

Integrated Transcriptomic Analysis Reveals Molecular Mechanisms Underlying Albinism in Schima superba Seedlings
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials and Samplings
2.2. Library Construction and Sequencing
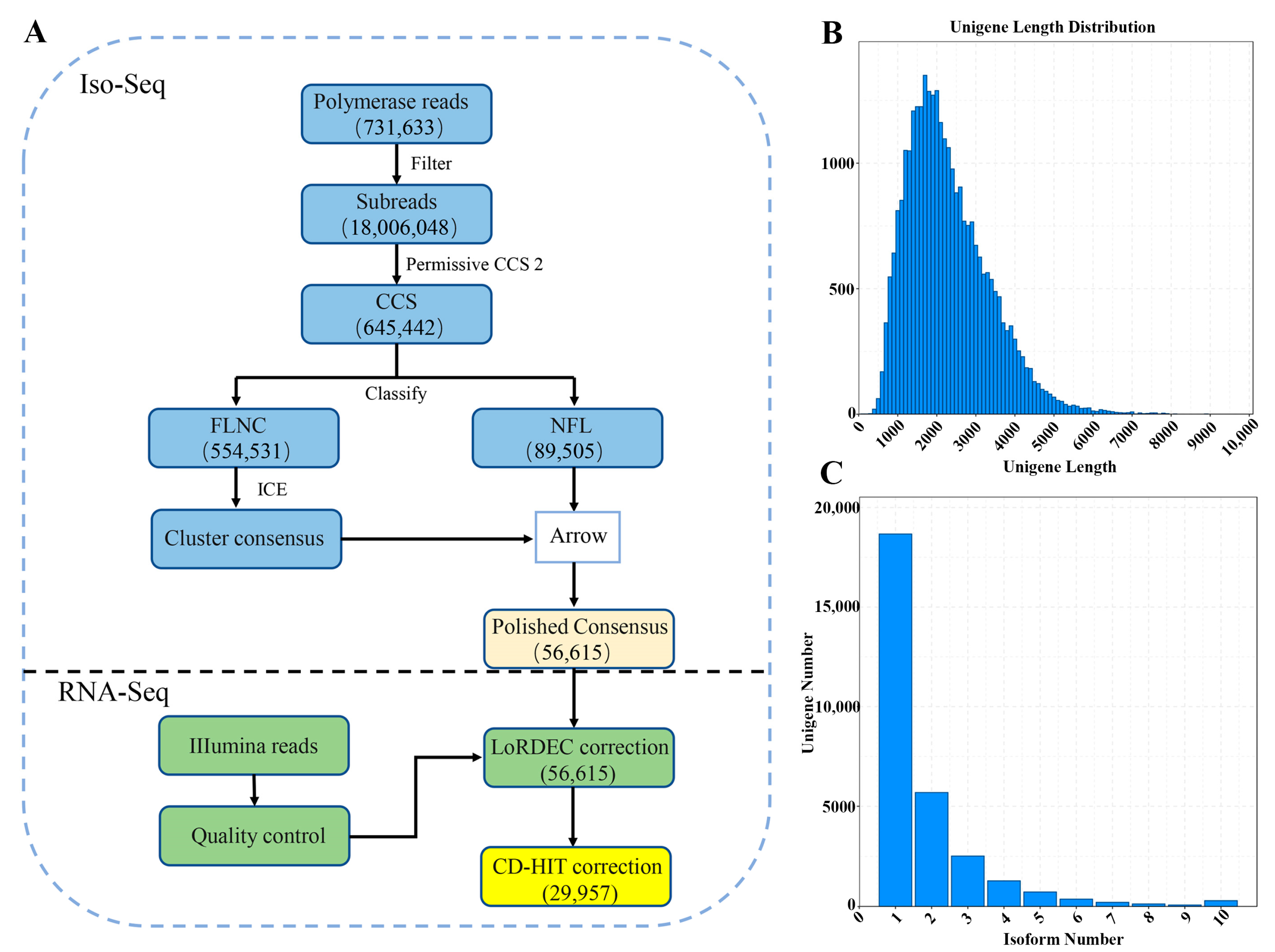
2.3. PacBio Iso-Seq Data Processing
2.4. Transcript Structure and Functional Annotation
2.5. Analysis of Gene Expression Levels
2.6. Differential Expression and Pathway Analysis
3. Results
3.1. Sequencing Data Overview
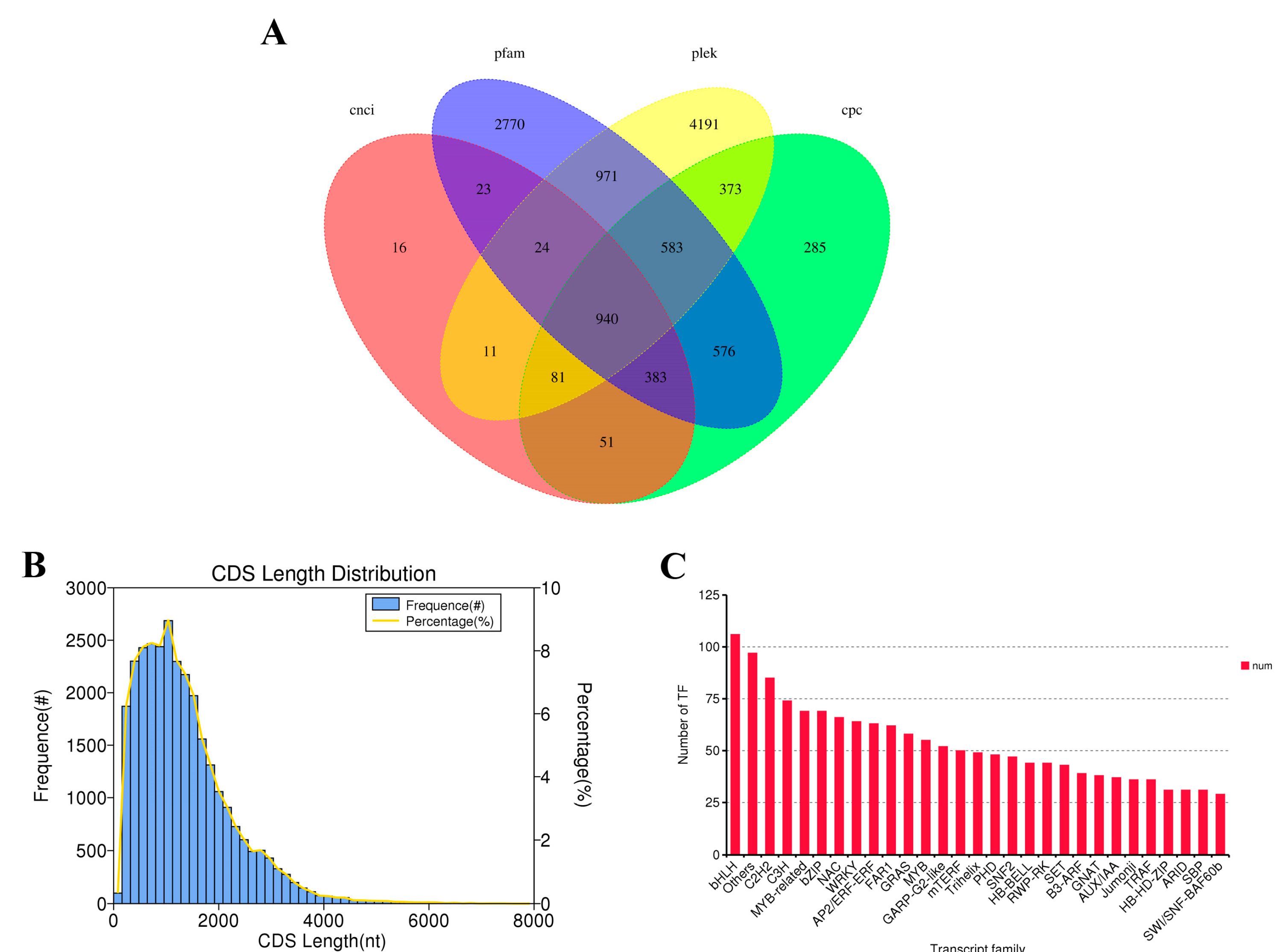
3.2. Full-Length Transcript Coding Sequences, LncRNA and Transcription Factor Prediction
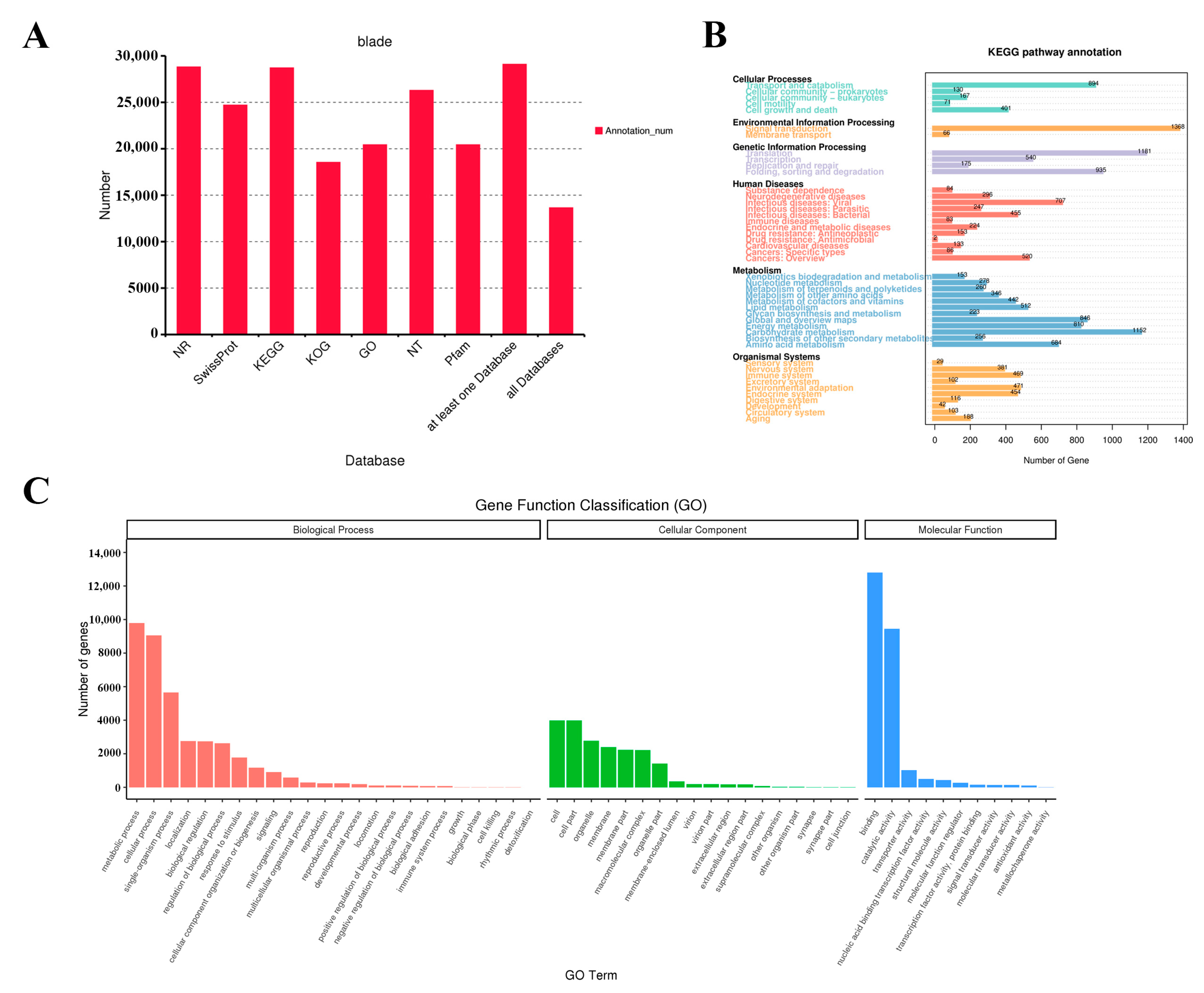
3.3. Functional Annotation Analysis of Full-Length Transcripts
3.4. Reference Sequence Comparison
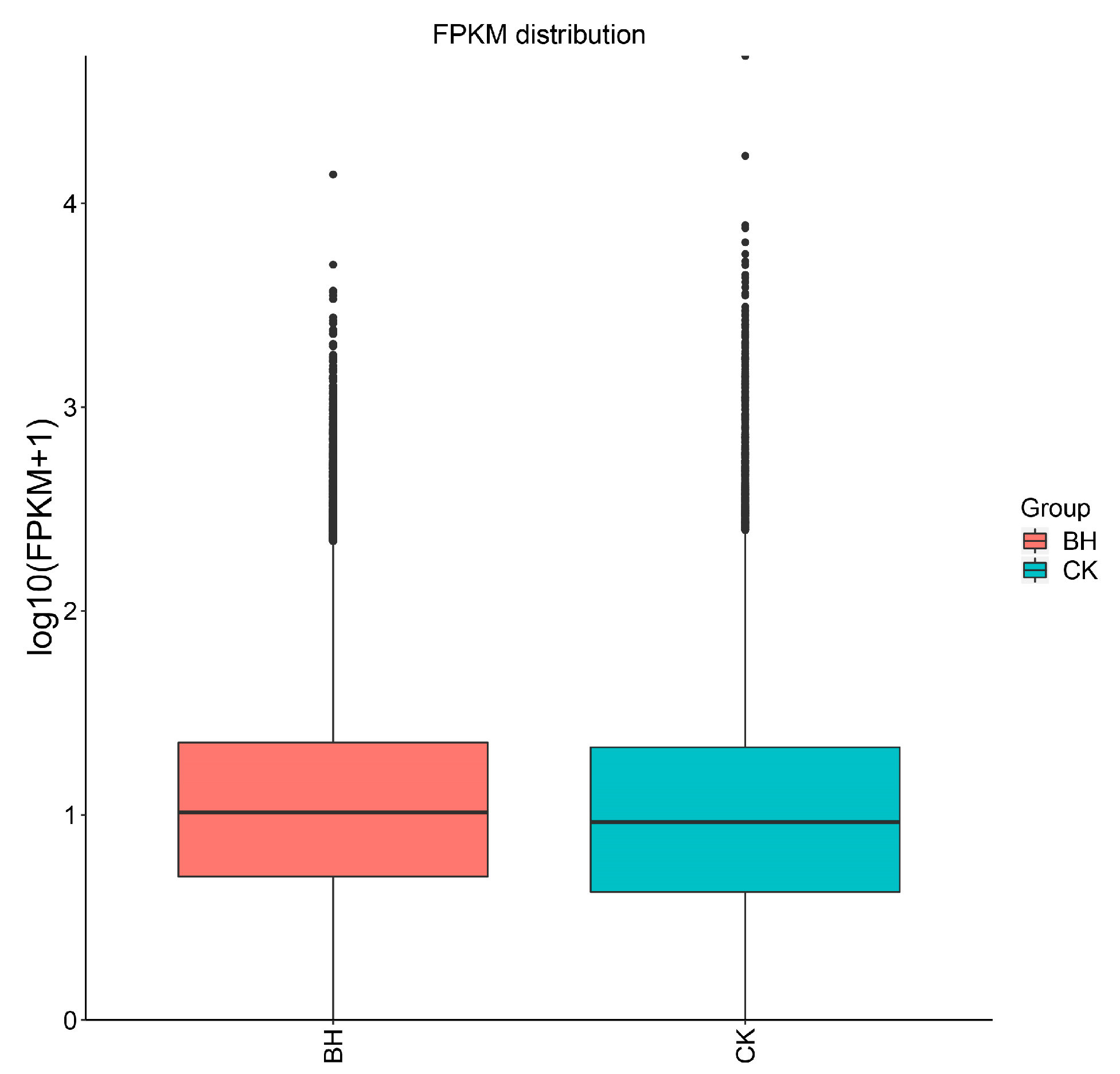
3.5. Analysis of Gene Expression and DEGs
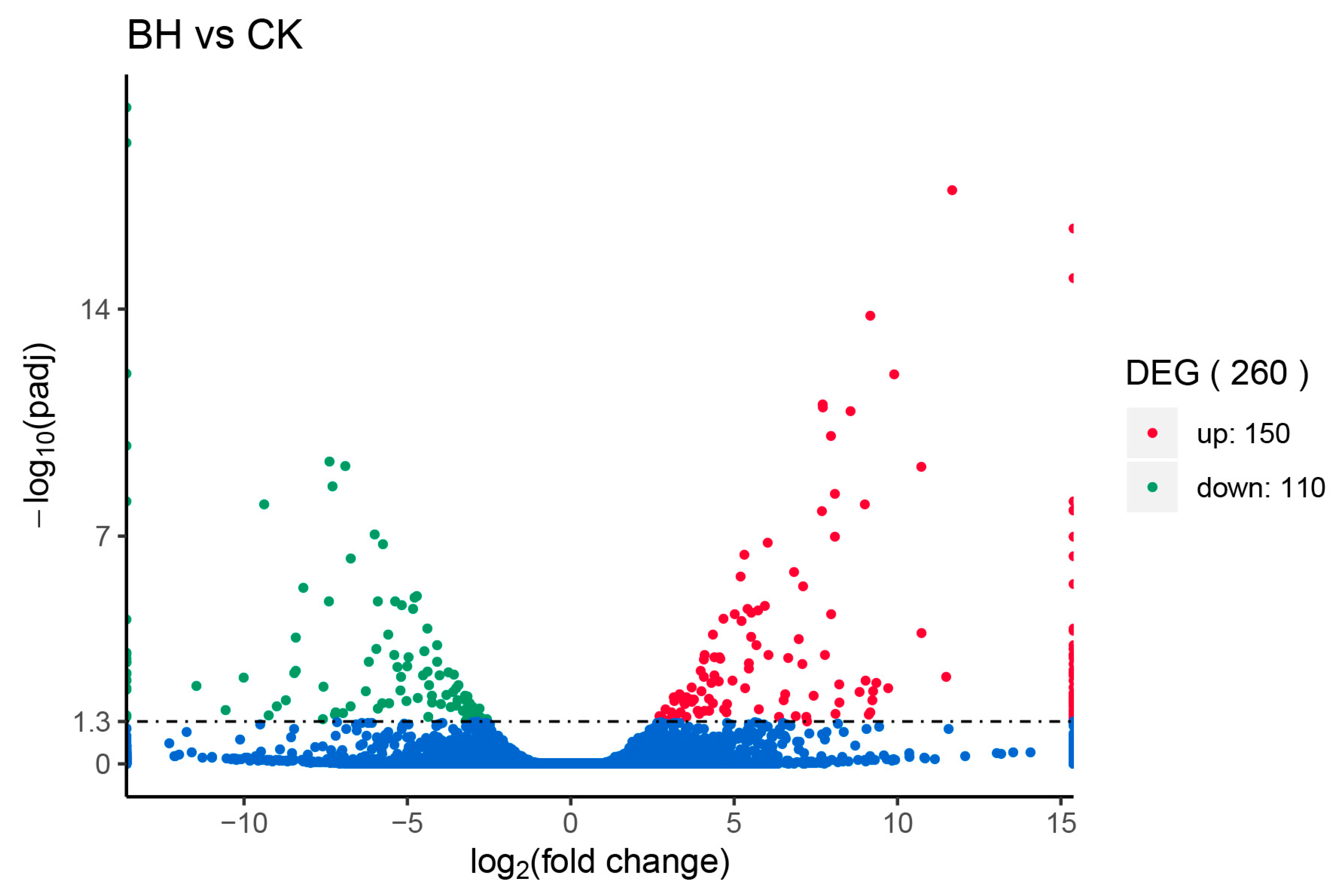
3.6. DEGs Profiling
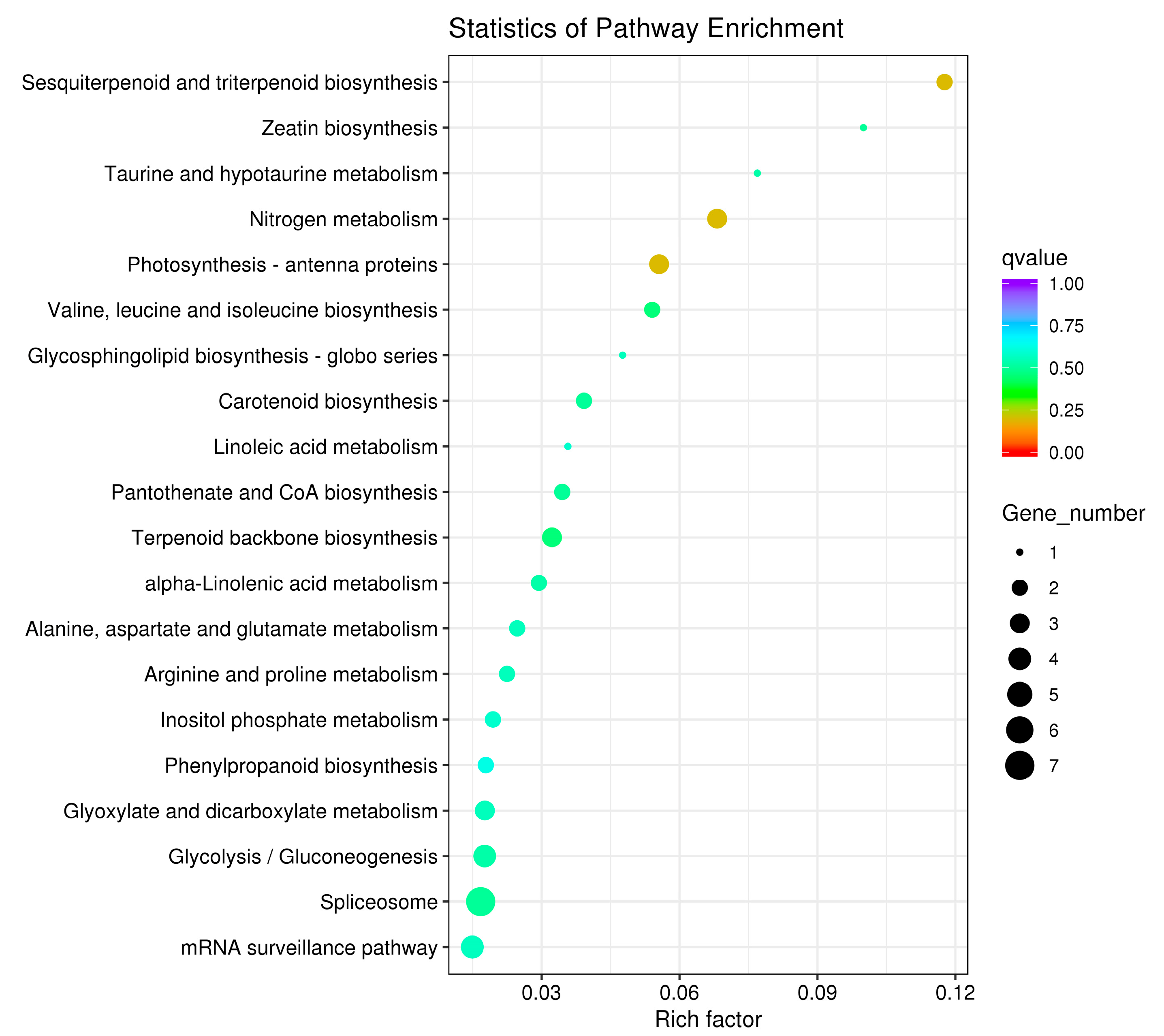
3.7. Functional Annotation of DEGs
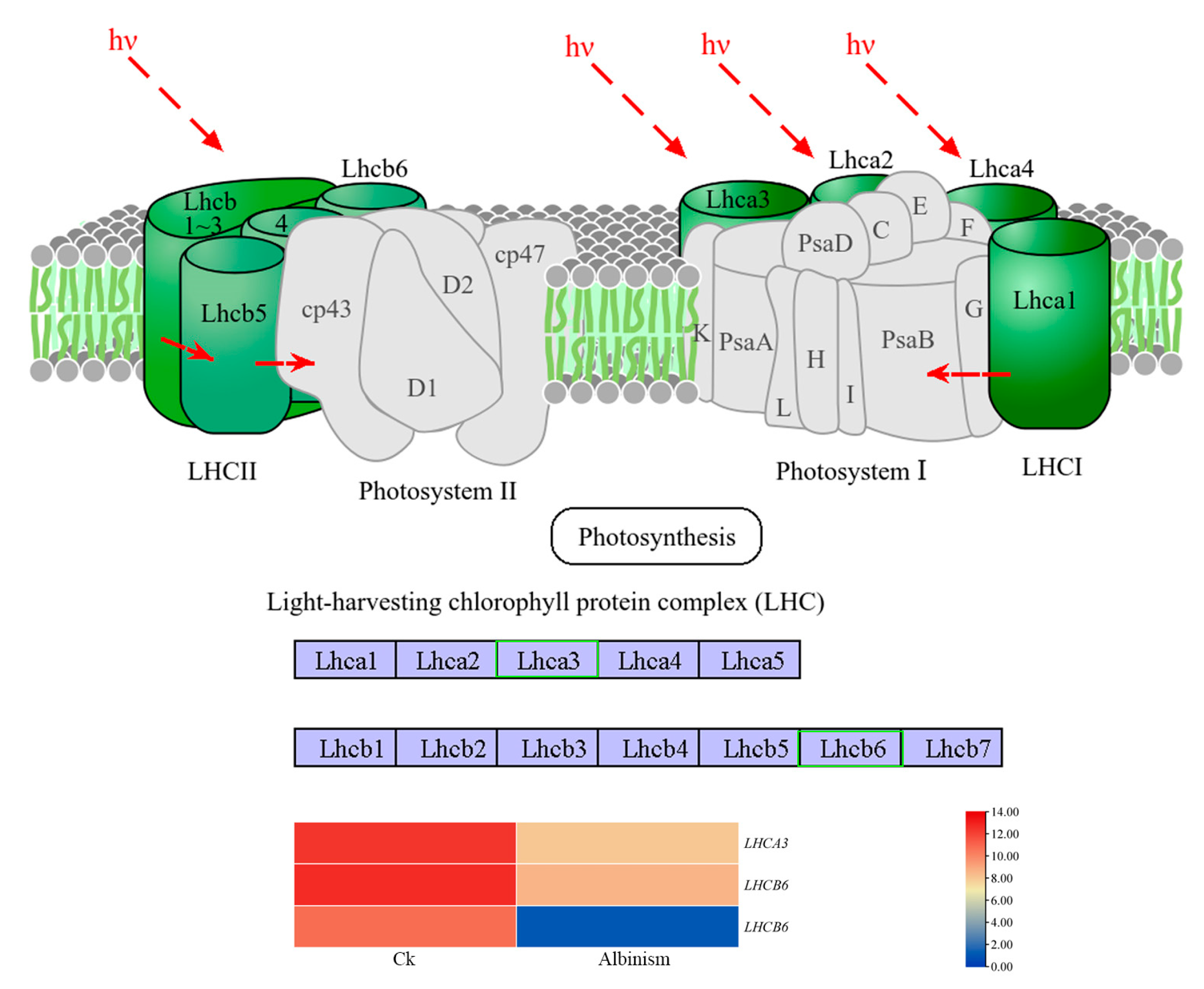
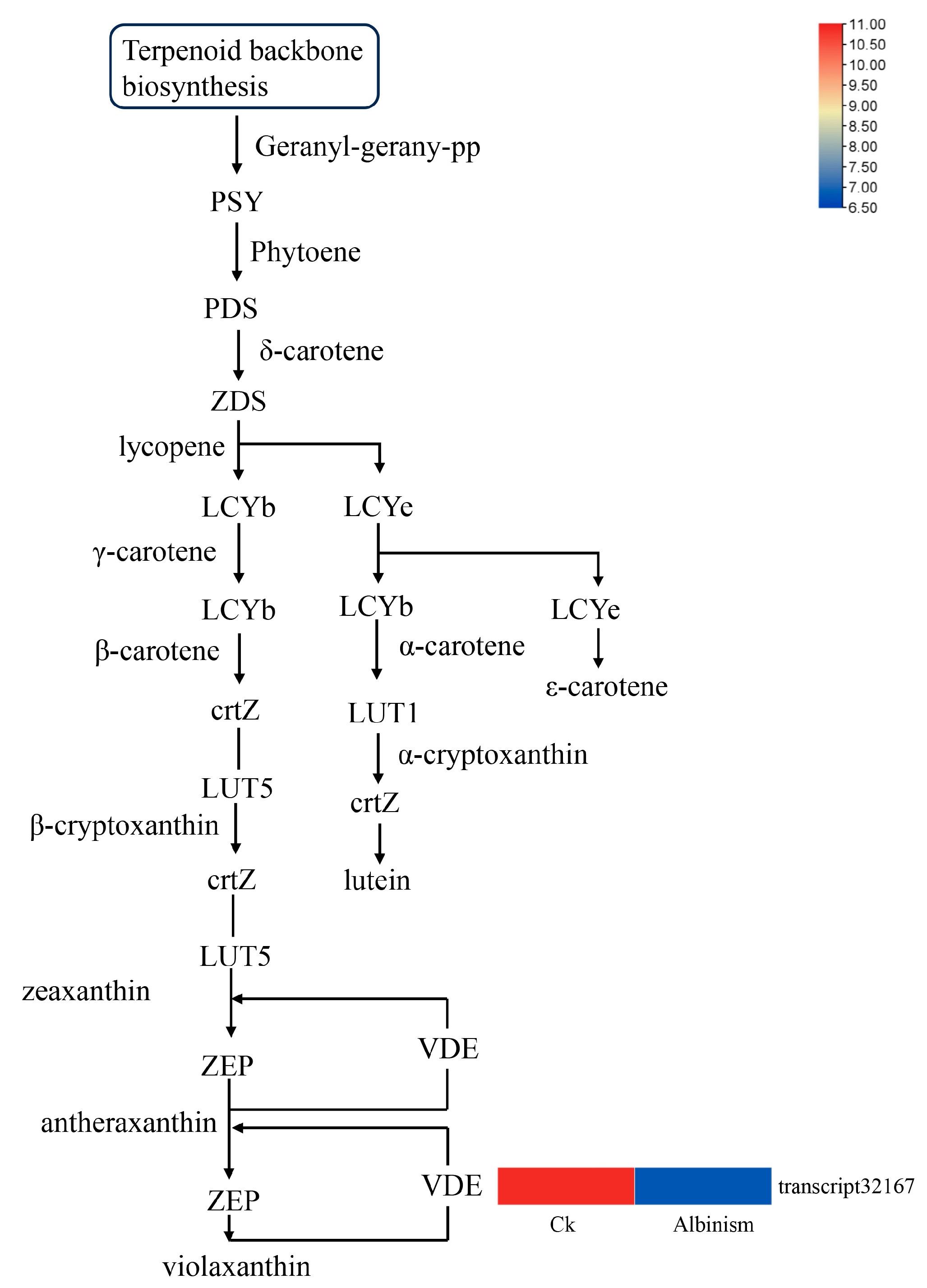
3.8. Differential Analysis of Genes Encoding Genes Related to the Photosynthesis-Antenna Protein Pathway and the Carotenoid Biosynthesis Pathway
3.9. Analysis of TFs in S. superba Albinism Seedlings
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, J.; Jia, X.C.; Liu, L.Y.; Cao, X.M.; Xiong, Y.F.; Yang, Y.D.; Zhou, H.Q.; Yi, M.; Li, M. Comparative biochemical and transcriptome analysis provides insights into the regulatory mechanism of striped leaf albinism in arecanut (Areca catechu L.). Ind. Crops Prod. 2020, 154, 112734. [Google Scholar] [CrossRef]
- Pandalai, K.M.; Pillai, R.V. Albinism in Coconut Seedlings. Nature 1959, 184, 1163. [Google Scholar] [CrossRef]
- Silva, L.a.S.; Sampaio, V.F.; Barbosa, L.C.S.; Machado, M.; Flores-Borges, D.N.A.; Sales, J.F.; De Oliveira, D.C.; Mayer, J.L.S.; Kuster, V.C.; Rocha, D.I. Albinism in plants-far beyond the loss of chlorophyll: Structural and physiological aspects of wild-type and albino royal poinciana (Delonix regia) seedlings. Plant Biol. 2020, 22, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.X.; Xu, J.H.; Zhang, M.N.; Du, R.Y.; Zhao, W.X.; Zeng, Q.; Tu, Z.H.; Chen, J.H.; Chen, B.B. Third-generation sequencing and metabolome analysis reveal candidate genes and metabolites with altered levels in albino jackfruit seedlings. BMC Genom. 2021, 22, 543. [Google Scholar] [CrossRef] [PubMed]
- Xiong, B.; Ye, S.; Qiu, X.; Liao, L.; Sun, G.C.; Luo, J.Y.; Dai, L.; Rong, Y.; Wang, Z.H. Transcriptome Analyses of Two Citrus Cultivars (Shiranuhi and Huangguogan) in Seedling Etiolation. Sci. Rep. 2017, 7, 46245. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.T.; Zhang, J.K.; Zhang, Q.; Li, X.L.; Li, M.J.; Yang, Y.Z.; Zhou, J.; Wei, Q.P.; Zhou, B.B. Methylome and transcriptome analyses of three different degrees of albinism in apple seedlings. BMC Genom. 2022, 23, 310. [Google Scholar] [CrossRef] [PubMed]
- Ke, X.B.; Shen, J.J.; Niu, Y.Q.; Zhao, H.J.; Guo, Y.L.; Sun, P.Y.; Yang, T.W.; Jiang, Y.X.; Zhao, B.S.; Wang, Z.; et al. Cucumber NUCLEAR FACTOR-YC2/-YC9 target translocon component CsTIC21 in chloroplast photomorphogenesis. Plant Physiol. 2023, 192, 2822–2837. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.W.; Shao, C.X.; Liu, L.B.; Wang, Y.; An, Y.Q.; Li, H.; Ding, Y.W.; Jing, Y.P.; Li, X.J.; Xiao, J.W. PtrDJ1C, an atypical member of the DJ-1 superfamily, is essential for early chloroplast development and lignin deposition in poplar. Hortic. Plant J. 2023, 9, 1039–1054. [Google Scholar] [CrossRef]
- Wu, C.; Wu, H.T.; Wang, Q.; Wang, G.H.; Yi, X.; Chen, Y.P.; Zhou, G.X. Anticandidal Potential of Stem Bark Extract from Schima superba and the Identification of Its Major Anticandidal Compound. Molecules 2019, 24, 1587. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.W.; Xu, D.T.; Chen, F.C.; Wang, Z.P.; Luo, J.D.; He, Y.H.; Zheng, Q.M.; Liu, C.Y. Integrated Cytological, Physiologicaland Transcriptome Analyses Provide Insight into the Albino Phenotype of Chinese Plum (Prunus salicina). Int. J. Mol. Sci. 2023, 24, 14457. [Google Scholar] [CrossRef] [PubMed]
- Qian, Q.X.; Ye, Q.F.; Xu, Y.; Vasupalli, N.; Lu, H.W.; Hu, Q.T.; Hou, D. Comparative Physiology and Transcriptome Analysis Provides Insights into the Regulatory Mechanism of Albinotic Bambusa oldhamii. Plants 2023, 12, 4090. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.J.; Liu, K.Y.; Zhang, X.; Tang, Q.H.; Zeng, L. CsNYC1a Mediates Chlorophyll Degradation and Albino Trait Formation in the Arbor-Type Tea Plant Camellia nanchuanica. J. Agric. Food Chem. 2024, 72, 14087–14098. [Google Scholar] [CrossRef] [PubMed]
- Bai, Q.S.; He, B.X.; Cai, Y.L.; Lian, H.M.; Zhang, Q.; Liang, D.C.; Wang, Y.L. Genetic Diversity and Population Structure of Schima superba From Southern China. Front. Ecol. Evol. 2022, 10, 879512. [Google Scholar] [CrossRef]
- Liu, B.; Liu, Q.Q.; Daryanto, S.; Guo, S.; Huang, Z.J.; Wang, Z.N.; Wang, L.X.; Ma, X.Q. Responses of Chinese fir and Schima superba seedlings to light gradients: Implications for the restoration of mixed broadleaf-conifer forests from Chinese fir monocultures. For. Ecol. Manag. 2018, 419, 51–57. [Google Scholar] [CrossRef]
- Wang, Y.P.; Wu, G.L.; Huang, S.H.; Ma, L.Z.; Fan, H.H.; Zhang, R.; Zhou, Z.C. Analysis of genetic parameters of growth and wood traits provides insight into the genetic improvement of Schima superba. Tree Genet. Genomes 2024, 20, 8. [Google Scholar] [CrossRef]
- Wu, C.P.; Fan, J.Z.; Xu, Y.H.; Jiang, B.; Jiao, J.J.; Yao, L.J.; Wieser, G. Adaptability Analysis of the Evergreen Pioneer Tree Species Schima superba to Climate Change in Zhejiang Province. Forests 2023, 14, 2438. [Google Scholar] [CrossRef]
- Thomas, S.; Underwood, J.G.; Tseng, E.; Holloway, A.K.; Bench to Basinet CvDC Informatics Subcommittee. Long-Read Sequencing of Chicken Transcripts and Identification of New Transcript Isoforms. PLoS ONE 2014, 9, e94650. [Google Scholar] [CrossRef] [PubMed]
- Salmela, L.; Rivals, E. LoRDEC: Accurate and efficient long read error correction. Bioinformatics 2014, 30, 3506–3514. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Luo, H.T.; Bu, D.C.; Zhao, G.G.; Yu, K.T.; Zhang, C.H.; Liu, Y.N.; Chen, R.S.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef] [PubMed]
- Li, A.M.; Zhang, J.Y.; Zhou, Z.Y. PLEK: A tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC Bioinf. 2014, 15, 311. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.J.; Yang, D.C.; Kong, L.; Hou, M.; Meng, Y.Q.; Wei, L.P.; Gao, G. CPC2: A fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Res. 2017, 45, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Adachi, J.; Muraoka, Y. ANGLE: A sequencing errors resistant program for predicting protein coding regions in unfinished cDNA. J. Bioinf. Comput. Biol. 2006, 4, 649–664. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Jiao, C.; Sun, H.H.; Rosli, H.G.; Pombo, M.A.; Zhang, P.F.; Banf, M.; Dai, X.B.; Martin, G.B.; Giovannoni, J.J.; et al. ITAK: A Program for Genome-Wide Prediction and Classification of Plant Transcription Factors, Transcriptional Regulators and Protein Kinases. Mol. Plant 2016, 9, 1667–1670. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.M.; Niu, B.F.; Zhu, Z.W.; Wu, S.T.; Li, W.Z. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate Transcript Quantification from RNA-Seq Data with or Without a Reference Genome. BMC Bioinformatics 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.Z.; Mcanulla, C.; Mcwilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Gao, M.J.; Hou, R.Y.; Hu, X.Y.; Zhang, L.; Wan, X.C.; Wei, S. Determination of quality constituents in the young leaves of albinism tea cultivars. Food Chem. 2014, 155, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Yang, L.H.; Wang, X.; Cao, Z.F.; Wang, C.J.; Cheng, H.W.; Luo, B.; Wei, Z.W.; Min, X.Y. Characterization of a novel green-revertible albinism mutant in alfalfa (Medicago sativa L.). J. Plant Physiol. 2025, 306, 154452. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ma, Q.L.; Lin, L.J.; Zhang, H.F.; Luo, X.; Wang, J.; Lv, X.L.; Deng, Q.X. Integrated cytological, physiological and transcriptome analysis of the bud mutant of jujube (Ziziphus jujuba Mill.) with non-lethal albinism phenotype. Ind. Crops Prod. 2023, 201, 116964. [Google Scholar] [CrossRef]
- Bae, C.H.; Abe, T.; Matsuyama, T.; Fukunishi, N.; Nagata, N.; Nakano, T.; Kaneko, Y.; Miyoshi, K.; Matsushima, H.; Yoshida, S. Regulation of chloroplast gene expression is affected in ali, a novel tobacco albino mutant. Ann. Bot. 2001, 88, 545–553. [Google Scholar] [CrossRef]
- Spoehr, H.A. The Culture of Albino Maize. Plant Physiol. 1942, 17, 397–410. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.H.; Zhu, H.T.; Xing, Y.; Tan, J.J.; Chen, X.H.; Zhang, J.J.; Peng, H.F.; Xie, Q.J.; Zhang, Z.M. Albino Leaf 2 is involved in the splicing of chloroplast group I and II introns in rice. J. Exp. Bot. 2016, 67, 5339–5347. [Google Scholar] [CrossRef] [PubMed]
- Roberts, R.J.; Carneiro, M.O.; Schatz, M.C. The advantages of SMRT sequencing. Genome Biol. 2013, 14, 405. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.J.; Xu, D.P.; Zhuo, Z.H.; Hu, J.M.; Lu, B.Q. SMRT sequencing of the full-length transcriptome of the Rhynchophorus ferrugineus (Coleoptera: Curculionida). PeerJ 2020, 8, e9133. [Google Scholar] [CrossRef] [PubMed]
- Au, K.F.; Underwood, J.G.; Lee, L.; Wong, W.H. Improving PacBio Long Read Accuracy by Short Read Alignment. PLoS ONE 2012, 7, e46679. [Google Scholar] [CrossRef] [PubMed]
- Zhong, F.R.; Huang, L.; Qi, L.M.; Ma, Y.T.; Yan, Z.Y. Full-length transcriptome analysis of Coptis deltoidea and identification of putative genes involved in benzylisoquinoline alkaloids biosynthesis based on combined sequencing platforms. Plant Mol. Biol. 2020, 102, 477–499. [Google Scholar] [CrossRef] [PubMed]
- Zaib, P.; Ahmad, H.M.; Attacha, S.; Rahman, M.U.; Shafiq, M.R.; Parveen, K.; Fiaz, S.; Attia, K.A.; Ishaq, S.; Arif, S.; et al. Comparative genomics of light harvesting chlorophyll (LHC) gene family and impact of chlorophyll-A contents under drought stress in Helianthus annuus. J. Plant Physiol. 2023, 291, 154136. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.M.; Hu, X.J.; Zhao, Y.Z.; Song, W.B.; Zhang, M.; Chen, Z.L.; Chen, W.; Dong, Y.B.; Wang, Z.H.; Lai, J.S. Map-Based Cloning of zb7 Encoding an IPP and DMAPP Synthase in the MEP Pathway of Maize. Mol. Plant 2012, 5, 1100–1112. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, W. Leaf-variegated mutations and their responsible genes in Arabidopsis thaliana. Genes Genet. Syst. 2003, 78, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Yamatani, H.; Ito, T.; Nishimura, K.; Yamada, T.; Sakamoto, W.; Kusaba, M. Genetic analysis of chlorophyll synthesis and degradation regulated by BALANCE of CHLOROPHYLL METABOLISM. Plant Physiol. 2022, 189, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Gong, J.; Zhang, L.C.; Shen, H.; Chen, G.P.; Xie, Q.L.; Hu, Z.L. Overexpression of SlPRE5, an atypical bHLH transcription factor, affects plant morphology and chlorophyll accumulation in tomato. J. Plant Physiol. 2022, 273, 153698. [Google Scholar] [CrossRef] [PubMed]
- Frangedakis, E.; Yelina, N.E.; Billakurthi, K.; Hua, L.; Schreier, T.; Dickinson, P.J.; Tomaselli, M.; Haseloff, J.; Hibberd, J.M. MYB-related transcription factors control chloroplast biogenesis. Cell 2024, 187, 4859–4876. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.B.; Xu, X.; Hu, X.W.; Liu, Y.D.; Cao, H.H.; Chan, H.E.; Gong, Z.H.; Yuan, Y.J.; Luo, Y.Q.; Feng, B.H.; et al. SlMYB72 Regulates the Metabolism of Chlorophylls, Carotenoids and Flavonoids in Tomato Fruit. Plant Physiol. 2020, 183, 854–868. [Google Scholar] [CrossRef] [PubMed]
- Cackett, L.; Luginbuehl, L.H.; Schreier, T.B.; Lopez-Juez, E.; Hibberd, J.M. Chloroplast development in green plant tissues: The interplay between light, hormone and transcriptional regulation. New Phytol. 2021, 233, 2000–2016. [Google Scholar] [CrossRef] [PubMed]
- Ali, N.; Chen, H.; Zhang, C.; Khan, S.A.; Gandeka, M.; Xie, D.; Zhuang, W. Ectopic expression of AhGLK1b (GOLDEN2-like transcription factor) in Arabidopsis confers dual resistance to fungal and bacterial pathogens. Genes 2020, 11, 343. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Muramatsu, M.; Hakata, M.; Ueno, O.; Nagamura, Y.; Hirochika, H.; Takano, M.; Ichikawa, H. Ectopic overex- pression of the transcription factor OsGLK1 induces chloroplast development in non-green rice cells. Plant Cell Physiol. 2009, 50, 1933–1949. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, C.V.; Vrebalov, J.T.; Gapper, N.E.; Zheng, Y.; Zhong, S.; Fei, Z.; Giovannoni, J.J. Tomato GOLDEN2-LIKE transcription factors reveal molecular gradients that function during fruit development and ripening. Plant Cell 2014, 26, 585–601. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.Y.; Wu, S.; Li, X.Y.; Deng, J.H.; Liu, P.; Wang, W.Q.; Yin, X.R.; Liu, X.F. Transcriptional factors AcNAC1/2 synergistically orchestrate kiwifruit chlorophyll degradation. Postharvest Biol. Technol. 2025, 227, 113600. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | Total Reads | Total Mapped |
|---|---|---|
| CK-1 | 54,089,180 | 45,545,316 (84.20%) |
| CK-2 | 62,614,986 | 50,384,258 (80.47%) |
| CK-3 | 73,948,040 | 60,144,360 (81.33%) |
| Albinism-1 | 69,103,854 | 55,065,662 (79.69%) |
| Albinism-2 | 56,452,222 | 44,111,492 (78.14%) |
| Albinism-3 | 61,578,328 | 50,147,544 (81.44%) |
| FPKM | CK-1 | CK-2 | CK-3 | Albinism-1 | Albinism-2 | Albinism-3 |
|---|---|---|---|---|---|---|
| FPKM ≤ 1 | 6636 (22.15%) | 5832 (19.47%) | 6148 (20.53%) | 5654 (18.87%) | 4983 (16.64%) | 5858 (19.55%) |
| 1 < FPKM ≤ 15 | 15,084 (50.35%) | 15,108 (50.43%) | 14,737 (49.2%) | 14,948 (49.9%) | 15,422 (51.49%) | 14,981 (50.01%) |
| FPKM > 15 | 8237 (27.5%) | 9017 (30.09%) | 9072 (30.29%) | 9355 (31.23%) | 9552 (31.89%) | 9118 (30.43%) |
| Transcript ID | Gene ID | log2 (FC) | Expression |
|---|---|---|---|
| transcript_HQ_blade_transcript24945 | bHLH | 9.8885 | up |
| transcript_HQ_blade_transcript30262 | bHLH | 9.1724 | up |
| transcript_HQ_blade_transcript37875 | bHLH | −5.0077 | down |
| transcript_HQ_blade_transcript38898 | bHLH | 7.9627 | up |
| transcript_HQ_blade_transcript49724 | bHLH | 7.7079 | up |
| transcript_HQ_blade_transcript237 | MYB | 3.4657 | up |
| transcript_HQ_blade_transcript3381 | MYB | 5.2246 | up |
| transcript_HQ_blade_transcript14434 | MYB | 6.8876 | up |
| transcript_HQ_blade_transcript26491 | GLK | 2.9225 | up |
| transcript_HQ_blade_transcript43928 | GLK | 7.7787 | up |
| transcript_HQ_blade_transcript44162 | GLK | −2.9006 | down |
| transcript_HQ_blade_transcript21143 | NAC | −3.8109 | down |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jia, J.; Chen, M.; Feng, Y.; Yang, Z.; Yan, P. Integrated Transcriptomic Analysis Reveals Molecular Mechanisms Underlying Albinism in Schima superba Seedlings. Forests 2025, 16, 1201. https://doi.org/10.3390/f16081201
Jia J, Chen M, Feng Y, Yang Z, Yan P. Integrated Transcriptomic Analysis Reveals Molecular Mechanisms Underlying Albinism in Schima superba Seedlings. Forests. 2025; 16(8):1201. https://doi.org/10.3390/f16081201
Chicago/Turabian StyleJia, Jie, Mengdi Chen, Yuanheng Feng, Zhangqi Yang, and Peidong Yan. 2025. "Integrated Transcriptomic Analysis Reveals Molecular Mechanisms Underlying Albinism in Schima superba Seedlings" Forests 16, no. 8: 1201. https://doi.org/10.3390/f16081201
APA StyleJia, J., Chen, M., Feng, Y., Yang, Z., & Yan, P. (2025). Integrated Transcriptomic Analysis Reveals Molecular Mechanisms Underlying Albinism in Schima superba Seedlings. Forests, 16(8), 1201. https://doi.org/10.3390/f16081201