Evidence of Ash Tree (Fraxinus spp.) Specific Associations with Soil Bacterial Community Structure and Functional Capacity

 , ,
, ,
Abstract

1. Introduction
2. Materials and Methods
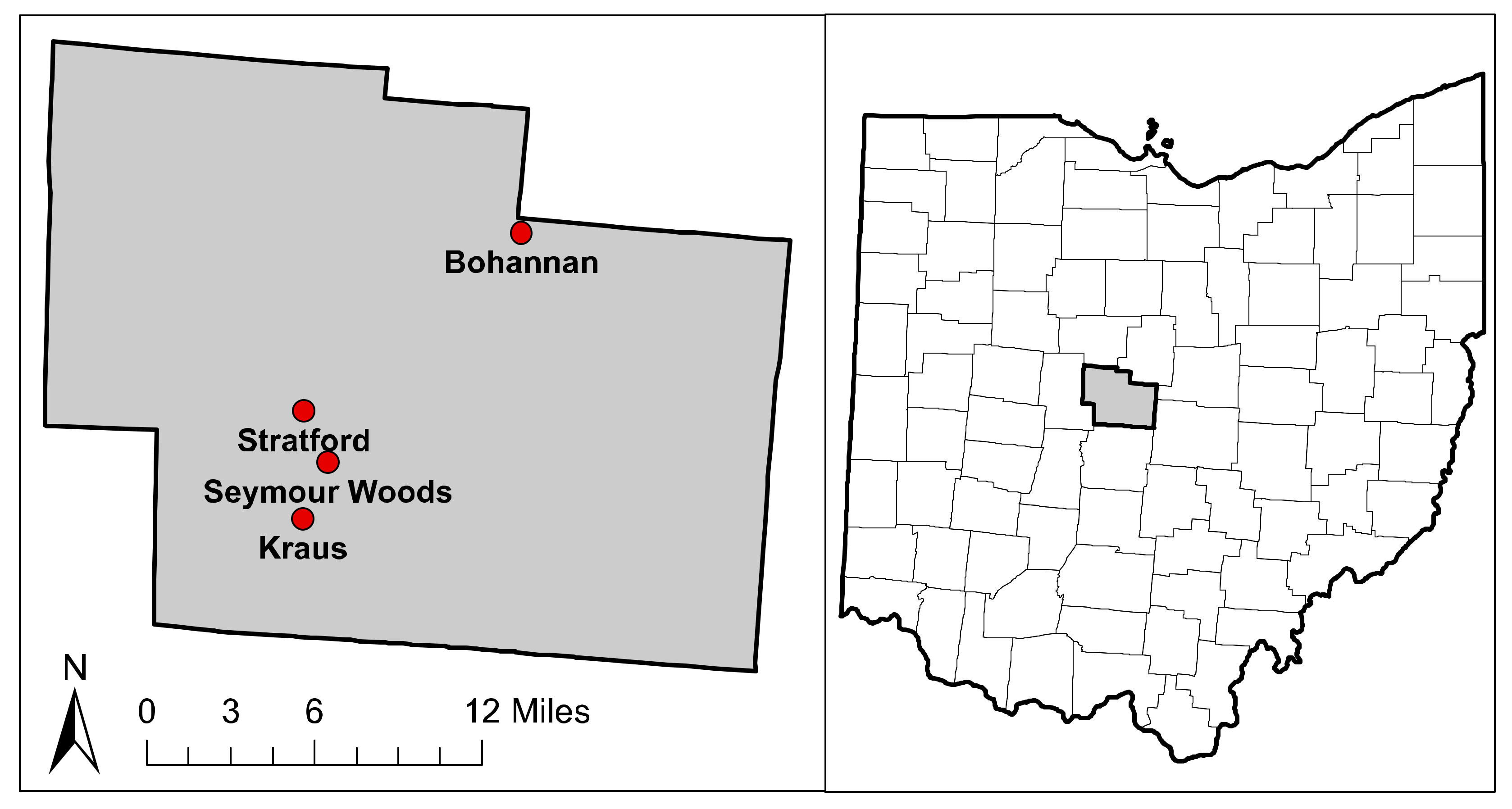
2.1. Site Description
2.2. Soil Collection and Characterization
2.3. DNA Extraction, Sequencing, Quality Control and Bioinformatics
2.4. Statistical Analyses
3. Results
3.1. Environmental and Site Differences
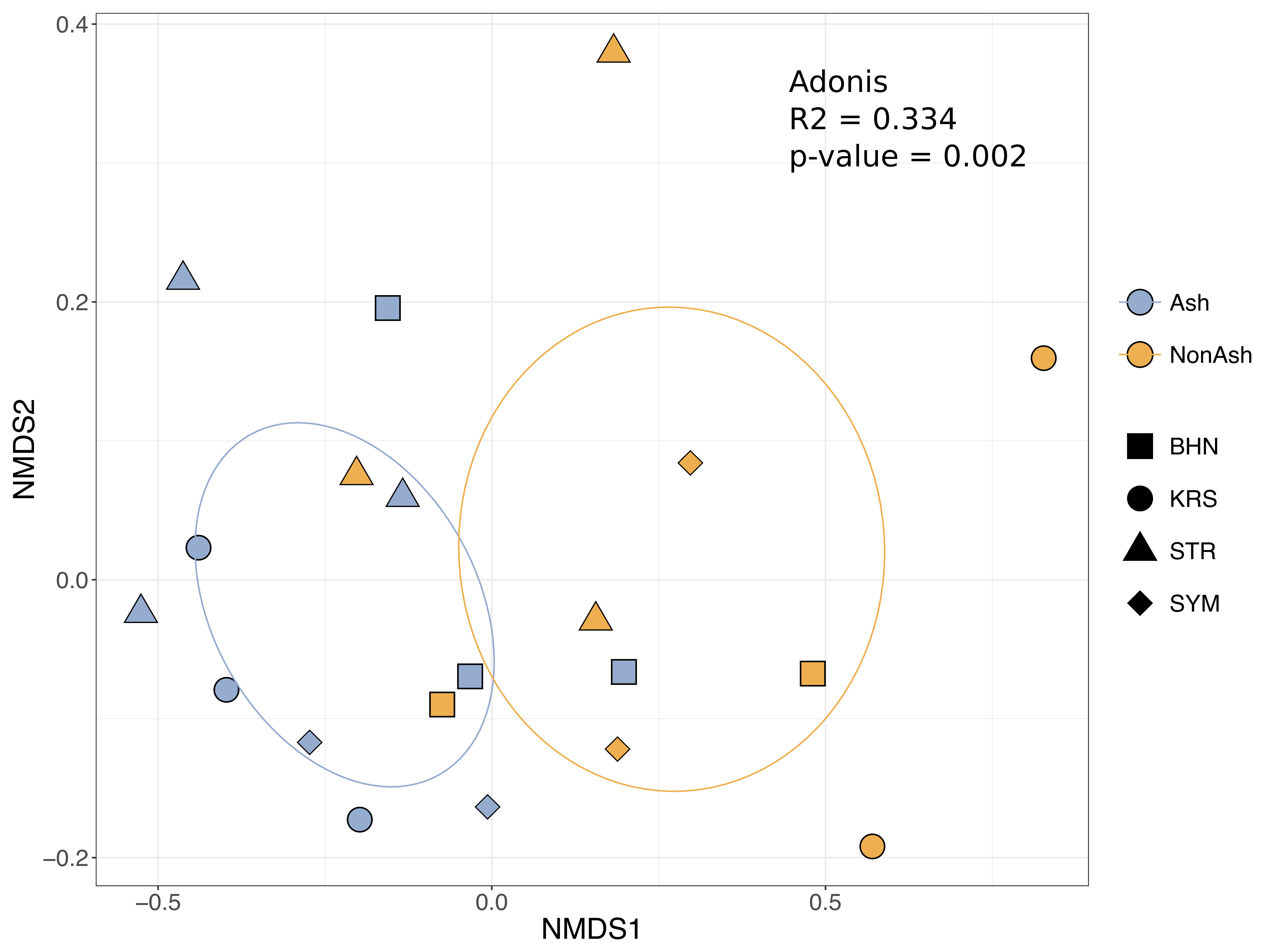
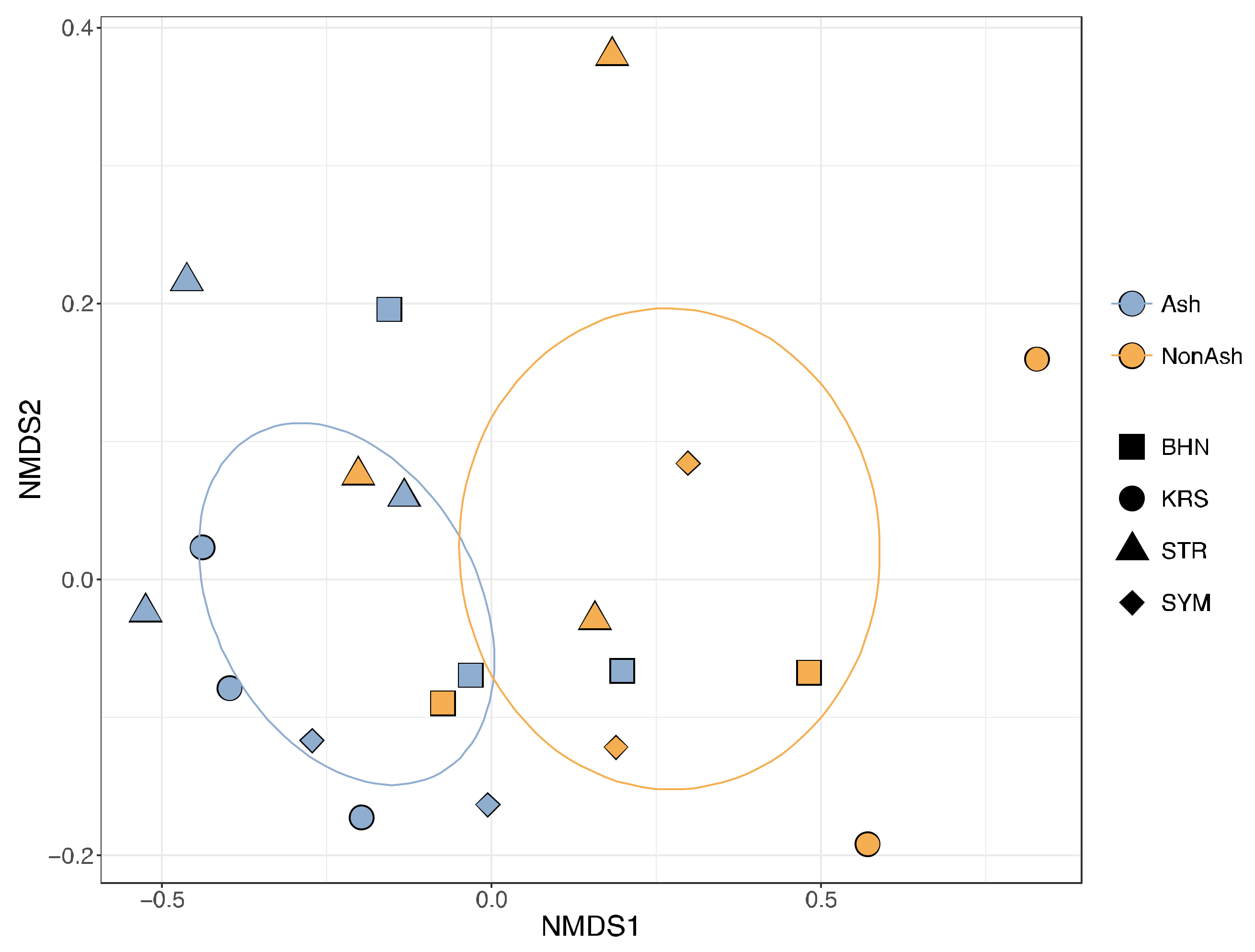
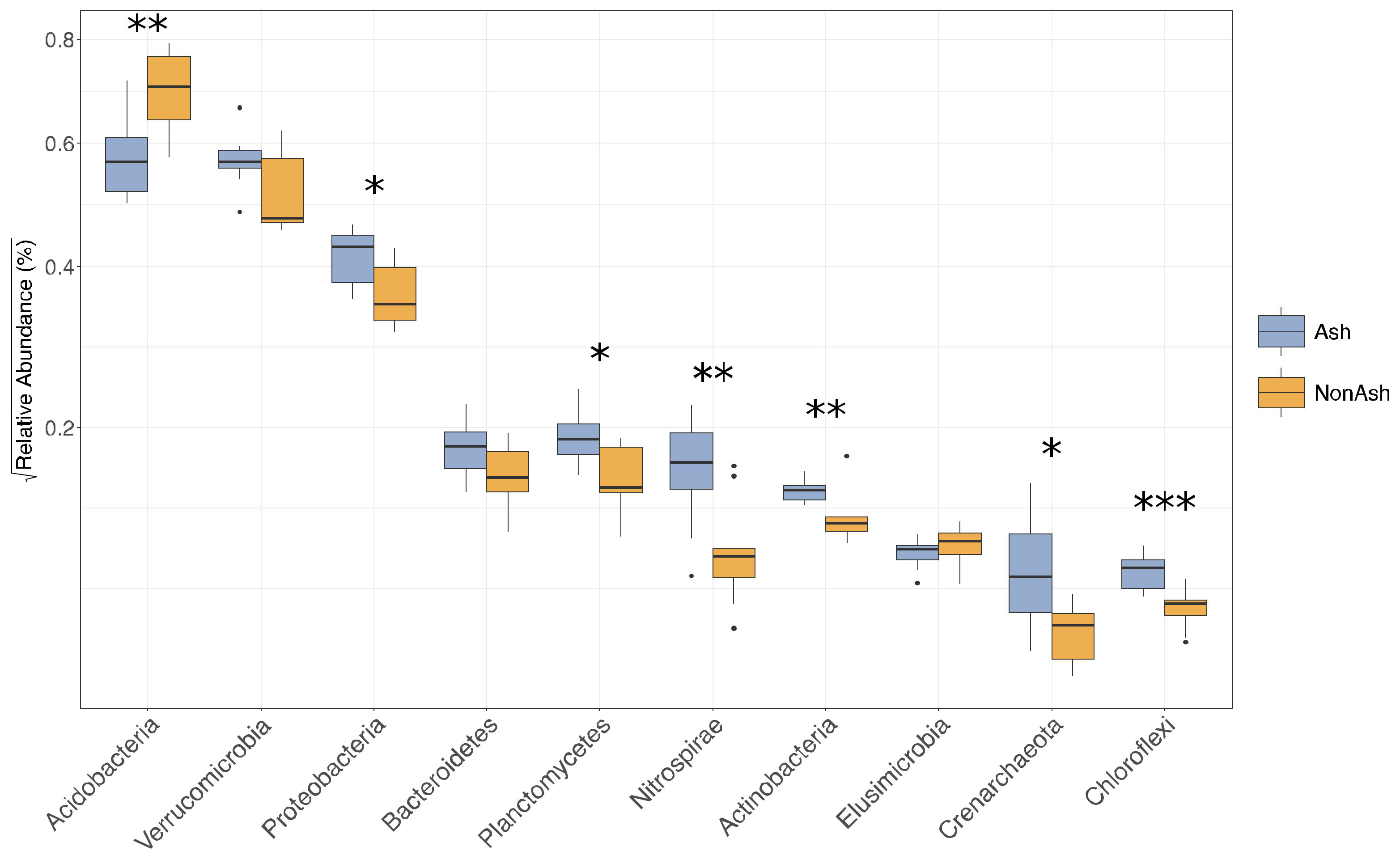
3.2. Bacterial Community Differences
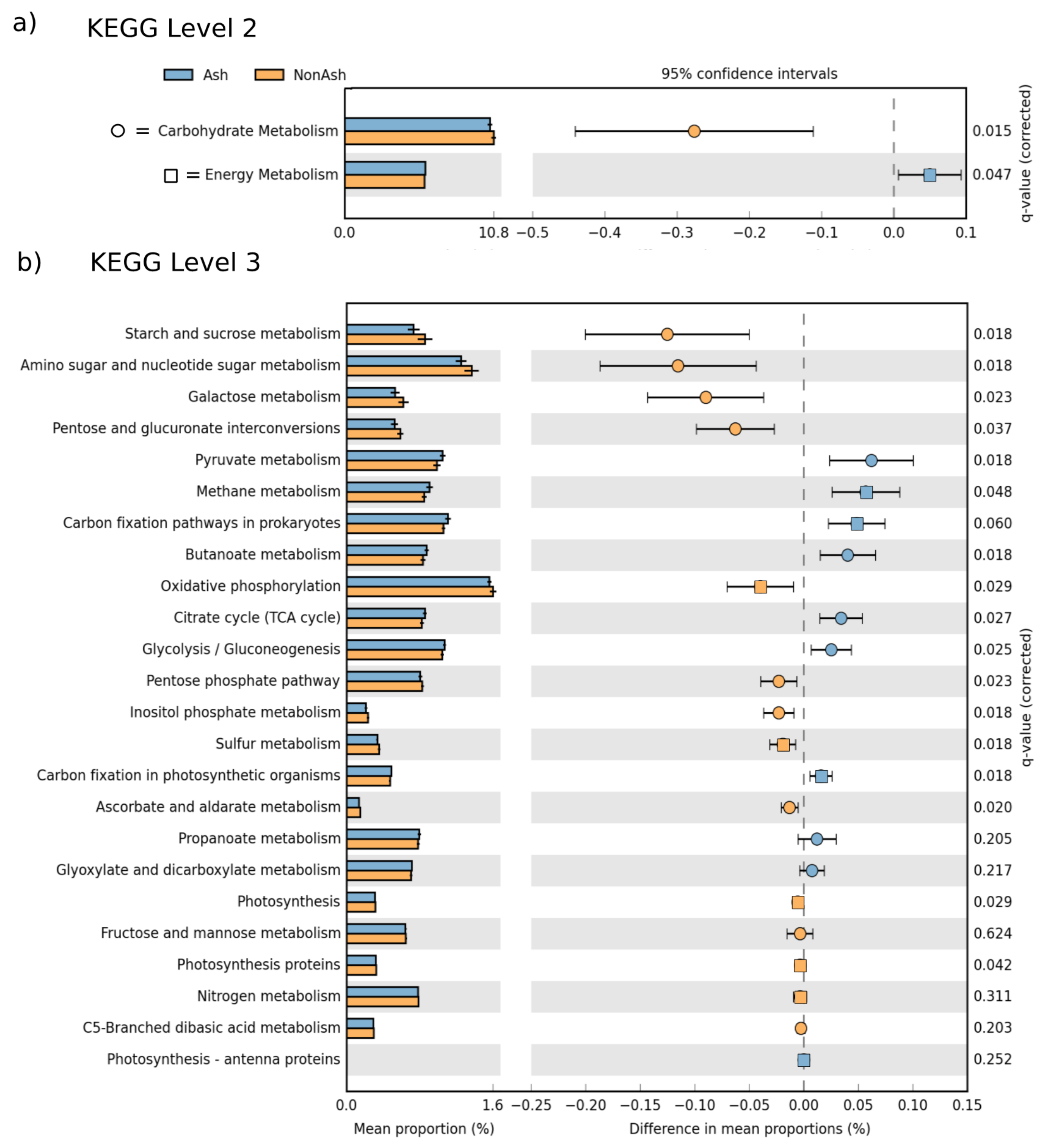
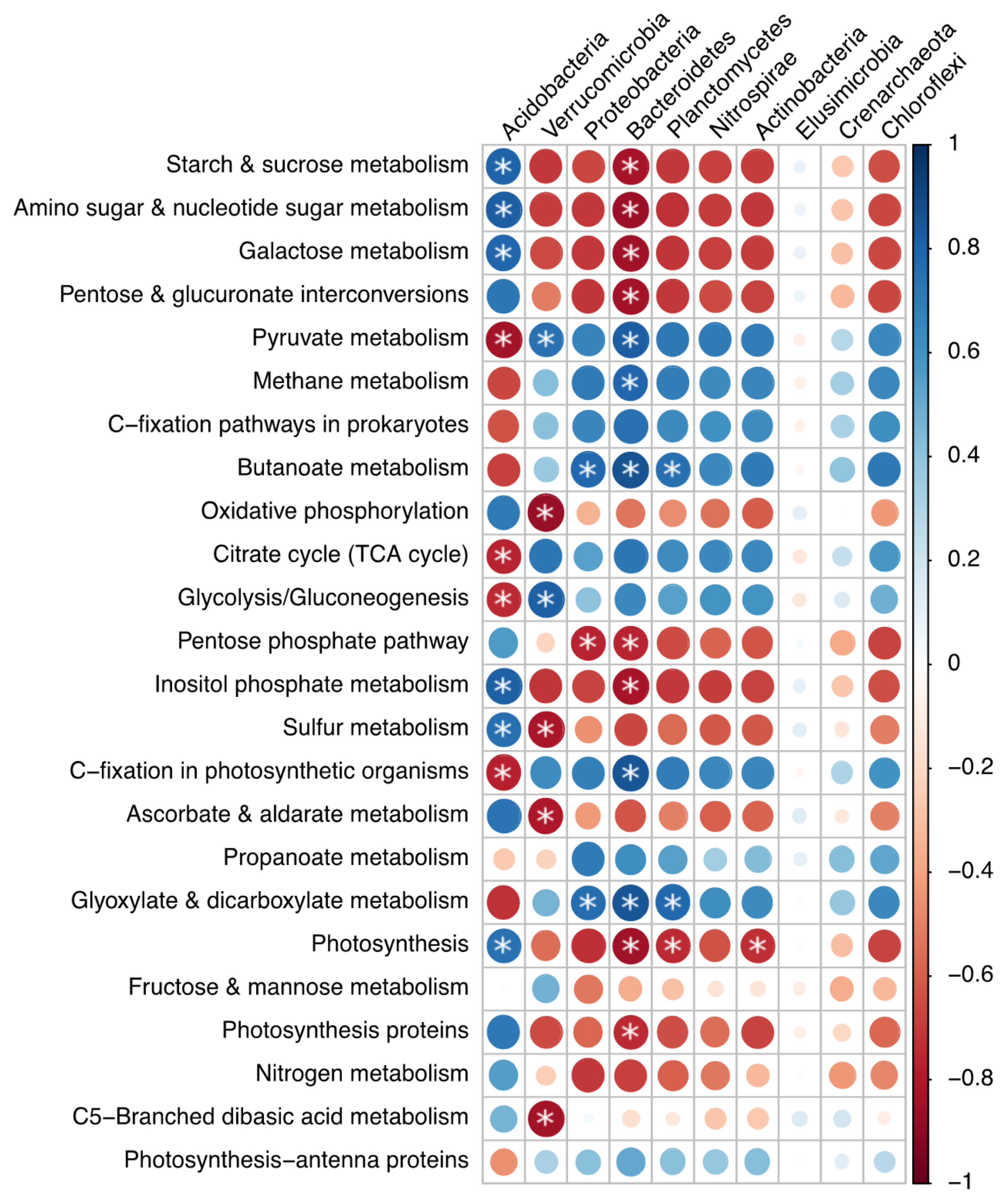
3.3. Bacterial Functional Differences
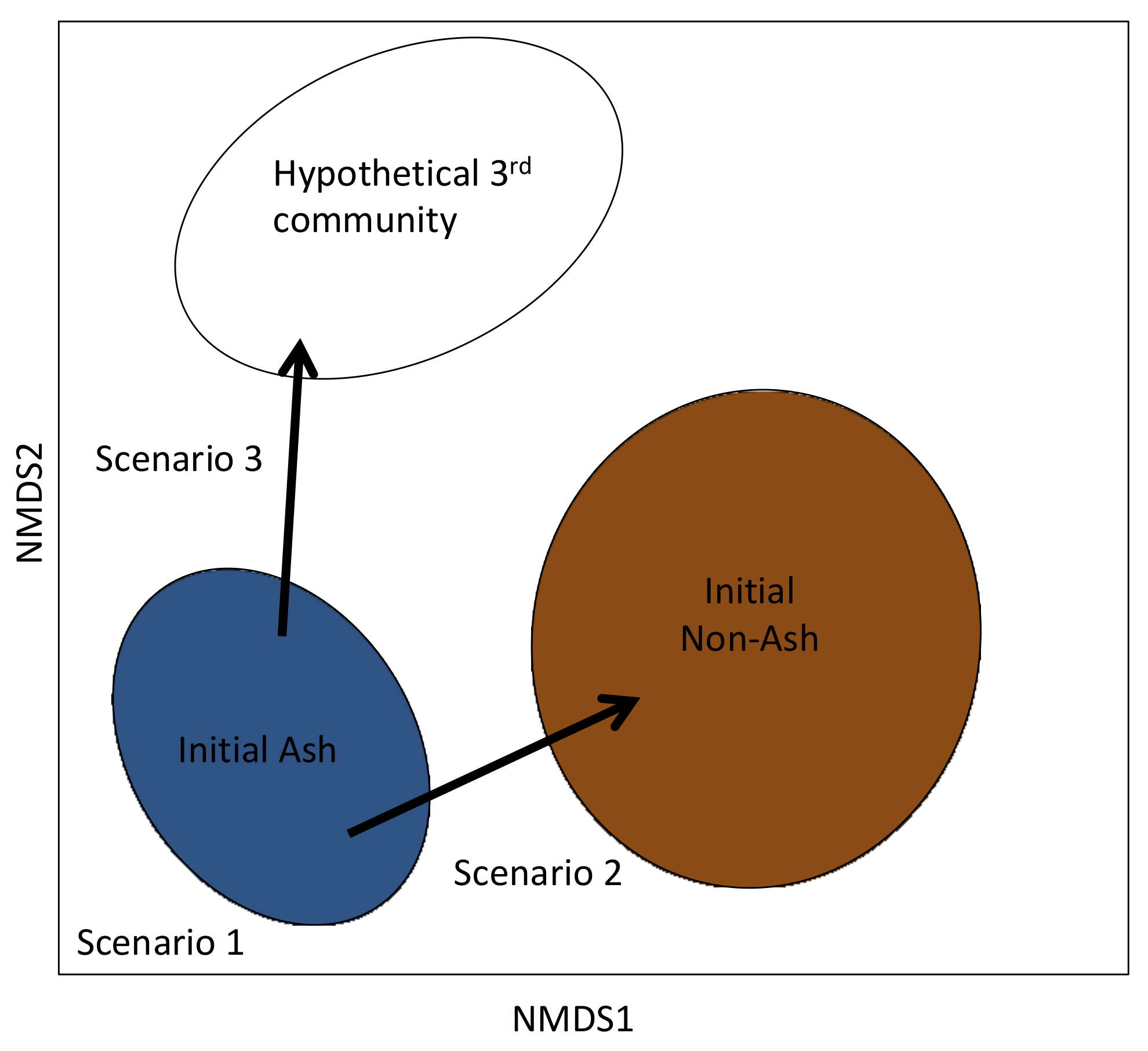
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Parmesan, C.; Yohe, G. A globally coherent fingerprint of climate change impacts across natural systems. Nature 2003, 421, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Settele, J.; Scholes, R.J.; Betts, R.A.; Bunn, S.; Leadley, P.; Nepstad, D.; Overpeck, J.T.; Toboada, M.A. Chapter 4—Terrestrial and inland water systems. In Climate Change 2014: Impacts, Adaptation, and Vulnerability. Part A: Global and Sectoral Aspects; Contribution of Working Group II to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change; Field, C.B., Barros, V.R., Dokken, D.J., Mach, K.J., Mastrandrea, M.D., Bilir, T.E., Chatterjee, M., Ebi, K.L., Estrada, Y.O., Genova, R.C., et al., Eds.; Cambridge University Press: Cambridge, UK; New York, NY, USA, 2014; pp. 271–359. ISBN 1102700991936. [Google Scholar]
- Cramer, W.; Bondeau, A.; Woodward, F.I.; Prentice, I.C.; Betts, R.A.; Brovkin, V.; Cox, P.M.; Fisher, V.; Foley, J.A.; Friend, A.D.; et al. Global response of terrestrial ecosystem structure and function to CO2 and climate change: Results from six dynamic global vegetation models. Glob. Chang. Biol. 2001, 7, 357–373. [Google Scholar] [CrossRef]
- Drewniak, B.; Gonzalez-Meler, M.A. Earth system model needs for including the interactive representation of nitrogen deposition and drought effects on forested ecosystems. Forests 2017, 8, 267. [Google Scholar] [CrossRef]
- Gonzalez-Meler, M.A.; Rucks, J.S.; Aubanell, G. Mechanistic insights on the responses of plant and ecosystem gas exchange to global environmental change: Lessons from Biosphere 2. Plant Sci. 2014, 226, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Mcnickle, G.G.; Gonzalez-Meler, M.A.; Lynch, D.J.; Baltzer, J.L.; Brown, J.S. The world’s biomes and primary production as a triple tragedy of the commons foraging game played among plants. Proc. R. Soc. B Biol. Sci. 2016, 283, 20161993. [Google Scholar] [CrossRef] [PubMed]
- Cappaert, D.; McCullough, D.G.; Poland, T.M.; Siegert, N.W. Emerald Ash Borer in North America: A Research and Regulatory Challenge. Am. Entomol. 2005, 51, 152–165. [Google Scholar] [CrossRef]
- Wang, X.Y.; Yang, Z.Q.; Gould, J.R.; Zhang, Y.N.; Liu, G.J.; Liu, E. The Biology and Ecology of the Emerald Ash Borer, Agrilus planipennis, in China. J. Insect Sci. 2010, 10, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Flower, C.E.; Lynch, D.J.; Knight, K.S.; Gonzalez-Meler, M.A. Biotic and abiotic drivers of sap flux in mature green ash trees (Fraxinus pennsylvanica) experiencing varying levels of emerald ash borer (Agrilus planipennis) infestation. Forests 2018, in press. [Google Scholar]
- McCullough, D.G.; Katovich, S.A. Emerald ash borer. Pest Alert 2004, 4–5. [Google Scholar] [CrossRef]
- Knight, K.S.; Robert, P.; Rebbeck, J. How Fast Will Trees Die? A Transition Matrix Model of Ash Decline in Forest Stands Infested by Emerald Ash Borer; U.S. Department of Agriculture, Forest Service: Washington, DC, USA, 2008; pp. 28–29.
- Costilow, K.C.; Knight, K.S.; Flower, C.E. Disturbance severity and canopy position control the radial growth response of maple trees (Acer spp.) in forests of northwest Ohio impacted by emerald ash borer (Agrilus planipennis). Ann. For. Sci. 2017, 74. [Google Scholar] [CrossRef]
- Birdsey, R.A. Carbon Storage and Accumulation in United States Forest Ecosystems; General Technical Report WO-59; United States Department of Agriculture, Forest Service: Washington, DC, USA, 1992.
- Birdsey, R.A.; Heath, L.S. Carbon Changes in U.S. Forests. In Productivity of America’s Forests and Climate Change; General Technical Report RM-GTR-271; Joyce, L.A., Ed.; United States Department of Agriculture, Forest Service, Rocky Mountain Forest and Experiment Station: Fort Collins, CO, USA, 1995; pp. 56–70. [Google Scholar]
- Goodale, C.L.; Apps, M.J.; Birdsey, R.A.; Field, C.B.; Heath, L.S.; Houghton, R.A.; Jenkins, J.C.; Kohlmaier, G.H.; Liu, S.; Nabuurs, G.; et al. Forest Carbon Sinks in the Northern Hemisphere. Ecol. Appl. 2002, 12, 891–899. [Google Scholar] [CrossRef]
- Flower, C.E.; Knight, K.S.; Gonzalez-Meler, M.A. Impacts of the emerald ash borer (Agrilus planipennis Fairmaire) induced ash (Fraxinus spp.) mortality on forest carbon cycling and successional dynamics in the eastern United States. Biol. Invasions 2013, 15, 931–944. [Google Scholar] [CrossRef]
- Higham, M.; Hoven, B.M.; Gorchov, D.L.; Knight, K.S. Patterns of Coarse Woody Debris in Hardwood Forests across a Chronosequence of Ash Mortality Due to the Emerald Ash Borer (Agrilus planipennis). Nat. Areas J. 2017, 37, 406–411. [Google Scholar] [CrossRef]
- Lovett, G.M.; Canham, C.D.; Arthur, M.A.; Weathers, K.C.; Fitzhugh, R.D. Forest ecosystem responses to exotic pests and pathogens in eastern North America. Bioscience 2006, 56, 395–405. [Google Scholar] [CrossRef]
- Telander, A.C.; Slesak, R.A.; D’Amato, A.W.; Palik, B.J.; Brooks, K.N.; Lenhart, C.F. Sap flow of black ash in wetland forests of northern Minnesota, USA: Hydrologic implications of tree mortality due to emerald ash borer. Agric. For. Meteorol. 2015, 206, 4–11. [Google Scholar] [CrossRef]
- Flower, C.E.; Gonzalez-Meler, M.A. Responses of Temperate Forest Productivity to Insect and Pathogen Disturbances. Annu. Rev. Plant Biol. 2015, 66, 547–569. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, F.; Gonzalez-Meler, M.A.; Flower, C.E.; Lynch, D.J.; Czimczik, C.; Tang, J.; Subke, J.A. Ecosystem-level controls on root-rhizosphere respiration. New Phytol. 2013, 199, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.; Parton, W.J.; Gonzalez-Meler, M.A.; Phillips, R.; Asao, S.; McNickle, G.G.; Brzostek, E.; Jastrow, J.D. Synthesis and modeling perspectives of rhizosphere priming. New Phytol. 2014, 201, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Van Der Heijden, M.G.A.; Bardgett, R.D.; Van Straalen, N.M. The unseen majority: Soil microbes as drivers of plant diversity and productivity in terrestrial ecosystems. Ecol. Lett. 2008, 11, 296–310. [Google Scholar] [CrossRef] [PubMed]
- Allison, S.S.D.; Martiny, J.B.H. Resistance, resilience, and redundancy in microbial communities. Proc. Natl. Acad. Sci. USA 2008, 105, 11512–11519. [Google Scholar] [CrossRef] [PubMed]
- Ricketts, M.P.; Poretsky, R.S.; Welker, J.M.; Gonzalez-Meler, M. Soil bacterial community and functional shifts in response to altered snowpack in moist acidic tundra of Northern Alaska. Soil 2016, 2, 459–474. [Google Scholar] [CrossRef]
- Bailey, V.L.; Fansler, S.J.; Stegen, J.C.; McCue, L.A. Linking microbial community structure to β-glucosidic function in soil aggregates. ISME J. 2013, 7, 2044–2053. [Google Scholar] [CrossRef] [PubMed]
- Schimel, J.P.; Schaeffer, S.M. Microbial control over carbon cycling in soil. Front. Microbiol. 2012, 3, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Fierer, N. Embracing the unknown: Disentangling the complexities of the soil microbiome. Nat. Rev. Microbiol. 2017, 15, 579–590. [Google Scholar] [CrossRef] [PubMed]
- Fierer, N.; Jackson, R.B. The diversity and biogeography of soil bacterial communities. Proc. Natl. Acad. Sci. USA 2006, 103, 626–631. [Google Scholar] [CrossRef] [PubMed]
- Lauber, C.L.; Hamady, M.; Knight, R.; Fierer, N. Pyrosequencing-Based Assessment of Soil pH as a Predictor of Soil Bacterial Community Structure at the Continental Scale. Appl. Environ. Microbiol. 2009. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.J.; Kim, M.H.; Lee, Y.O. Effect of pH on soil bacterial diversity. J. Ecol. Environ. 2016, 40, 10. [Google Scholar] [CrossRef]
- Schlatter, D.C.; Bakker, M.G.; Bradeen, J.M.; Kinkel, L.L. Plant community richness and microbial interactions structure bacterial communities in soil. Ecology 2015, 96, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Bakker, M.G.; Bradeen, J.M.; Kinkel, L.L. Effects of plant host species and plant community richness on streptomycete community structure. FEMS Microbiol. Ecol. 2013, 83, 596–606. [Google Scholar] [CrossRef] [PubMed]
- Prescott, C.E.; Grayston, S.J. Tree species influence on microbial communities in litter and soil: Current knowledge and research needs. For. Ecol. Manag. 2013, 309, 19–27. [Google Scholar] [CrossRef]
- Flower, C.E.; Knight, K.S.; Rebbeck, J.; Gonzalez-Meler, M.A. The relationship between the emerald ash borer (Agrilus planipennis) and ash (Fraxinus spp.) tree decline: Using visual canopy condition assessments and leaf isotope measurements to assess pest damage. For. Ecol. Manage. 2013, 303, 143–147. [Google Scholar] [CrossRef]
- Smith, A. Effects of Community Structure on Forest Susceptibility and Response to the Emerald Ash Borer Invasion of the Huron River Watershed in Southeast Michigan. Doctor’s Dissertation, Ohio State University, Columbus, OH, USA, 2006. [Google Scholar]
- Gilbert, J.A.; Jansson, J.K.; Knight, R. The Earth Microbiome project: Successes and aspirations. BMC Biol. 2014, 12, 69. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.; Kuczynski, J.; Stombaugh, J. QIIME allows analysis of high—Throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. 2013. Available online: http://www.R-project.org/ (accessed on 3 March 2018).
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; Mcglinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package. R Package Version 2.4-5. 2017. Available online: https://CRAN.R-project.org/package=vegan (accessed on 3 March 2018).
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, R.J.; Huenneke, L.F. Disturbance, Diversity, and Invasion: Implications for Conservation. Conserv. Biol. 1992, 6, 324–337. [Google Scholar] [CrossRef]
- Kaiser, C.; Koranda, M.; Kitzler, B.; Fuchslueger, L.; Schnecker, J.; Schweiger, P.; Rasche, F.; Zechmeister-Boltenstern, S.; Sessitsch, A.; Richter, A. Belowground carbon allocation by trees drives seasonal patterns of extracellular enzyme activities by altering microbial community composition in a beech forest soil. New Phytol. 2010, 187, 843–858. [Google Scholar] [CrossRef] [PubMed]
- Urbanová, M.; Šnajdr, J.; Baldrian, P. Composition of fungal and bacterial communities in forest litter and soil is largely determined by dominant trees. Soil Biol. Biochem. 2015, 84, 53–64. [Google Scholar] [CrossRef]
- Lejon, D.P.H.; Chaussod, R.; Ranger, J.; Ranjard, L. Microbial Community Structure and Density under Different Tree Species in an Acid Forest Soil (Morvan, France). Microb. Ecol. 2005, 50, 614–625. [Google Scholar] [CrossRef] [PubMed]
- Ward, N.L.; Challacombe, J.F.; Janssen, P.H.; Henrissat, B.; Coutinho, P.M.; Wu, M.; Xie, G.; Haft, D.H.; Sait, M.; Badger, J.; et al. Three Genomes from the Phylum Acidobacteria Provide Insight into the Lifestyles of These Microorganisms in Soils. Appl. Environ. Microbiol. 2009, 75, 2046–2056. [Google Scholar] [CrossRef] [PubMed]
- Burns, R.M.; Honkala, B.H. Fraxinus. In Silvics of North America: Volume 2, Hardwoods; Agriculture Handbook 654; United States Department of Agriculture, Forest Service: Washington, DC, USA, 1990; Volume 2, pp. 333–357. ISBN 1800553684. [Google Scholar]
- Kielak, A.M.; Barreto, C.C.; Kowalchuk, G.A.; van Veen, J.A.; Kuramae, E.E. The ecology of Acidobacteria: Moving beyond genes and genomes. Front. Microbiol. 2016, 7, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Margalef, O.; Sardans, J.; Fernández-Martínez, M.; Molowny-Horas, R.; Janssens, I.A.; Ciais, P.; Goll, D.; Richter, A.; Obersteiner, M.; Asensio, D.; et al. Global patterns of phosphatase activity in natural soils. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Turner, B.L.; Lambers, H.; Condron, L.M.; Cramer, M.D.; Leake, J.R.; Richardson, A.E.; Smith, S.E. Soil microbial biomass and the fate of phosphorus during long-term ecosystem development. Plant Soil 2013, 367, 225–234. [Google Scholar] [CrossRef]
- Richardson, A.E.; Simpson, R.J. Soil Microorganisms Mediating Phosphorus Availability Update on Microbial Phosphorus. Plant Physiol. 2011, 156, 989–996. [Google Scholar] [CrossRef] [PubMed]
- Ferrenberg, S.; Knelman, J.E.; Jones, J.M.; Beals, S.C.; Bowman, W.D.; Nemergut, D.R. Soil bacterial community structure remains stable over a 5-year chronosequence of insect-induced tree mortality. Front. Microbiol. 2014, 5, 1–11. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Forest | Soil Type 1 | Number of Plots (Ash/Non-ash) | BA (m2/ha) | Relative BA of Ash Trees (%) |
|---|---|---|---|---|
| Bohannan (BHN) | Cardington silt loam | 3/2 | 37.7 ± 2.5 | 49.3 ± 5.7 |
| Kraus (KRS) | Glynwood silt loam | 3/2 | 34.7 ± 3.0 | 63.2 ± 4.4 |
| Seymour (SYM) | Blount silt loam | 2/2 | 26.0 ± 3.0 | 46.5 ± 6.0 |
| Stratford (STR) | Glynwood silt loam | 3/3 | 33.9 ± 5.0 | 35.5 ± 13.0 |
| (a) | Adonis Test | |||||||||||
| Bacterial Community | Soil Environment | |||||||||||
| Categorical Variables | R2 | p-value | R2 | p-value | ||||||||
| Ash vs. Non-ash | 0.334 | 0.002 | 0.098 | 0.066 | ||||||||
| Forest site | 0.140 | 0.502 | 0.301 | 0.003 | ||||||||
| (b) | Mann-Whitney U Test (Ash vs. Non-Ash) | Kruskal-Wallis Test (Forest Site; df = 3) | Mantel Test (Bacterial Community) | |||||||||
| Continuous Variables | W | p-value | H | p-value | r-statistic | p-value | ||||||
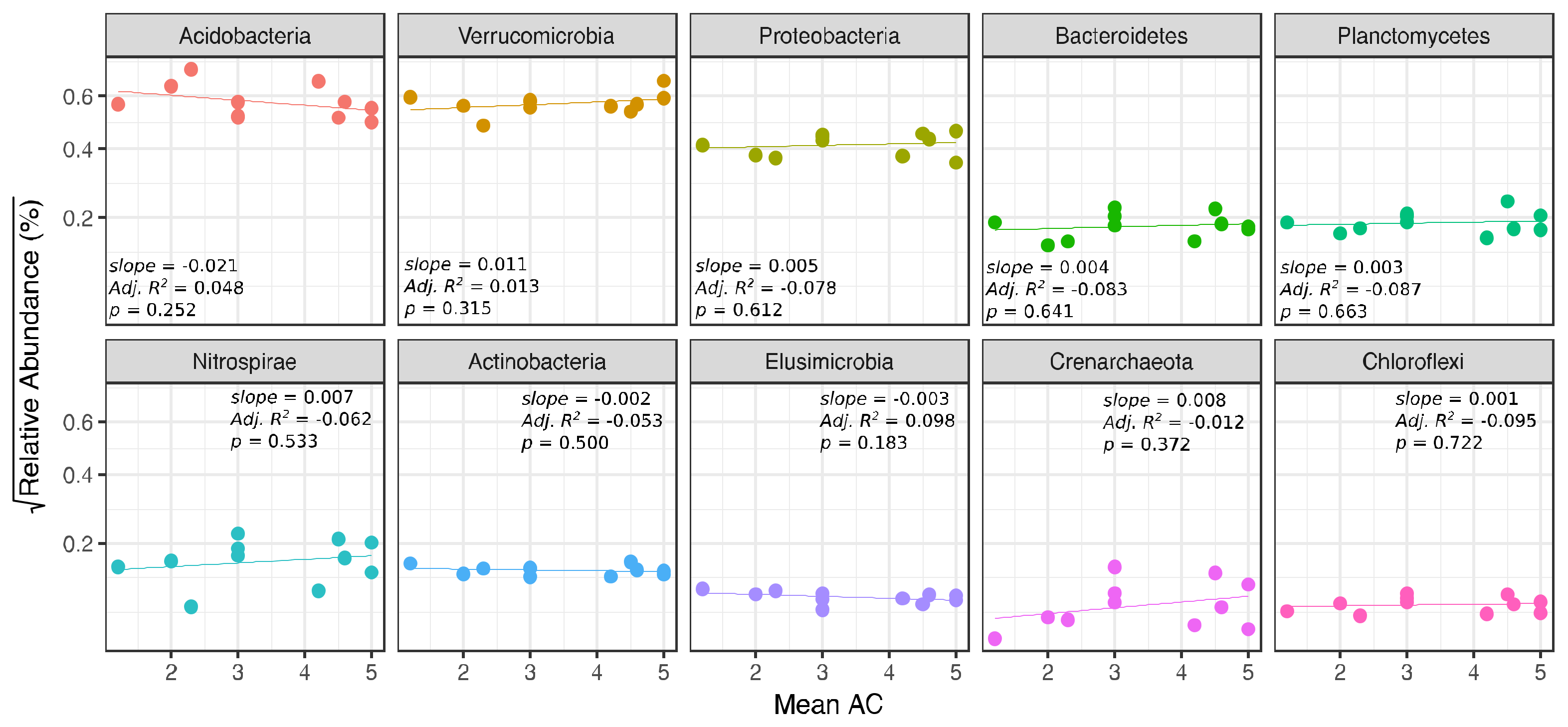
| Mean AC (ash only) | - | - | 9.24 | 0.026 | −0.060 | 0.620 | ||||||
| Mean Stems (#/ha) | 75.5 | 0.051 | 8.78 | 0.032 | −0.127 | 0.870 | ||||||
| Mean BA (m2/ha) | 69 | 0.152 | 5.23 | 0.156 | 0.060 | 0.261 | ||||||
| Ash (%) | - | - | 1.19 | 0.755 | 0.264 | 0.007 | ||||||
| Maple (%) | 49 | 1.000 | 4.42 | 0.220 | 0.041 | 0.306 | ||||||
| Oak (%) | 11 | 0.003 | 4.13 | 0.247 | 0.030 | 0.338 | ||||||
| Beech (%) | 57 | 0.570 | 12.09 | 0.007 | 0.182 | 0.097 | ||||||
| Hickory (%) | 53.5 | 0.743 | 3.03 | 0.387 | 0.028 | 0.334 | ||||||
| α-diversity (tree) | 60.5 | 0.425 | 5.67 | 0.129 | −0.048 | 0.631 | ||||||
| α-diversity (bacteria) | 54 | 0.766 | 4.07 | 0.254 | 0.039 | 0.329 | ||||||
| Soil pH | 73 | 0.080 | 3.88 | 0.275 | 0.289 | 0.006 | ||||||
| %C | 49.5 | 1.000 | 11.51 | 0.009 | −0.173 | 0.981 | ||||||
| %N | 58 | 0.541 | 12.96 | 0.005 | −0.175 | 0.986 | ||||||
| C:N | 34.5 | 0.270 | 10.15 | 0.017 | −0.134 | 0.911 | ||||||
| Ca | 69 | 0.152 | 4.53 | 0.210 | 0.304 | 0.007 | ||||||
| K | 42 | 0.603 | 3.71 | 0.295 | −0.030 | 0.594 | ||||||
| Mg | 67 | 0.201 | 4.81 | 0.186 | 0.274 | 0.011 | ||||||
| P | 49 | 1.000 | 12.35 | 0.006 | −0.088 | 0.846 | ||||||
| Al | 29 | 0.131 | 9.71 | 0.021 | 0.177 | 0.045 | ||||||
| B | 40 | 0.494 | 3.64 | 0.303 | −0.075 | 0.708 | ||||||
| Cu | 12.5 | 0.006 | 0.32 | 0.957 | 0.047 | 0.304 | ||||||
| Fe | 18 | 0.016 | 1.49 | 0.685 | 0.273 | 0.010 | ||||||
| Mn | 48 | 0.941 | 2.11 | 0.550 | −0.143 | 0.921 | ||||||
| Na | 56 | 0.656 | 6.37 | 0.095 | 0.002 | 0.439 | ||||||
| S | 24 | 0.056 | 0.88 | 0.831 | −0.143 | 0.924 | ||||||
| Zn | 40 | 0.503 | 9.79 | 0.020 | 0.083 | 0.241 | ||||||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ricketts, M.P.; Flower, C.E.; Knight, K.S.; Gonzalez-Meler, M.A. Evidence of Ash Tree (Fraxinus spp.) Specific Associations with Soil Bacterial Community Structure and Functional Capacity. Forests 2018, 9, 187. https://doi.org/10.3390/f9040187
Ricketts MP, Flower CE, Knight KS, Gonzalez-Meler MA. Evidence of Ash Tree (Fraxinus spp.) Specific Associations with Soil Bacterial Community Structure and Functional Capacity. Forests. 2018; 9(4):187. https://doi.org/10.3390/f9040187
Chicago/Turabian StyleRicketts, Michael P., Charles E. Flower, Kathleen S. Knight, and Miquel A. Gonzalez-Meler. 2018. "Evidence of Ash Tree (Fraxinus spp.) Specific Associations with Soil Bacterial Community Structure and Functional Capacity" Forests 9, no. 4: 187. https://doi.org/10.3390/f9040187
APA StyleRicketts, M. P., Flower, C. E., Knight, K. S., & Gonzalez-Meler, M. A. (2018). Evidence of Ash Tree (Fraxinus spp.) Specific Associations with Soil Bacterial Community Structure and Functional Capacity. Forests, 9(4), 187. https://doi.org/10.3390/f9040187