
Chromosome-Level Genome Assembly of Myricaria laxiflora and Identification of Candidate MylAP2/ERF Genes Involved in Flooding Stress

 , , ,
, , ,
Abstract
1. Introduction
2. Results
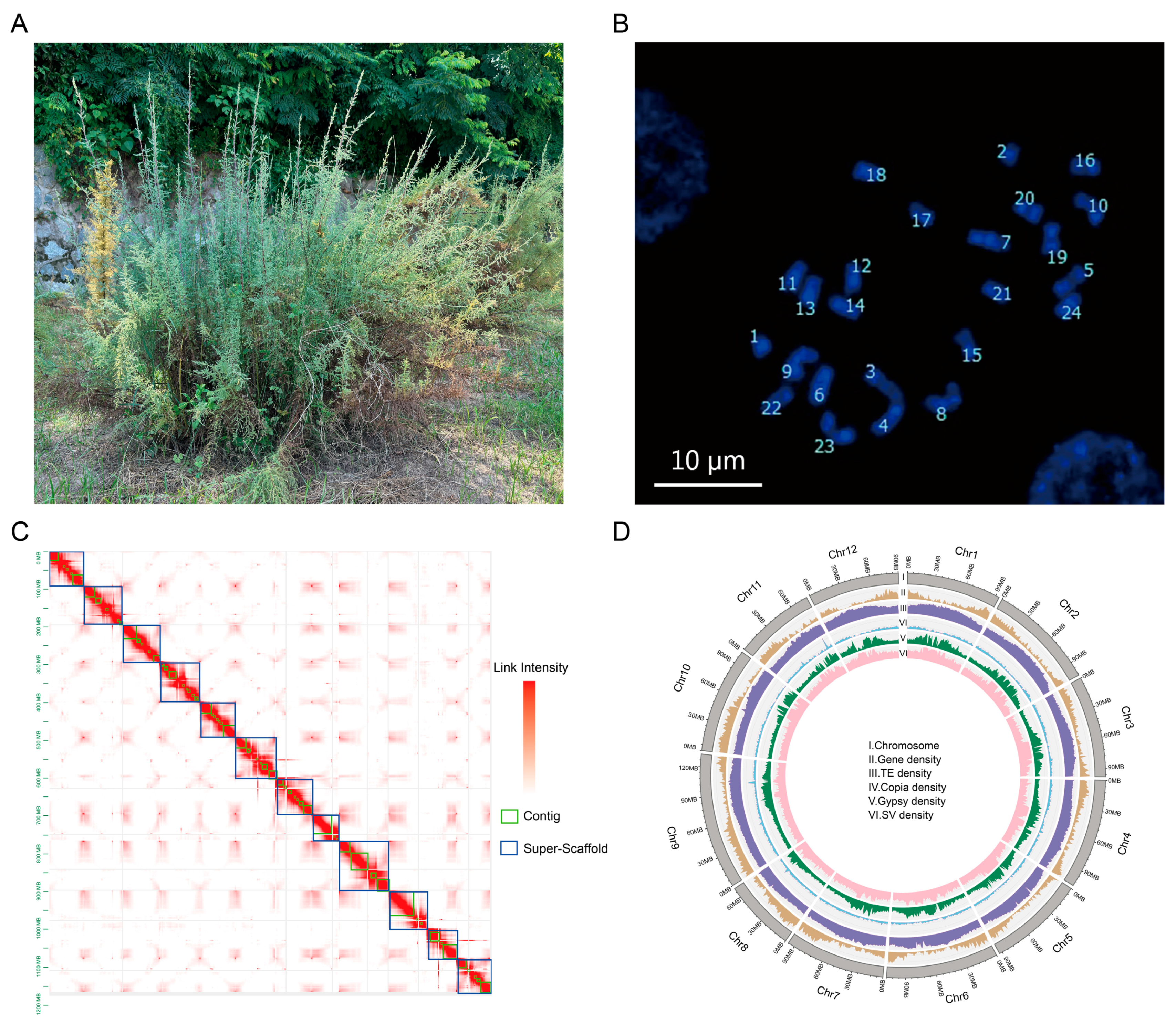
2.1. M. laxiflora Genome Survey, Sequencing, and Assembly
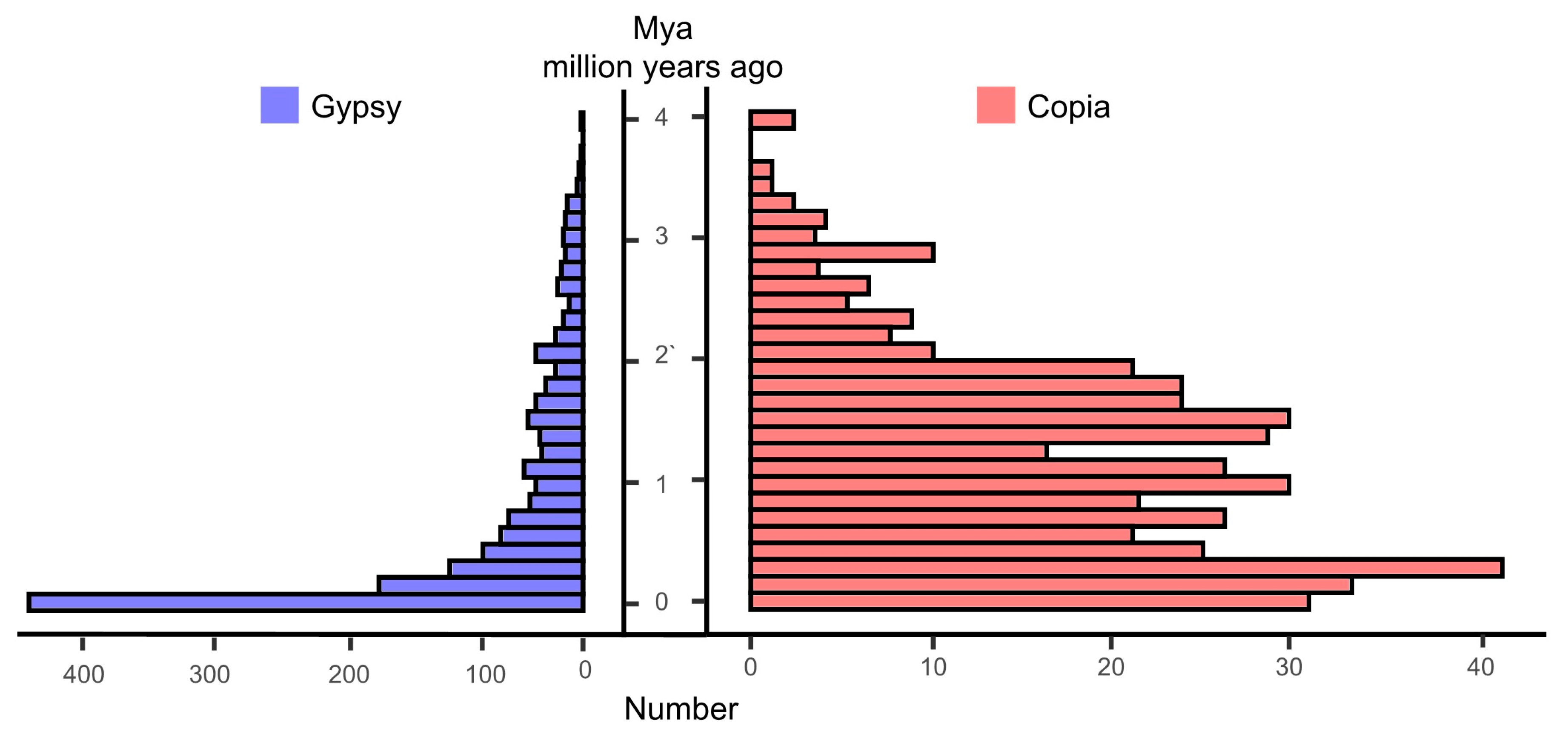
2.2. Genome Annotation
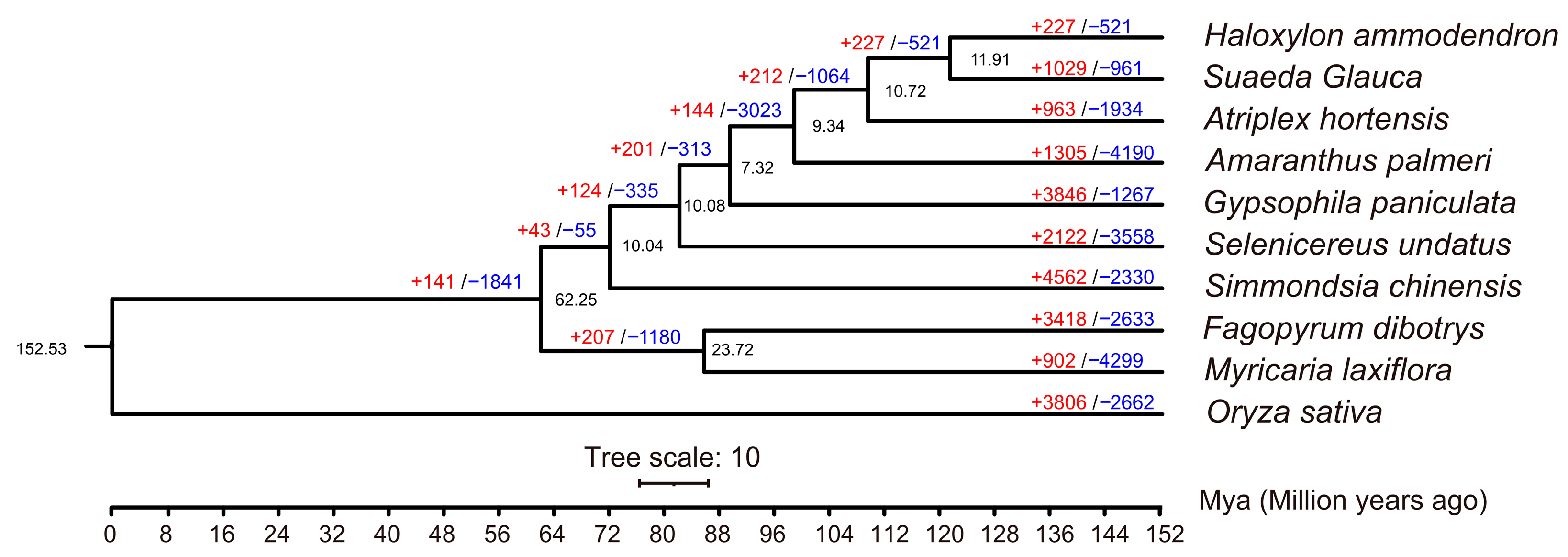
2.3. Analysis of the Expression and Contraction of Gene Families
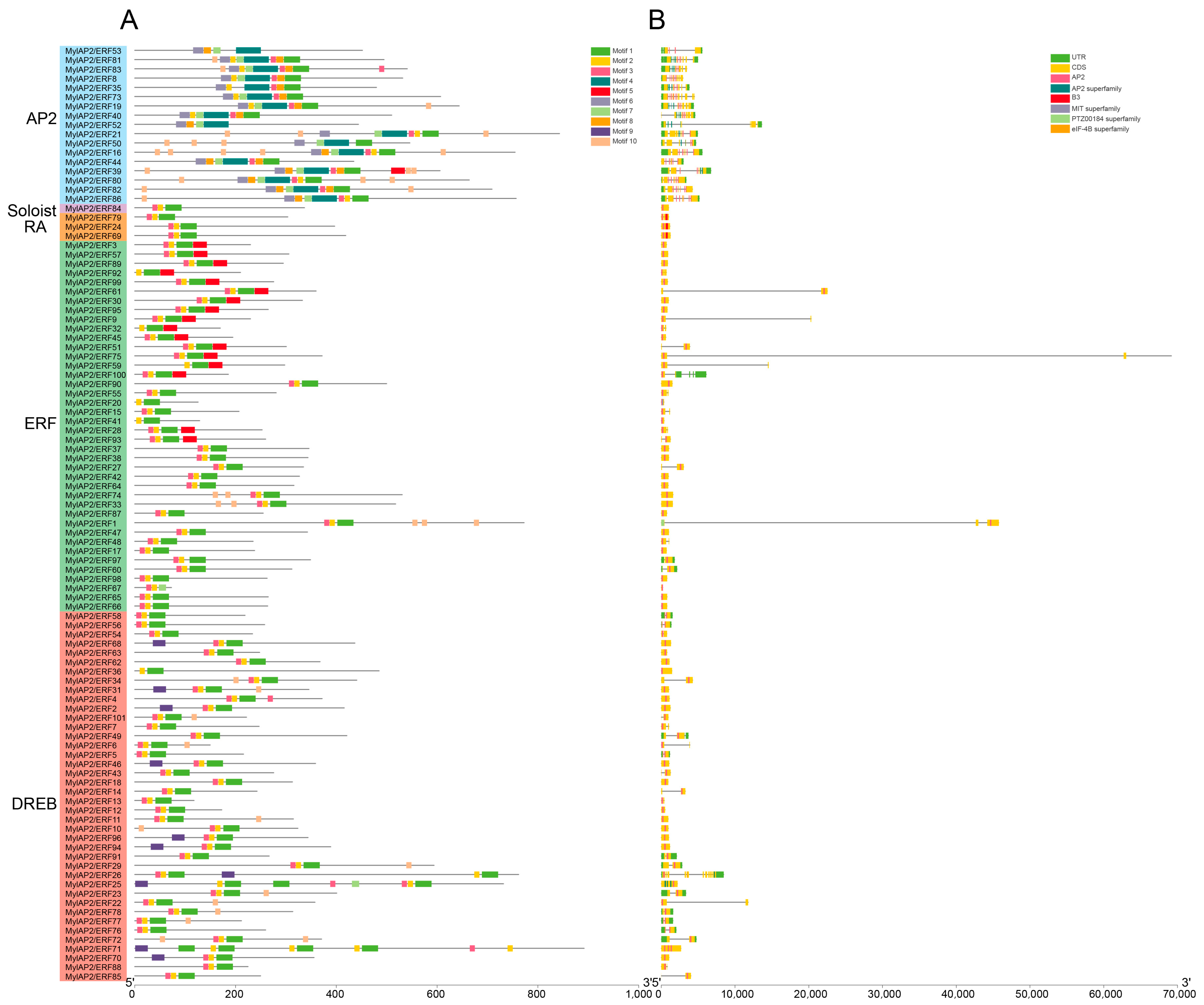
2.4. AP2/ERF Gene Families Identified in the M. laxiflora Genome
2.5. Phylogenetic and Motif Analyses of the MylAP2/ERFs
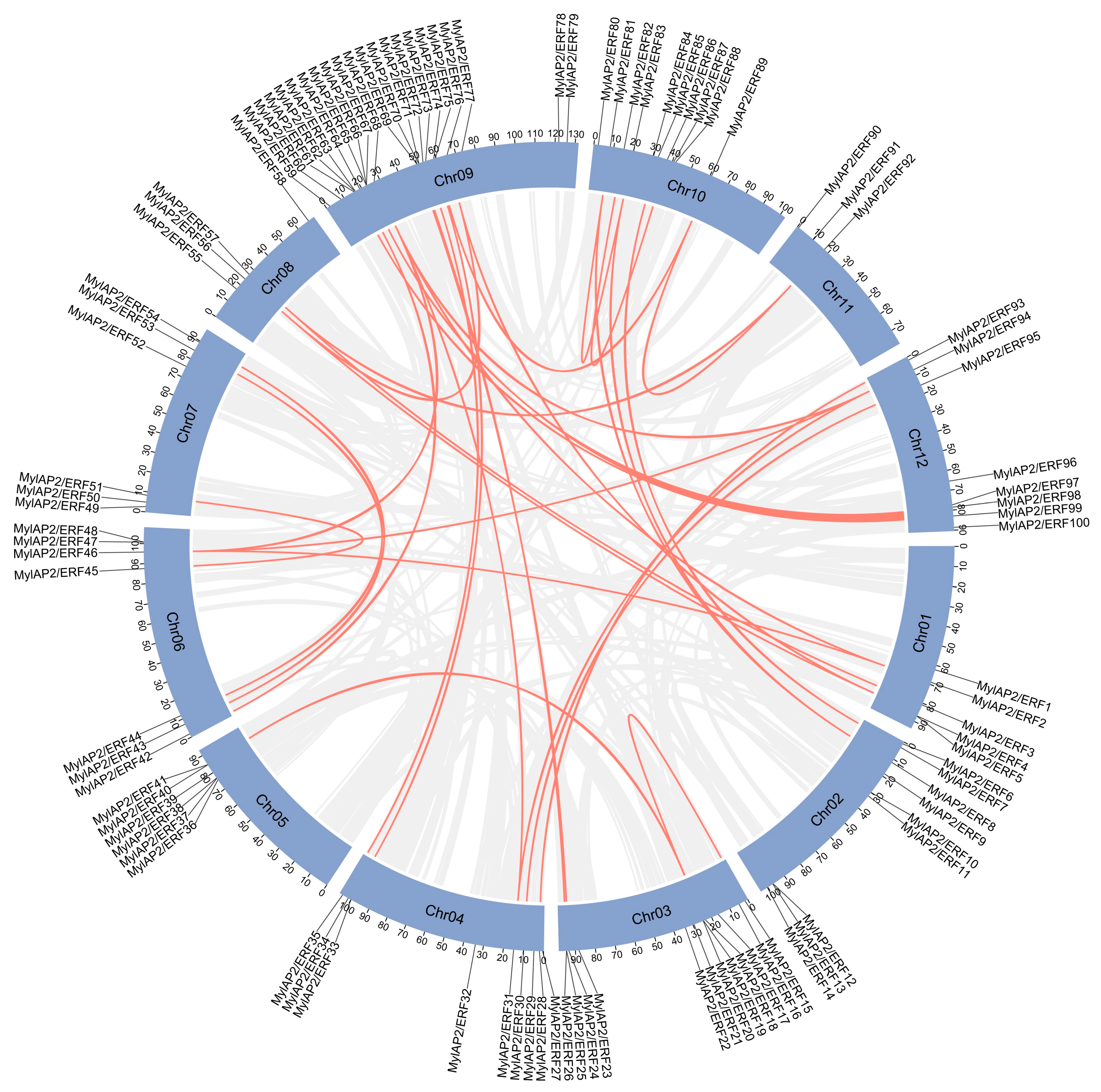
2.6. Gene Duplication Analysis of the MylAP2/ERF Genes
2.7. Analysis of Cis-Acting Elements in the Promoter Region of MylAP2/ERFs
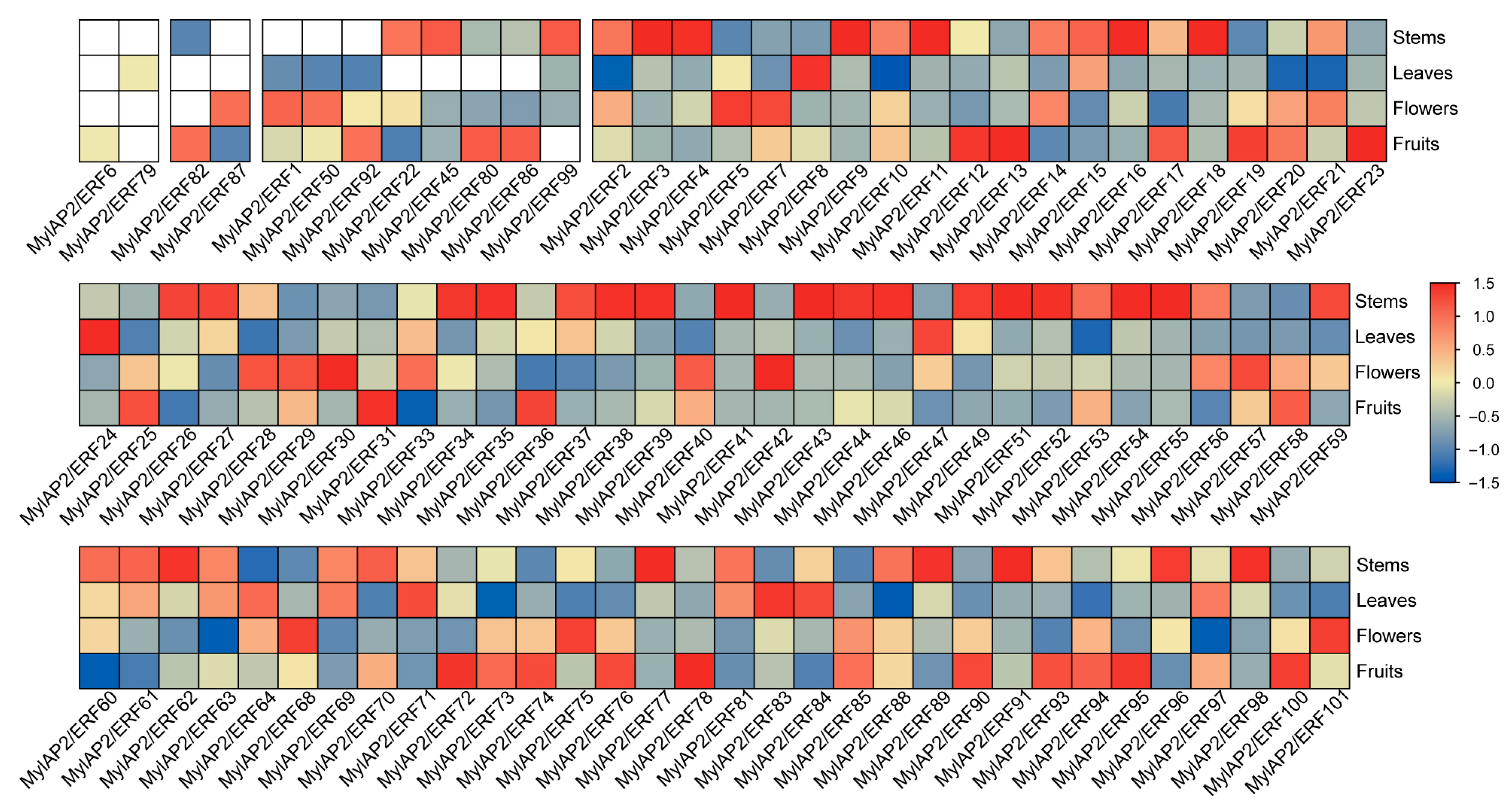
2.8. Expression Patterns of MylAP2/ERF Members in Different Tissues
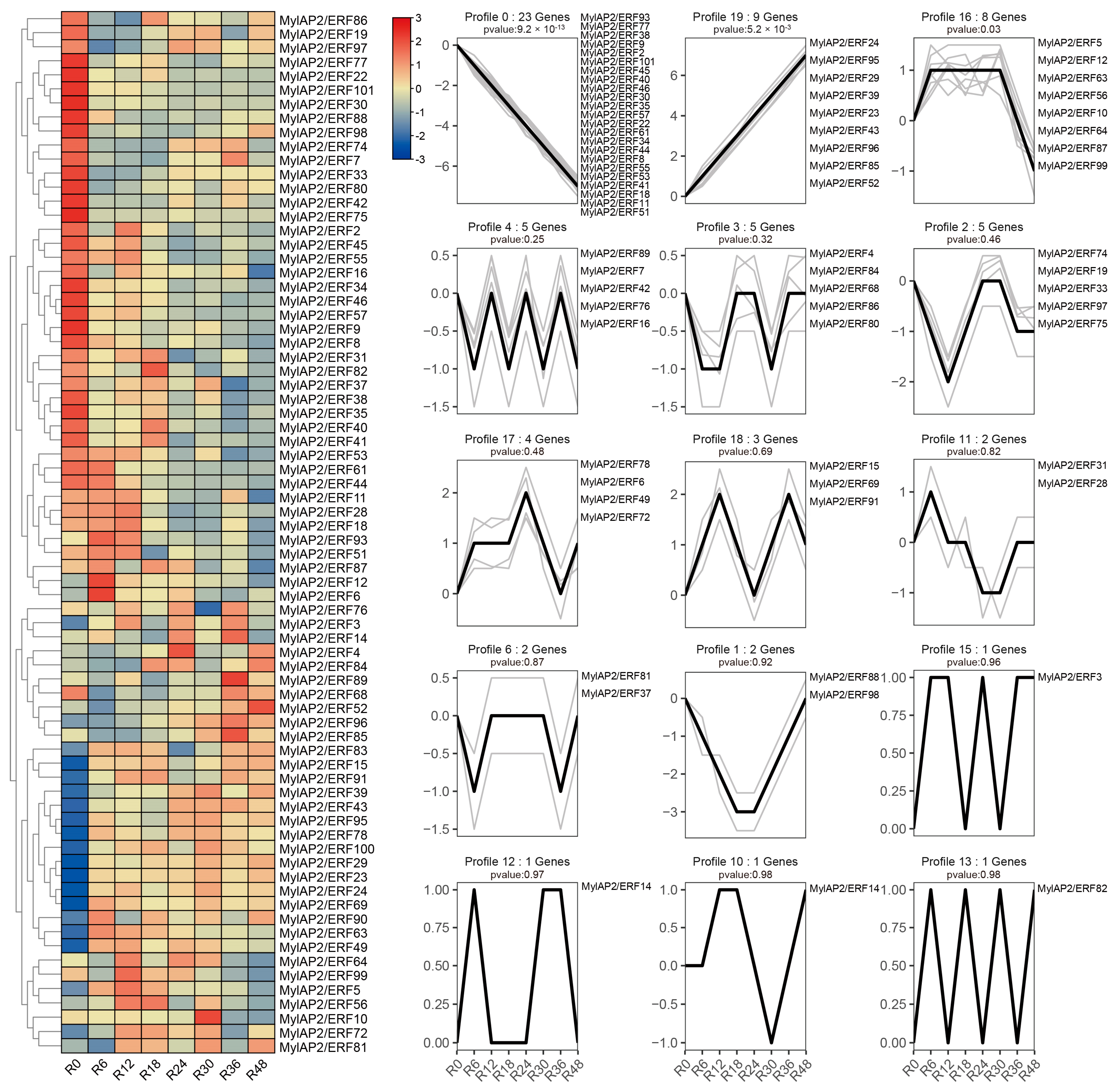
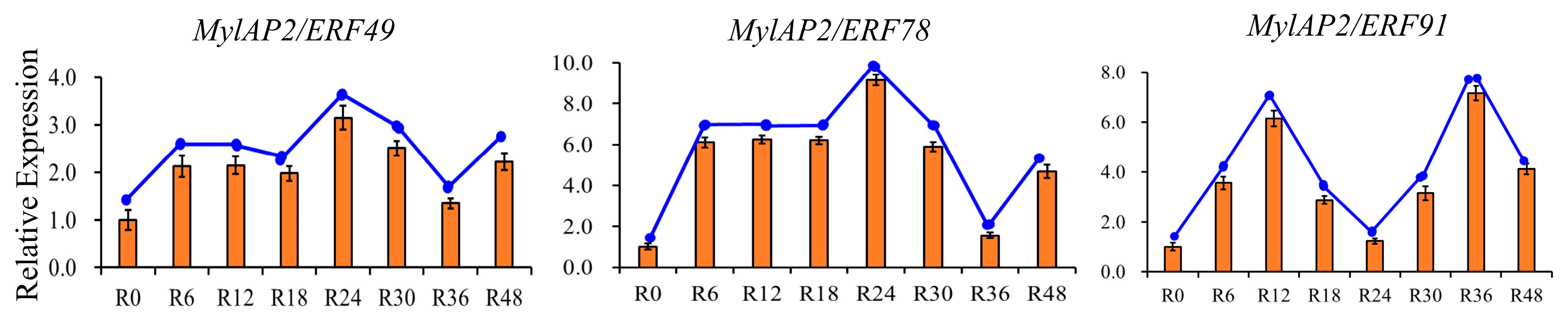
2.9. Analysis of the Expression Patterns of MylAP2/ERF Under Flooding Stress
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Sequencing and Genome Assembly
4.3. Genome Annotation and Repeat Analysis
4.4. Identification and Chromosomal Distribution of the AP2/ERF Gene Family in M. laxiflora
4.5. Phylogenetic and Motif Analyses of MylAP2/ERFs
4.6. Gene Duplication Event and Cis-Acting Element Analysis
4.7. Expression Analysis of MylAP2/ERF Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Li, L.; Wu, D.; Zhen, Q.; Zhang, J.; Qiu, L.; Huang, G.; Yang, C. Morphological structures and histochemistry of roots and shoots in Myricaria laxiflora (Tamaricaceae). Open Life Sci. 2021, 16, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Huang, G.; Xiang, W.; Zhu, H.; Zhang, H.; Zhang, J.; Ding, Z.; Liu, J.; Wu, D. Integrated Transcriptomic and Proteomic Analyses Uncover the Regulatory Mechanisms of Myricaria laxiflora Under Flooding Stress. Front. Plant Sci. 2022, 13, 924490. [Google Scholar] [CrossRef]
- Wu, J.; Zhao, Z.; Jin, Y.; Shen, Z. Investigation and study on the endemic plant Myricaria laxiflora in the Three-Gorge reservoir area. Wuhan Bot. Res. 1998, 16, 111–116. [Google Scholar]
- Wang, Y.; Wu, J.; Tao, Y.; Li, Z.; Huang, H. Natural distribution and ex situ conservation of endemic species Myricaria laxiflora in water-level-fluctuation Zone within Three-Gorges reservoir area of Changjiang River. Wuhan Bot. Res. 2003, 21, 415–422. [Google Scholar]
- Bao, D.; Lu, Z.; Jiang, M.; Xu, S.; Yao, Q.; Liu, Q.; Wang, Q. Population structure and dynamics of remanent Myricaria laxiflora downstream from the Three Gorges dam. J. Wuhan Bot. Res. 2010, 28, 711–716. [Google Scholar]
- Chen, F.Q.; Xie, Z.Q. Survival and growth responses of Myricaria laxiflora seedlings to summer flooding. Aquat. Bot. 2009, 90, 333–338. [Google Scholar] [CrossRef]
- Chen, F.; Guan, S.; Ma, Y.; Xie, Z.; Lv, K.; Huang, Y.; Jia, G. Impact of regulated water level fluctuations on the sexual reproduction of remnant Myricaria laxiflora populations. Glob. Ecol. Conserv. 2019, 18, e00628. [Google Scholar] [CrossRef]
- Guan, S.; Chen, F.; Zhou, J.; Xie, Z.; Huang, Y. Spatiotemporal photosynthetic physiology responses of remnant Myricaria laxiflora populations to regulated water level fluctuations. Conserv. Physiol. 2020, 8, coaa020. [Google Scholar] [CrossRef]
- Zhou, J.; Chen, F.; Lv, K.; Wu, Y.; Huang, Y. Summer dormancy in an endangered riparian shrub Myricaria laxiflora: Changes in branches, leaves, and nonstructural carbohydrates. Glob. Ecol. Conserv. 2021, 31, e01809. [Google Scholar] [CrossRef]
- Jia, W.; Ma, M.; Chen, J.; Wu, S. Plant Morphological, Physiological and Anatomical Adaptation to Flooding Stress and the Underlying Molecular Mechanisms. Int. J. Mol. Sci. 2021, 22, 1088. [Google Scholar] [CrossRef]
- Fukao, T.; Barrera-Figueroa, B.E.; Juntawong, P.; Pena-Castro, J.M. Submergence and waterlogging stress in plants: A review highlighting research opportunities and understudied aspects. Front. Plant Sci. 2019, 10, 340. [Google Scholar] [CrossRef]
- Bailey-Serres, J.; Voesenek, L.A.C.J. Flooding stress: Acclimations and genetic diversity. Annu. Rev. Plant Biol. 2008, 59, 313–339. [Google Scholar] [CrossRef]
- Bailey-Serres, J.; Lee, S.C.; Brinton, E. Waterproofing crops: Effective flooding survival strategies. Plant Physiol. 2012, 160, 1698–1709. [Google Scholar] [CrossRef]
- Nishiuchi, S.; Yamauchi, T.; Takahashi, H.; Kotula, L.; Nakazono, M. Mechanisms for coping with submergence and waterlogging in rice. Rice 2012, 5, 2. [Google Scholar] [CrossRef]
- Setter, T.L.; Waters, I. Review of prospects for germplasm improvement for waterlogging tolerance in wheat, barley and oats. Plant Soil. 2013, 253, 1–34. [Google Scholar] [CrossRef]
- Voesenek, L.A.C.J.; Bailey-Serres, J. Flood adaptive traits and processes: An overview. New Phytol. 2015, 206, 57–73. [Google Scholar] [CrossRef]
- Loreti, E.; van Veen, H.; Perata, P. Plant responses to flooding stress. Curr. Opin. Plant Biol. 2016, 33, 64–71. [Google Scholar] [CrossRef]
- Booker, M.A.; DeLong, A. Producing the Ethylene Signal: Regulation and Diversification of Ethylene Biosynthetic Enzymes. Plant Physiol. 2015, 169, 42–50. [Google Scholar] [CrossRef]
- Zhou, Z.; Vriezen, W.; Van Caeneghem, W.; Van Montagu, M.; Van Der Straeten, D. Rapid induction of a novel ACC synthase gene in deepwater rice seedlings upon complete submergence. Euphytica 2001, 121, 137–143. [Google Scholar] [CrossRef]
- Lee, S.C.; Mustroph, A.; Sasidharan, R.; Vashisht, D.; Pedersen, O.; Oosumi, T.; Voesenek, L.A.C.J.; Bailey-Serres, J. Molecular characterization of the submergence response of the Arabidopsis thaliana ecotype Columbia. New Phytol. 2011, 190, 457–471. [Google Scholar] [CrossRef]
- Sasidharan, R.; Voesenek, L.A.C.J. Ethylene-mediated acclimations to flooding stress. Plant Physiol. 2015, 169, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Bailey-Serres, J.; Fukao, T.; Gibbs, D.J.; Holdsworth, M.J.; Lee, S.C.; Licausi, F.; Perata, P.; Voesenek, L.A.C.J.; van Dongen, J.T. Making sense of low oxygen sensing. Trends Plant Sci. 2012, 17, 129–138. [Google Scholar] [CrossRef]
- Hattori, Y.; Nagai, K.; Furukawa, S.; Song, X.J.; Kawano, R.; Sakakibara, H.; Wu, J.; Matsumoto, T.; Yoshimura, A.; Kitano, H.; et al. The ethylene response factors SNORKEL1 and SNORKEL2 allow rice to adapt to deep water. Nature 2009, 460, 1026–1030. [Google Scholar] [CrossRef]
- Niroula, R.K.; Pucciariello, C.; Ho, V.T.; Novi, G.; Fukao, T.; Perata, P. SUB1A-dependent and -independent mechanisms are involved in the flooding tolerance of wild rice species. Plant J. 2012, 72, 282–293. [Google Scholar] [CrossRef]
- Jung, K.H.; Seo, Y.S.; Walia, H.; Cao, P.; Fukao, T.; Canlas, P.E.; Amonpant, F.; Bailey-Serres, J.; Ronald, P.C. The submergence tolerance regulator Sub1A mediates stress-responsive expression of AP2/ERF transcription factors. Plant Physiol. 2010, 152, 1674–1692. [Google Scholar] [CrossRef]
- Yang, L.; Li, N.; Liu, Y.; Miao, P.; Liu, J.; Wang, Z. Updates and Prospects: Morphological, Physiological, and Molecular Regulation in Crop Response to Waterlogging Stress. Agronomy 2023, 13, 2599. [Google Scholar] [CrossRef]
- Wei, X.; Xu, H.; Rong, W.; Ye, X.; Zhang, Z. Constitutive expression of a stabilized transcription factor group VII ethylene response factor enhances waterlogging tolerance in wheat without penalizing grain yield. Plant Cell Environ. 2019, 42, 1471–1485. [Google Scholar] [CrossRef]
- Yu, F.; Liang, K.; Fang, T.; Zhao, H.; Han, X.; Cai, M.; Qiu, F. A group VII ethylene response factor gene, ZmEREB180, coordinates waterlogging tolerance in maize seedlings. Plant Biotechnol. J. 2019, 17, 2286–2298. [Google Scholar] [CrossRef]
- Hinz, M.; Wilson, I.W.; Yang, J.; Buerstenbinder, K.; Llewellyn, D.; Dennis, E.S.; Sauter, M.; Dolferus, R. Arabidopsis RAP2.2: An ethylene response transcription factor that is important for hypoxia survival. Plant Physiol. 2010, 153, 757–772. [Google Scholar] [CrossRef]
- Licausi, F.; van Dongen, J.T.; Giuntoli, B.; Novi, G.; Santaniello, A.; Geigenberger, P.; Perata, P. HRE1 and HRE2, two hypoxia-inducible ethylene response factors, affect anaerobic responses in Arabidopsis thaliana. Plant J. 2010, 62, 302–315. [Google Scholar] [CrossRef]
- Hartman, S.; Liu, Z.; Van Veen, H.; Vicente, J.; Reinen, E.; Martopawiro, S.; Zhang, H.; Van Dongen, N.; Bosman, F.; Bassel, G.W.; et al. Ethylene-mediated nitric oxide depletion preadapts plants to hypoxia stress. Nat. Commun. 2019, 10, 4020. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, D.J.; Tedds, H.M.; Labandera, A.M.; Bailey, M.; White, M.D.; Hartman, S.; Sprigg, C.; Mogg, S.L.; Osborne, R.; Dambire, C.; et al. Oxygen-dependent proteolysis regulates the stability of angiosperm polycomb repressive complex 2 subunit VERNALIZATION 2. Nat. Commun. 2018, 9, 5438. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, D.J.; Md Isa, N.; Movahedi, M.; Lozano-Juste, J.; Mendiondo, G.M.; Berckhan, S.; Marin-De La Rosa, N.; Vicente Conde, J.; Sousa Correia, C.; Pearce, S.P.; et al. Nitric oxide sensing in plants is mediated by proteolytic control of group VII ERF transcription factors. Mol. Cell 2014, 53, 369–379. [Google Scholar] [CrossRef]
- Yan, Z.; Sang, L.; Ma, Y.; He, Y.; Sun, J.; Ma, L.; Li, S.; Miao, F.; Zhang, Z.; Huang, J.; et al. A de novo assembled high-quality chromosome-scale Trifolium pratense genome and fine-scale phylogenetic analysis. BMC Plant Biol. 2022, 22, 332. [Google Scholar] [CrossRef]
- Whibley, A.; Kelley, J.L.; Narum, S.R. The changing face of genome assemblies: Guidance on achieving high-quality reference genomes. Mol. Ecol. Resour. 2021, 21, 641–652. [Google Scholar] [CrossRef]
- Marks, R.A.; Hotaling, S.; Frandsen, P.B.; VanBuren, R. Representation and participation across 20 years of plant genome sequencing. Nat. Plants 2021, 7, 1571–1578. [Google Scholar] [CrossRef]
- Ma, Z.; Hu, L.; Jiang, W. Understanding AP2/ERF Transcription Factor Responses and Tolerance to Various Abiotic Stresses in Plants: A Comprehensive Review. Int. J. Mol. Sci. 2024, 25, 893. [Google Scholar] [CrossRef]
- Hu, F.; Zhang, Y.; Guo, J. Identification and Characterization of AP2/ERF Transcription Factors in Yellow Horn. Int. J. Mol. Sci. 2022, 23, 14991. [Google Scholar] [CrossRef]
- Nakano, T.; Suzuki, K.; Fujimura, T.; Shinshi, H. Genome-wide analysis of the ERF gene family in Arabidopsis and rice. Plant Physiol. 2006, 140, 411–432. [Google Scholar] [CrossRef]
- Zhang, J.; Liao, J.; Ling, Q.; Xi, Y.; Qian, Y. Genome-wide identification and expression profiling analysis of maize AP2/ERF superfamily genes reveal essential roles in abiotic stress tolerance. BMC Genom. 2022, 23, 125. [Google Scholar] [CrossRef]
- Liu, X.; Zhou, G.; Chen, S.; Jia, Z.; Zhang, S.; Ren, M.; He, F. Genome-wide analysis of the AP2/ERF gene family in Tritipyrum and the response of TtERF_B2-50 in salt-tolerance. BMC Genom. 2023, 24, 541. [Google Scholar] [CrossRef]
- Lynch, M.; Conery, J.S. The evolutionary fate and consequences of duplicate genes. Science 2000, 290, 1151–1155. [Google Scholar] [CrossRef]
- Cannon, S.B.; Mitra, A.; Baumgarten, A.; Young, N.D.; May, G. The roles of segmental and tandem gene duplication in the evolution of large gene families in Arabidopsis thaliana. BMC Plant Biol. 2004, 4, 10. [Google Scholar] [CrossRef]
- Lieberman-Aiden, E.; van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. Gigascience 2012, 1, 18. [Google Scholar] [CrossRef]
- Marçais, G.; Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.S.; Peluso, P.; Sedlazeck, F.J.; Nattestad, M.; Concepcion, G.T.; Clum, A.; Dunn, C.; O’Malley, R.; Figueroa-Balderas, R.; Morales-Cruz, A.; et al. Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods 2016, 13, 1050. [Google Scholar] [CrossRef]
- Chaisson, M.J.; Tesler, G. Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): Application and theory. BMC Bioinform. 2012, 13, 238. [Google Scholar] [CrossRef]
- Servant, N.; Varoquaux, N.; Lajoie, B.R.; Viara, E.; Chen, C.J.; Vert, J.P.; Heard, E.; Dekker, J.; Barillot, E. HiC-Pro: An optimized and flexible pipeline for Hi-C data processing. Genome Biol. 2015, 16, 259. [Google Scholar] [CrossRef]
- Durand, N.C.; Robinson, J.T.; Shamim, M.S.; Machol, I.; Mesirov, J.P.; Lander, E.S.; Aiden, E.L. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 2016, 3, 99–101. [Google Scholar] [CrossRef]
- Durand, N.C.; Shamim, M.S.; Machol, I.; Rao, S.S.; Huntley, M.H.; Lander, E.S.; Aiden, E.L. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 2016, 3, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Stanke, M.; Steinkamp, R.; Waack, S.; Morgenstern, B. AUGUSTUS: A web server for gene finding in eukaryotes. Nucleic Acids Res. 2004, 32, W309–W312. [Google Scholar] [CrossRef] [PubMed]
- Salamov, A.A.; Solovyev, V.V. Ab initio gene finding in drosophila genomic DNA. Genome Res. 2000, 10, 516–522. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290. [Google Scholar] [CrossRef]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, H.; Debarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Section | M. laxiflora |
|---|---|
| Size (bp) | 1,291,671,364 |
| Contig N50 (Mb) | 29.52 |
| Contig L50 | 39 |
| Contig N90 (Mb) | 0.96 |
| Contig L90 | 190 |
| Chromosome number | 12 |
| Scaffold number | 618 |
| Busco | 93.00% |
| Repeat percentage | 76.16% |
| Section | Results |
|---|---|
| Genome size (bp) | 1,291,671,364 |
| Repeat sequence (bp) | 996,660,267 |
| Number of genes | 23,666 |
| Gene average length (CDS) | 1311 |
| Gene average length (DNA) | 6996 |
| Exon number per gene | 5.79 |
| Exon average length (bp) | 378 |
| Genome GC content (%) | 34.32% |
| Element | Number of Elements | Length Occupied (bp) | Percentage |
|---|---|---|---|
| Retroelements | 399,663 | 383,047,690 | 29.66% |
| SINEs | 355 | 2,029,456 | 0.16% |
| Penelope | 0 | 0 | 0.00% |
| LINEs | 30,131 | 18,800,839 | 1.46% |
| CRE/SLACS | 630 | 140,222 | 0.01% |
| L2/CR1/Rex | 276 | 116,525 | 0.01% |
| R1/LOA/Jockey | 0 | 0 | 0.00% |
| R2/R4/NeSL | 996 | 600,492 | 0.05% |
| RTE/Bov-B | 1846 | 359,132 | 0.03% |
| L1/CIN4 | 26,383 | 17,584,468 | 1.36% |
| LTR elements | 369,177 | 362,217,395 | 28.04% |
| BEL/Pao | 798 | 234,561 | 0.02% |
| Ty1/Copia | 79,472 | 58,072,541 | 4.50% |
| Ty3-retrotransposons/DIRS1 | 267,560 | 281,010,280 | 21.76% |
| Retroviral | 0 | 0 | 0.00% |
| DNA transposons | 161,684 | 92,770,623 | 7.18% |
| obo-Activator | 78,533 | 38,677,231 | 2.99% |
| Tc1-IS630-Pogo | 484 | 70,890 | 0.01% |
| En-Spm | 0 | 0 | 0.00% |
| MuDR-IS905 | 0 | 0 | 0.00% |
| PiggyBac | 0 | 0 | 0.00% |
| Tourist/Harbinger | 7997 | 3,859,662 | 0.30% |
| Other (Mirage, P-element, Transib) | 0 | 0 | 0.00% |
| Rolling circles | 6129 | 5,289,121 | 0.41% |
| Unclassified | 820,451 | 357,710,559 | 27.69% |
| Total interspersed repeats | 833,528,872 | 64.53% | |
| Small RNA | 4845 | 10,899,526 | 0.84% |
| Satellites: | 4113 | 14,453,354 | 1.12% |
| Simple repeats | 222,441 | 119,244,549 | 9.23% |
| Low complexity | 33,662 | 13,244,845 | 1.03% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiang, W.; Li, L.; Li, G.; Huang, G.; Dun, B.; Wang, J.; Chen, H.; Ma, X.; Zhang, H.; Xiao, Z.; et al. Chromosome-Level Genome Assembly of Myricaria laxiflora and Identification of Candidate MylAP2/ERF Genes Involved in Flooding Stress. Forests 2025, 16, 295. https://doi.org/10.3390/f16020295
Xiang W, Li L, Li G, Huang G, Dun B, Wang J, Chen H, Ma X, Zhang H, Xiao Z, et al. Chromosome-Level Genome Assembly of Myricaria laxiflora and Identification of Candidate MylAP2/ERF Genes Involved in Flooding Stress. Forests. 2025; 16(2):295. https://doi.org/10.3390/f16020295
Chicago/Turabian StyleXiang, Weibo, Linbao Li, Guoli Li, Guiyun Huang, Bicheng Dun, Junchen Wang, Huiyuan Chen, Xiaobo Ma, Haibo Zhang, Zhiqiang Xiao, and et al. 2025. "Chromosome-Level Genome Assembly of Myricaria laxiflora and Identification of Candidate MylAP2/ERF Genes Involved in Flooding Stress" Forests 16, no. 2: 295. https://doi.org/10.3390/f16020295
APA StyleXiang, W., Li, L., Li, G., Huang, G., Dun, B., Wang, J., Chen, H., Ma, X., Zhang, H., Xiao, Z., Liu, J., Yang, Z., & Wu, D. (2025). Chromosome-Level Genome Assembly of Myricaria laxiflora and Identification of Candidate MylAP2/ERF Genes Involved in Flooding Stress. Forests, 16(2), 295. https://doi.org/10.3390/f16020295