 647–652. [Google Scholar] [CrossRef] [PubMed]
- Fiorentini, S.; Riboldi, E.; Facchetti, F.; Avolio, M.; Fabbri, M.; Tosti, G.; Becker, PD.; Guzman, CA.; Sozzani, S.; Caruso, A. HIV-1 matrix protein p17 induces human plasmacytoid dendritic cells to acquire a migratory immature cell phenotype. Proc. Natl. Acad. Sci. USA 2008, 105, 3867–3872. [Google Scholar] [CrossRef]
- Fitzgerald-Bocarsly, P.; Feng, D. The role of type I interferon production by dendritic cells in host defense. Biochimie 2007, 89, 843–855. [Google Scholar] [CrossRef] [PubMed]
- Gabriele, L.; Ozato, K. The role of the interferon regulatory factor (IRF) family in dendritic cell development and function. Cytokine Growth Factor Rev. 2007, 18, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Gendelman, H.E.; Baca, L.M.; Turpin, J.; Kalter, D.C.; Hansen, B.; Orenstein, J.M.; Dieffenbach, C.W.; Friedman, R.M.; Meltzer, M.S. Regulation of HIV replication in infected monocytes by IFN-α. Mechanisms for viral restriction. J. Immunol. 1990, 145, 2669–2676. [Google Scholar] [PubMed]
- Gerlach, N.; Schimmer, S.; Weiss, S.; Kalinke, U.; Dittmer, U. Effects of type I interferons on Friend retrovirus infection. J. Virol. 2006, 80, 3438–3444. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Lucia, E.; Tejerizo, G.; Doménech, A. Effect of steroid hormones on retroviruses. In Oncogenic Viruses. Research Trends. 2007; Johannes, L.T.; Nova Biomedical: New York, USA. [Google Scholar]
- Goodbourn, S.; Didcock, L.; Randall, R.E. Interferons: cell signalling, immune modulation, antiviral response and virus countermeasures. J. Gen. Virol. 2000, 81, 2341–2364. [Google Scholar] [PubMed]
- Göttlinger, H.G.; Dorfman, T.; Sodroski, J.G.; Haseltine, W.A. Effect of mutations affecting the p6 gag protein on human immunodeficiency virus particle release. Proc. Natl. Acad. Sci. USA 1991, 88, 3195–3199. [Google Scholar] [CrossRef]
- Haller, O.; Kochs, G.; Weber, F. The interferon response circuit: induction and suppression by pathogenic viruses. Virology 2006, 344, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Hansen, B.D.; Nara, P.L.; Maheshwari, R.K.; Sidhu, G.S.; Bernbaum, J.G.; Hoekzema, D.; Meltzer, M.S.; Gendelman, H.E. Loss of infectivity by progeny virus from α interferon-treated human immunodeficiency virus type 1-infected T cells is associated with defective assembly of envelope gp120. J. Virol. 1992, 66, 7543–7548. [Google Scholar] [PubMed]
- Hardy, A.W.; Graham, D.R.; Shearer, G.M.; Herbeuval, J.P. HIV turns plasmacytoid dendritic cells (pDC) into TRAIL-expressing killer pDC and down-regulates HIV coreceptors by Toll-like receptor 7-induced IFN-α. Proc. Natl. Acad. Sci. U S A. 2007, 104, 17453–17458. [Google Scholar] [CrossRef] [PubMed]
- Haupt, S.; Donhauser, N.; Chaipan, C.; Schuster, P.; Puffer, B.; Daniels, R.S.; Greenough, T.C.; Kirchhoff, F.; Schmidt, B. CD4 binding affinity determines human immunodeficiency virus type 1-induced α interferon production in plasmacytoid dendritic cells. J. Virol. 2008, 82, 8900–8905. [Google Scholar] [CrossRef] [PubMed]
- Herbeuval, J.P.; Shearer, G.M. HIV-1 immunopathogenesis: how good interferon turns bad. Clin Immunol. 2007, 123, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Herbeuval, J.P.; Grivel, J.C.; Boasso, A.; Hardy, A.W.; Chougnet, C.; Dolan, M.J.; Yagita, H.; Lifson, J.D.; Shearer, G.M. CD4+ T-cell death induced by infectious and noninfectious HIV-1: role of type 1 interferon-dependent, TRAIL/DR5-mediated apoptosis. Blood 2005, 106, 3524–3531. [Google Scholar] [CrossRef] [PubMed]
- Herbeuval, J.P.; Nilsson, J.; Boasso, A.; Hardy, A.W.; Kruhlak, M.J.; Anderson, S.A.; Dolan, M.J.; Dy, M.; Andersson, J.; Shearer, G.M. Differential expression of IFN-α and TRAIL/DR5 in lymphoid tissue of progressor versus nonprogressor HIV-1-infected patients. Proc. Natl. Acad. Sci. U S A. 2006, 103, 7000–7005. [Google Scholar] [CrossRef] [PubMed]
- Hishizawa, M.; Imada, K.; Kitawaki, T.; Ueda, M.; Kadowaki, N.; Uchiyama, T. Depletion and impaired interferon-α-producing capacity of blood plasmacytoid dendritic cells in human T-cell leukaemia virus type I-infected individuals. Br. J. Haematol. 2004, 125, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Yanai, H.; Negishi, H.; Asagiri, M.; Sato, M.; Mizutani, T.; Shimada, N.; Ohba, Y.; Takaoka, A.; Yoshida, N.; Taniguchi, T. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 2005, 434, 772–777. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Yanai, H.; Takaoka, A.; Taniguchi, T. Regulation of the type I IFN induction: a current view. Int. Immunol. 2005, 17, 1367–1378. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.S.; Bhatnagar ,N.; Ballmaier, M.; Schubert, U.; Henklein, P.; Volgmann, T.; Heiken, H.; Schmidt, R.E.; Meyer-Olson, D. Exogenous HIV-1 Vpr disrupts IFN-α response by plasmacytoid dendritic cells (pDCs) and subsequent pDC/NK interplay . Immunol. Lett. 2009, 125, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Huthoff, H.; Towers, G.J. Restriction of retroviral replication by APOBEC3G/F and TRIM5α. Trends Microbiol. 2008, 16, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Imbeault, M.; Ouellet, M.; Tremblay, M.J. Microarray study reveals that HIV-1 induces rapid type-I interferon-dependent p53 mRNA up-regulation in human primary CD4+ T cells . Retrovirology 2009, 6:5, 14. [Google Scholar]
- Jameson, P.; Essex, M. Inhibition of feline leukemia virus replication by human leukocyte interferon. Antiviral Res. 1983, 3, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Jarrett,O. Strategies of retrovirus survival in the cat . Vet. Microbiol. 1999, 69, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Juste, R.A.; Ott, T.L.; Kwang, J.; Bazer, F.W.; de la Concha-Bermejillo, A. Effects of recombinant interferon-tau on ovine lentivirus replication. J. Interferon Cytokine Res. 1996, 16, 989–994. [Google Scholar] [PubMed]
- Juste, R.A.; Ott, T.L.; Kwang, J.; Bazer, F.W.; de La Concha-Bermejillo, A. Effects of recombinant ovine interferon-tau on ovine lentivirus replication and progression of disease. J. Gen. Virol. 2000, 81, 525–532. [Google Scholar] [PubMed]
- Kamga, I.; Kahi, S.; Develioglu, L.; Lichtner, M.; Marañón, C.; Deveau, C.; Meyer, L.; Goujard, C.; Lebon, P.; Sinet, M.; Hosmalin, A. Type I interferon production is profoundly and transiently impaired in primary HIV-1 infection. J. Infect. Dis. 2005, 192, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Karpov, A.V. Endogenous and exogenous interferons in HIV-infection. Eur. J. Med. Res. 2001, 6, 507–524. [Google Scholar] [PubMed]
- Kawaguchi, Y; Norimine, J; Miyazawa, T; Kai, C; Mikami, T. Sequences within the feline immunodeficiency virus long terminal repeat that regulate gene expression and respond to activation by feline herpesvirus type 1 . Virology 1992, 190, 465–468. [Google Scholar] [CrossRef] [PubMed]
- Keir, M.E.; Stoddart, C.A.; Linquist-Stepps, V.; Moreno, M.E.; McCune, J.M. IFN-α secretion by type 2 predendritic cells up-regulates MHC class I in the HIV-1-infected thymus. J. Immunol. 2002, 168, 325–331. [Google Scholar] [PubMed]
- Keir, M.E.; Rosenberg, M.G.; Sandberg, J.K.; Jordan, K.A.; Wiznia, A.; Nixon, D.F.; Stoddart, C.A.; McCune, J.M. Generation of CD3+CD8low thymocytes in the HIV type 1-infected thymus. J. Immunol. 2002, 169, 2788–2796. [Google Scholar] [PubMed]
- Kiermer, V.; Van Lint, C.; Briclet, D.; Vanhulle, C.; Kettmann, R.; Verdin, E.; Burny, A.; Droogmans, L. An interferon regulatory factor binding site in the U5 region of the bovine leukemia virus long terminal repeat stimulates Tax-independent gene expression. J. Virol. 1998, 72, 5526–5534. [Google Scholar] [PubMed]
- Kinpara, S.; Hasegawa, A.; Utsunomiya, A.; Nishitsuji, H.; Furukawa, H.; Masuda, T.; Kannagi, M. Stromal cell-mediated suppression of human T-cell leukemia virus type 1 expression in vitro and in vivo by type I interferon. J. Virol. 2009, 83, 5101–5108. [Google Scholar] [CrossRef] [PubMed]
- Kohara, J.; Yokomizo, Y. In vitro and in vivo effects of recombinant bovine interferon-tau on bovine leukemia virus. J. Vet. Med. Sci. 2007, 69, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Kornbluth, R.S.; Oh, P.S.; Munis, J.R.; Cleveland, P.H.; Richman, D.D. Interferons and bacterial lipopolysaccharide protect macrophages from productive infection by human immunodeficiency virus in vitro. J. Exp. Med. 1989, 169, 1137–1151. [Google Scholar] [CrossRef] [PubMed]
- Künzi, M.S.; Pitha, P.M. Role of interferon-stimulated gene ISG-15 in the interferon-omega-mediated inhibition of human immunodeficiency virus replication. J. Interferon Cytokine Res. 1996, 16, 919–927. [Google Scholar] [CrossRef] [PubMed]
- Künzi, M.S.; Farzadegan, H.; Margolick, J.B.; Vlahov, D.; Pitha, P.M. Identification of human immunodeficiency virus primary isolates resistant to interferon-α and correlation of prevalence to disease progression. J. Infect. Dis. 1995, 171, 822–828. [Google Scholar] [PubMed]
- Lehner, T.; Wang, Y.; Pido-Lopez, J.; Whittall, T.; Bergmeier, L.A.; Babaahmady, K. The emerging role of innate immunity in protection against HIV-1 infection. Vaccine 2008, 26, 2997–3001. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.E. Physiological significance of STAT proteins: investigations through gene disruption in vivo. Cell. Mol. Life Sci. 1999, 55, 1559–1567. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.A.; Scott, I.; Mackewicz, C. Protection from HIV/AIDS: the importance of innate immunity. Clin. Immunol. 2003, 108, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Lopez, C.; Fitzgerald, P.A.; Siegal, F.P. Severe acquired immune deficiency syndrome in male homosexuals: diminished capacity to make interferon-α in vitro associated with severe opportunistic infections. J. Infect. Dis. 1983, 148, 962–966. [Google Scholar] [PubMed]
- Luo, C.; Wang, K.; Liu de, Q.; Li, Y.; Zhao, Q.S. The functional roles of lipid rafts in T cell activation; immune diseases and HIV infection and prevention. Cell. Mol. Immunol. 2008, 5, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Macchi, B.; Faraoni, I.; Mastino, A.; D'Onofrio, C.; Romeo, G.; Bonmassar, E. Protective effect of interferon beta on human T cell leukaemia virus type I infection of CD4+ T cells isolated from human cord blood. Cancer Immunol. Immunother. 1993, 37, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Mandl, J.N.; Barry, A.P.; Vanderford, T.H.; Kozyr, N.; Chavan, R.; Klucking, S.; Barrat, F.J.; Coffman, R.L.; Staprans, S.I.; Feinberg, M.B. Divergent TLR7 and TLR9 signaling and type I interferon production distinguish pathogenic and nonpathogenic AIDS virus infections. Nat Med. 2008, 14, 1077–1087. [Google Scholar] [CrossRef] [PubMed]
- Maneglier, B.; Rogez-Kreuz, C.; Dereuddre-Bosquet, N.; Martal, J.; Devillier, P.; Dormont, D.; Clayette, P. Anti-HIV effects of IFN-tau in human macrophages: role of cellular antiviral factors and interleukin-6. Pathol. Biol. (Paris) 2008, 56, 492–503. [Google Scholar] [PubMed]
- Mangino, G.; Percario, Z.A.; Fiorucci, G.; Vaccari, G.; Manrique, S.; Romeo, G.; Federico, M.; Geyer, M.; Affabris, E. In vitro treatment of human monocytes/macrophages with myristoylated recombinant Nef of human immunodeficiency virus type 1 leads to the activation of mitogen-activated protein kinases, IkappaB kinases, and interferon regulatory factor 3 and to the release of beta interferon. J. Virol. 2007, 81, 2777–2791. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, M.; Monier, M.N.; Fradagrada, A.; Mitchell, K.; Baychelier, F.; Eid, P.; Johannes, L.; Lamaze, C. Stat-mediated signaling induced by type I and type II interferons (IFNs) is differentially controlled through lipid microdomain association and clathrin-dependent endocytosis of IFN receptors. Mol. Biol. Cell. 2006, 17, 2896–2909. [Google Scholar] [CrossRef] [PubMed]
- Marcondes, M.C.; Flynn, C.; Huitron-Rezendiz, S.; Watry, D.D.; Zandonatti, M.; Fox, H.S. Early antiretroviral treatment prevents the development of central nervous system abnormalities in simian immunodeficiency virus-infected rhesus monkeys. AIDS 2009, 23, 1187–1195. [Google Scholar] [CrossRef] [PubMed]
- Marié, I.; Durbin, J.E.; Levy, D.E. Differential viral induction of distinct interferon-α genes by positive feedback through interferon regulatory factor-7. EMBO J. 1998, 17, 6660–6669. [Google Scholar] [CrossRef] [PubMed]
- Marsili, G.; Borsetti, A.; Sgarbanti, M.; Remoli, A.L.; Ridolfi, B.; Stellacci, E.; Ensoli, B.; Battistini, A. On the role of interferon regulatory factors in HIV-1 replication. Ann. N Y Acad. Sci. 2003, 1010, 29–42. [Google Scholar] [CrossRef] [PubMed]
- Meylan, P.R.; Guatelli, J.C.; Munis, J.R.; Richman, D.D.; Kornbluth, R.S. Mechanisms for the inhibition of HIV replication by interferons-α, -beta, and -gamma in primary human macrophages. Virology 1993, 193, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, K.E.; Lewerenz, M.; Reboul, J.; Lutfalla, G.; Uzé, G. The type I interferon receptor: structure, function, and evolution of a family business. J. Interferon Cytokine Res. 1999, 19, 1069–1098. [Google Scholar] [PubMed]
- Munier, S.; Delcroix-Genête, D.; Carthagéna, L.; Gumez, A.; Hazan, U. Characterization of two candidate genes, NCoA3 and IRF8, potentially involved in the control of HIV-1 latency. Retrovirology 2005, 2, 73. [Google Scholar] [CrossRef] [PubMed]
- Murphy, F.A.; Gibbs, E.P.J.; Horzinek, M.C.; Studdert, M.J. Retroviridae, 3 edAcademic Press: San Diego, USA, 1999; pp. 363–389. [Google Scholar]
- Niermann, G.L.; Buehring, G.C. Hormone regulation of bovine leukemia virus via the long terminal repeat. Virology 1997, 239, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Oka, T.; Ohtsuki, Y.; Sonobe, H.; Furihata, M.; Miyoshi, I. Suppressive effects of interferons on the production and release of human T-lymphotropic virus type-I (HTLV-I). Arch. Virol. 1990, 115, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Okumura, A.; Lu, G.; Pitha-Rowe, I.; Pitha, P.M. Innate antiviral response targets HIV-1 release by the induction of ubiquitin-like protein ISG15. Proc. Natl. Acad. Sci. U S A. 2006, 103, 1440–1445. [Google Scholar] [CrossRef] [PubMed]
- Pacanowski, J.; Kahi, S.; Baillet, M.; Lebon, P.; Deveau, C.; Goujard, C.; Meyer, L.; Oksenhendler, E.; Sinet, M.; Hosmalin, A. Reduced blood CD123+ (lymphoid) and CD11c+ (myeloid) dendritic cell numbers in primary HIV-1 infection . Blood 2001, 98, 3016–3021. [Google Scholar] [CrossRef] [PubMed]
- Pacanowski, J.; Develioglu, L.; Kamga, I.; Sinet, M.; Desvarieux, M.; Girard, P. M.; Hosmalin, A. Early plasmacytoid dendritic cell changes predict plasma HIV load rebound during primary infection. J. Infect. Dis. 2004, 190, 1889–1892. [Google Scholar] [CrossRef] [PubMed]
- Pedretti, E.; Passeri, B.; Amadori, M.; Isola, P.; Di Pede, P.; Telera, A.; Vescovini, R.; Quintavalla, F.; Pistello, M. Low-dose interferon-α treatment for feline immunodeficiency virus infection. Vet. Immunol. Immunopathol. 2006, 109, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Pestka, S.; Krause, C.D.; Walter, M.R. Interferons, interferon-like cytokines, and their receptors. Immunol. Rev. 2004, 202, 8–32. [Google Scholar] [CrossRef]
- Poli, G.; Orenstein, J.M.; Kinte,r A.; Folks, T.M.; Fauci, A.S. Interferon-α but not AZT suppresses HIV expression in chronically infected cell lines . Science 1989, 244, 575–577. [Google Scholar] [PubMed]
- Pontzer, C.H.; Yamamoto, J.K.; Bazer, F.W.; Ott, T.L.; Johnson, H.M. Potent anti-feline immunodeficiency virus and anti-human immunodeficiency virus effect of IFN-tau. J. Immunol. 1997, 158, 4351–4357. [Google Scholar] [PubMed]
- Quaranta, M.G.; Mattioli, B.; Giordani, L.; Viora, M. HIV-1 Nef equips dendritic cells to reduce survival and function of CD8+ T cells: a mechanism of immune evasion. FASEB J. 2004, 18, 1459–1461. [Google Scholar] [PubMed]
- Ramji, D.P; Foka, P. CCAAT/enhancer-binding proteins: structure, function and regulation . Biochem. J. 2002, 365, 561–575. [Google Scholar] [PubMed]
- Randall, R.E.; Goodbourn, S. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J. Gen. Virol. 2008, 89, 1–47. [Google Scholar] [CrossRef]
- Rho, M.B.; Wesselingh, S.; Glass, J.D.; McArthur, J.C.; Choi, S.; Griffin, J.; Tyor, W.R. A potential role for interferon-α in the pathogenesis of HIV-associated dementia. Brain Behav. Immun. 1995, 9, 366–377. [Google Scholar] [CrossRef] [PubMed]
- Rogez, C.; Martin, M.; Dereuddre-Bosquet, N.; Martal, J.; Dormont, D.; Clayette, P. Anti-human immunodeficiency virus activity of tau interferon in human macrophages: involvement of cellular factors and beta-chemokines. J. Virol. 2003, 77, 12914–12920. [Google Scholar] [CrossRef] [PubMed]
- Rogez-Kreuz, C.; Manéglier, B.; Martin, M.; Dereuddre-Bosquet, N.; Martal, J.; Dormont, D.; Clayette, P. Involvement of IL-6 in the anti-human immunodeficiency virus activity of IFN-tau in human macrophages. Int. Immunol. 2005, 17, 1047–1057. [Google Scholar] [CrossRef] [PubMed]
- Rossol, S.; Voth, R.; Laubenstein, H.P.; Müller, W.E.; Schröder, H.C.; Meyer zum Büschenfelde, K.H.; Hess, G. Interferon production in patients infected with HIV-1. J. Infect. Dis. 1989, 159, 815–821. [Google Scholar] [PubMed]
- Saito, M.; Nakagawa, M.; Kaseda, S.; Matsuzaki, T.; Jonosono, M.; Eiraku, N.; Kubota, R.; Takenouchi, N.; Nagai, M.; Furukawa, Y.; Usuku, K.; Izumo, S.; Osame, M. Decreased human T lymphotropic virus type I (HTLV-I) provirus load and alteration in T cell phenotype after interferon-α therapy for HTLV-I-associated myelopathy/tropical spastic paraparesis. J. Infect. Dis. 2004, 189, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, R.; Mael, A.A.; Ikeda, Y. Α interferon enhances TRIM5α-mediated antiviral activities in human and rhesus monkey cells. J. Virol. 2007, 81, 10201–10206. [Google Scholar] [CrossRef] [PubMed]
- Samuel, C.E. Antiviral actions of interferons. Clin. Microbiol. Rev. 2001, 14, 778–809. [Google Scholar] [CrossRef] [PubMed]
- Sas, A.R.; Bimonte-Nelson, H.A.; Tyor, W.R. Cognitive dysfunction in HIV encephalitic SCID mice correlates with levels of Interferon-α in the brain. AIDS 2007, 21, 2151–2159. [Google Scholar] [CrossRef] [PubMed]
- Sas, A.R.; Bimonte-Nelson, H.; Smothers, C.T.; Woodward, J.; Tyor, W.R. Interferon-α causes neuronal dysfunction in encephalitis. J. Neurosci. 2009, 29, 3948–3955. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, B.; Ashlock, B. M.; Foster, H.; Fujimura, S. H.; Levy, J. A. HIV-infected cells are major inducers of plasmacytoid dendritic cell interferon production, maturation, and migration. Virology 2005, 343, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Severa, M.; Fitzgerald, K.A. TLR-mediated activation of type I IFN during antiviral immune responses: fighting the battle to win the war. Curr. Top. Microbiol. Immunol. 2007, 316, 167–192. [Google Scholar] [PubMed]
- Sgarbanti, M.; Borsetti, A.; Moscufo, N.; Bellocchi, MC.; Ridolfi, B.; Nappi, F.; Marsili, G.; Marziali, G.; Coccia, EM.; Ensoli, B.; Battistini, A. Modulation of human immunodeficiency virus 1 replication by interferon regulatory factors. J. Exp. Med. 2002, 195, 1359–1370. [Google Scholar] [CrossRef] [PubMed]
- Sgarbanti, M.; Marsili, G.; Remoli, A.L.; Ridolfi, B.; Stellacci, E.; Borsetti, A.; Ensoli, B.; Battistini, A. Analysis of the signal transduction pathway leading to human immunodeficiency virus-1-induced interferon regulatory factor-1 upregulation. Ann. N Y Acad. Sci. 2004, 1030, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Shirazi, Y.; Pitha, P.M. Α interferon inhibits early stages of the human immunodeficiency virus type 1 replication cycle. J. Virol. 1992, 66, 1321–1328. [Google Scholar] [PubMed]
- Shirazi, Y.; Pitha, P.M. Interferon α-mediated inhibition of human immunodeficiency virus type 1 provirus synthesis in T-cells. Virology 1993, 193, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Short, J.A.L. Viral evasion of interferon stimulated genes. Bioscience Horizons 2009, 2, 212–224. [Google Scholar] [CrossRef]
- Singh, B.; Ott, T.L.; Bazer, F.W.; de la Concha-Bermejillo, A. Phenotypic and ultrastructural characteristics of bronchoalveolar lavage cells of lentivirus-infected lambs treated with recombinant ovine IFN-tau. J. Interferon Cytokine Res. 2001, 21, 677–686. [Google Scholar] [PubMed]
- Skalka, A.M.; Katz, R.A. Retroviral DNA integration and the DNA damage response . Cell Death Differ. 2005, Suppl 1, 971–978. [Google Scholar] [CrossRef]
- Smith, M.S.; Thresher, R.J.; Pagano, J.S. Inhibition of human immunodeficiency virus type 1 morphogenesis in T cells by α interferon. Antimicrob. Agents Chemother. 1991, 35, 62–67. [Google Scholar] [PubMed]
- Soumelis, V.; Scott, I.; Gheyas, F.; Bouhour, D.; Cozon, G.; Cotte, L.; Huang, L.; Levy, J.A.; Liu, Y.J. Depletion of circulating natural type 1 interferon-producing cells in HIV-infected AIDS patients . Blood 2001, 98, 906–912. [Google Scholar] [CrossRef] [PubMed]
- Soumelis, V.; Scott, I.; Liu, Y.J.; Levy, J. Natural type 1 interferon producing cells in HIV infection. Hum. Immunol. 2002, 63, 1206–1212. [Google Scholar] [CrossRef] [PubMed]
- Sparger, E.E.; Shacklett, B.L.; Renshaw-Gegg, L.; Barry, P.A.; Pedersen, N.C.; Elder, J.H.; Luciw, P.A. Regulation of gene expression directed by the long terminal repeat of the feline immunodeficiency virus. Virology 1992, 187, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Stark, G.R.; Kerr, I.M.; Williams, B.R.; Silverman, R.H.; Schreiber, R.D. How cells respond to interferons. Annu. Rev. Biochem. 1998, 67, 227–264. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, T.; Yamamoto, J.K. Feline immunodeficiency virus lacks sensitivity to the antiviral activity of feline IFN-gamma. J. Interferon Cytokine Res. 2001, 21, 1039–46. [Google Scholar] [PubMed]
- Taylor, M.D.; Korth, M.J.; Katze, M.G. Interferon treatment inhibits the replication of simian immunodeficiency virus at an early stage: evidence for a block between attachment and reverse transcription. Virology 1998, 241, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.J.V.; Locarnini, S.A. Toll-like receptors, RIG-I-like RNA helicases and the antiviral innate immune response. Immunol. Cell. Biol. 2007, 85, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Thompson, F.J.; Elder, J.; Neil, J.C. Cis- and trans-regulation of feline immunodeficiency virus: identification of functional binding sites in the long terminal repeat. J. Gen. Virol. 1994, 75, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Tilton, J.C.; Johnson, A.J.; Luskin, M.R.; Manion, M.M.; Yang, J.; Adelsberger, J.W.; Lempicki, R.A.; Hallahan, C.W.; McLaughlin, M.; Mican, J.M.; Metcalf, J.A.; Iyasere, C.; Connors, M. Diminished production of monocyte proinflammatory cytokines during human immunodeficiency virus viremia is mediated by type I interferons. J. Virol. 2006, 80, 11486–11497. [Google Scholar] [CrossRef] [PubMed]
- Tilton, J.C.; Manion, M.M.; Luskin, M.R.; Johnson, A.J.; Patamawenu, A.A.; Hallahan, C.W.; Cogliano-Shutta, N.A.; Mican, J.M.; Davey Jr., R.T.; Kottilil, S.; Lifson, J.D.; Metcalf, J.A.; Lempicki, R.A.; Connors, M. Human immunodeficiency virus viremia induces plasmacytoid dendritic cell activation in vivo and diminished α interferon production in vitro. J. Virol. 2008, 82, 3997–4006. [Google Scholar] [CrossRef] [PubMed]
- Tsukahara, T.; Kim, S.; Taylor, M.W. REFINEMENT: a search framework for the identification of interferon-responsive elements in DNA sequences--a case study with ISRE and GAS. Comput. Biol. Chem. 2006, 30, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Uematsu, S.; Akira, S. Toll-like receptors and type I interferons. J. Biol. Chem. 2007, 282, 15319–15324. [Google Scholar] [CrossRef] [PubMed]
- Van Lint, C.; Amella, C.A.; Emiliani, S.; John, M.; Jie, T.; Verdin, E. Transcription factor binding sites downstream of the human immunodeficiency virus type 1 transcription start site are important for virus infectivity. J. Virol. 1997, 71, 6113–6127. [Google Scholar] [PubMed]
- Vieillard, V.; Costagliola, D.; Simon, A.; Debré, P; French Asymptomatiques à Long Terme (ALT) Study Group. Specific adaptive humoral response against a gp41 motif inhibits CD4 T-cell sensitivity to NK lysis during HIV-1 infection . AIDS 2006, 20, 1795–1804. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.R. Transcriptional regulation of interferon-stimulated genes. Eur. J. Biochem. 1991, 200, 1–11. [Google Scholar] [CrossRef]
- Yamamoto, J.K.; Ho, E.; Pedersen, N.C. A feline retrovirus induced T-lymphoblastoid cell-line that produces an atypical α type of interferon. Vet. Immunol. Immunopathol. 1986, 11, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Yonezawa, A.; Morita, R.; Takaori-Kondo, A.; Kadowaki, N.; Kitawaki, T.; Hori, T.; Uchiyama, T. Natural α interferon-producing cells respond to human immunodeficiency virus type 1 with α interferon production and maturation into dendritic cells. J. Virol. 2003, 77, 3777–3784. [Google Scholar] [CrossRef] [PubMed]
- Zeidner, N.S.; Myles, M.H.; Mathiason-DuBard, C.K.; Dreitz, M.J.; Mullins, J.I.; Hoover, E.A. Α interferon (2b) in combination with zidovudine for the treatment of presymptomatic feline leukemia virus-induced immunodeficiency syndrome. Antimicrob. Agents Chemother. 1990, 34, 1749–1756. [Google Scholar] [PubMed]
- Zhang, J.; Yamada, O.; Kawagishi, K.; Araki, H.; Yamaoka, S.; Hattori, T.; Shimotohno, K. Human T-cell leukemia virus type 1 Tax modulates interferon-α signal transduction through competitive usage of the coactivator CBP/p300. Virology 2008, 379, 306–313. [Google Scholar] [CrossRef] [PubMed]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Gómez-Lucía, E.; Collado, V.M.; Miró, G.; Doménech, A. Effect of Type-I Interferon on Retroviruses. Viruses 2009, 1, 545-573. https://doi.org/10.3390/v1030545
Gómez-Lucía E, Collado VM, Miró G, Doménech A. Effect of Type-I Interferon on Retroviruses. Viruses. 2009; 1(3):545-573. https://doi.org/10.3390/v1030545
Chicago/Turabian StyleGómez-Lucía, Esperanza, Victorio M. Collado, Guadalupe Miró, and Ana Doménech. 2009. "Effect of Type-I Interferon on Retroviruses" Viruses 1, no. 3: 545-573. https://doi.org/10.3390/v1030545
APA StyleGómez-Lucía, E., Collado, V. M., Miró, G., & Doménech, A. (2009). Effect of Type-I Interferon on Retroviruses. Viruses, 1(3), 545-573. https://doi.org/10.3390/v1030545