
Transcriptome Analysis and Novel EST-SSR Marker Development for Pinus tabuliformis Seedlings from Four Provenances

Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. RNA and DNA Extraction
2.3. Transcriptome Sequencing, Assembly, and Gene Annotation
2.4. Indentification of Simple Sequence Repeats
2.5. Genotyping and Genetic Diversity Analysis
2.6. Statistical Analysis
3. Results
3.1. Transcriptome Analysis
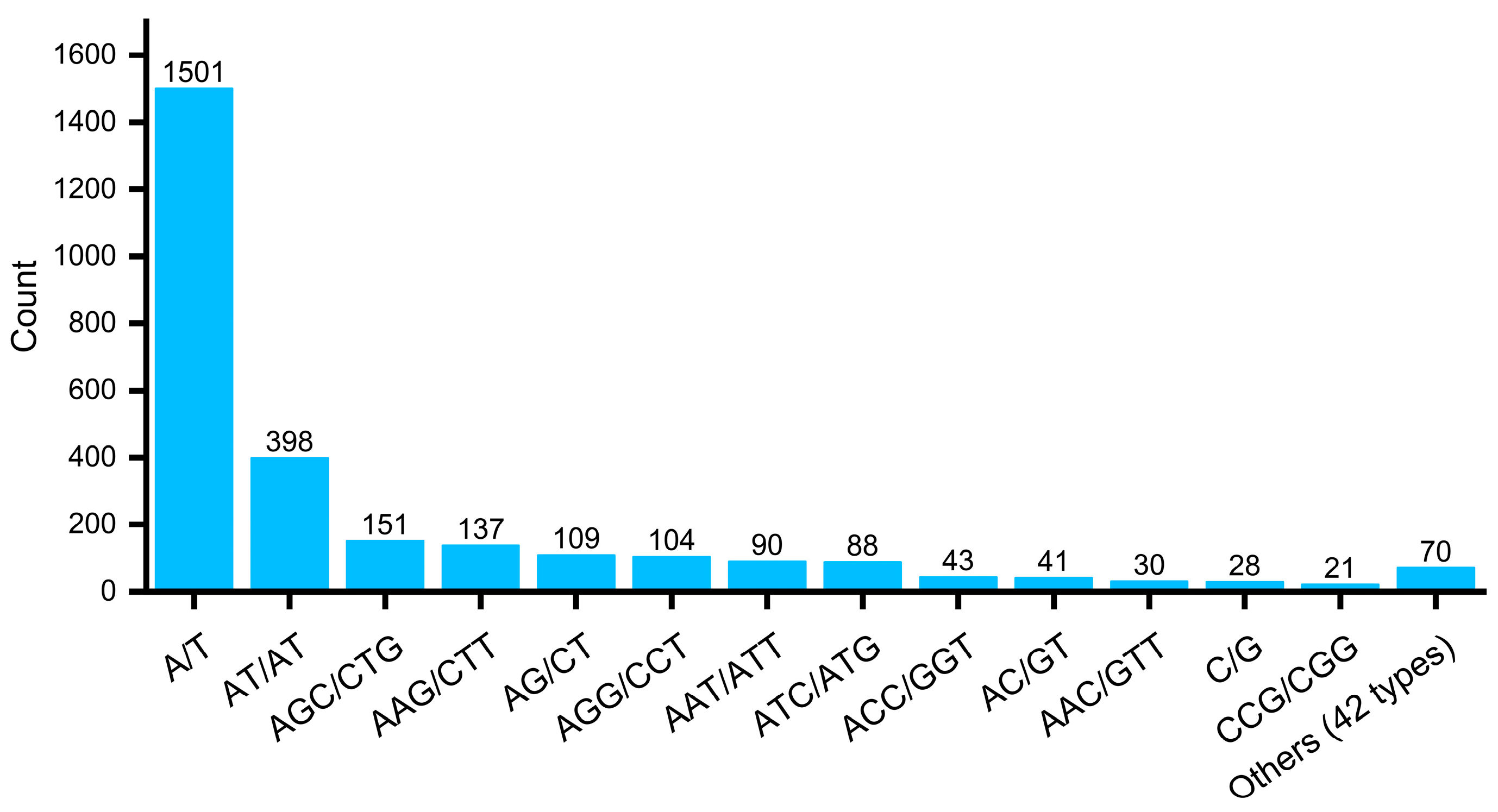
3.2. Identification and Analysis of EST-SSR Loci
3.3. Screening and Validation of Polymorphic Primers
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, K.; Abbott, R.J.; Milne, R.I.; Tian, X.M.; Liu, J. Phylogeography of Pinus tabulaeformis Carr. (Pinaceae), a dominant species of coniferous forest in northern China. Mol. Ecol. 2008, 17, 4276–4288. [Google Scholar] [CrossRef]
- Li, W.; Liu, S.W.; Ma, J.J.; Liu, H.M.; Han, F.X.; Li, Y.; Niu, S.H. Gibberellin signaling is required for far-red light-induced shoot elongation in Pinus tabuliformis seedlings. Plant Physiol. 2020, 182, 658–668. [Google Scholar] [CrossRef]
- Zhang, J.X.; Liu, H.M.; Yang, B.N.; Wang, H.L.; Niu, S.H.; El-Kassaby, Y.A.; Li, W. Phytohormone profiles and related gene expressions after endodormancy release in developing Pinus tabuliformis male strobili. Plant Sci. 2022, 316, 111167. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Niu, S.; El-Kassaby, Y.A.; Li, W. Transcriptomic and proteomic analyses of far-red light effects in inducing shoot elongation in the presence or absence of paclobutrazol in Chinese pine. J. For. Res. 2021, 33, 1033–1043. [Google Scholar] [CrossRef]
- Pervaiz, T.; Liu, S.W.; Uddin, S.; Amjid, M.W.; Niu, S.H.; Wu, H.X. The Transcriptional landscape and hub genes associated with physiological responses to drought stress in Pinus tabuliformis. Int. J. Mol. Sci. 2021, 22, 9604. [Google Scholar] [CrossRef]
- Niu, S.H.; Li, Z.X.; Yuan, H.W.; Chen, X.Y.; Li, Y.; Li, W. Transcriptome characterisation of Pinus tabuliformis and evolution of genes in the Pinus phylogeny. BMC Genom. 2013, 14, 263. [Google Scholar] [CrossRef]
- Balakrishnan, S.; Dev, S.A.; Sakthi, A.R.; Vikashini, B.; Bhasker, T.R.; Magesh, N.S.; Ramasamy, Y. Gene-ecological zonation and population genetic structure of Tectona grandis L.f. in India revealed by genome-wide SSR markers. Tree Genet. Genomes 2021, 17, 33. [Google Scholar] [CrossRef]
- Zhou, Y.; Wei, X.; Abbas, F.; Yu, Y.; Yu, R.; Fan, Y. Genome-wide identification of simple sequence repeats and assessment of genetic diversity in Hedychium. J. Appl. Res. Med. Aromat. Plants 2021, 24, 100312. [Google Scholar] [CrossRef]
- Zhang, G.; Sun, Z.; Zhou, D.; Xiong, M.; Wang, X.; Yang, J.; Wei, Z. Development and characterization of novel EST-SSRs from Larix gmelinii and their cross-species transferability. Molecules 2015, 20, 12469–12480. [Google Scholar] [CrossRef]
- Grover, A.; Sharma, P.C. Development and use of molecular markers: Past and present. Crit. Rev. Biotechnol. 2016, 36, 290–302. [Google Scholar] [CrossRef]
- Zhao, D.W.; Yang, J.B.; Yang, S.X.; Kato, K.; Luo, J.P. Genetic diversity and domestication origin of tea plant Camellia taliensis (Theaceae) as revealed by microsatellite markers. BMC Plant Biol. 2014, 14, 14. [Google Scholar] [CrossRef] [PubMed]
- Stephen, K.; Aparna, K.; Beena, R.; Sah, R.P.; Jha, U.C.; Behera, S. Identification of simple sequence repeat markers linked to heat tolerance in rice using bulked segregant analysis in F(2) population of NERICA-L 44 x Uma. Front. Plant Sci. 2023, 14, 1113838. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Shu, G.; Hu, Y.; Cao, G.; Wang, Y. Pattern and variation in simple sequence repeat (SSR) at different genomic regions and its implications to maize evolution and breeding. BMC Genom. 2023, 24, 136. [Google Scholar] [CrossRef] [PubMed]
- Ellis, J.R.; Burke, J.M. EST-SSRs as a resource for population genetic analyses. Heredity 2007, 99, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Liu, L.; Wang, L.; Wang, S.; Somta, P.; Cheng, X. Development and validation of EST-SSR markers from the transcriptome of adzuki bean (Vigna angularis). PLoS ONE 2015, 10, e0131939. [Google Scholar] [CrossRef]
- Zheng, J.Y.; Wang, H.; Chen, X.X.; Wang, P.; Gao, P.; Li, X.N.; Zhu, G.P. Microsatellite markers for assessing genetic diversity of the medicinal plant Paris. polyphylla var. chinensis (Trilliaceae). Genet. Mol. Res. 2012, 11, 1975–1980. [Google Scholar] [CrossRef]
- Koohi Dehkordi, M.; Beigzadeh, T.; Sorkheh, K. Novel in silico EST-SSR markers and bioinformatic approaches to detect genetic variation among peach (Prunus persica L.) germplasm. J. For. Res. 2019, 31, 1359–1370. [Google Scholar] [CrossRef]
- Li, S.; Ji, F.; Hou, F.; Cui, H.; Shi, Q.; Xing, G.; Weng, Y.; Kang, X. Characterization of Hemerocallis citrina transcriptome and development of EST-SSR markers for evaluation of genetic diversity and population structure of Hemerocallis collection. Front. Plant Sci. 2020, 11, 686. [Google Scholar] [CrossRef]
- Zhou, Y.; Yin, M.; Abbas, F.; Sun, Y.; Gao, T.; Yan, F.; Li, X.; Yu, Y.; Yue, Y.; Yu, R.; et al. Classification and association analysis of gerbera (Gerbera hybrida) flower color traits. Front. Plant Sci. 2021, 12, 779288. [Google Scholar] [CrossRef]
- Fang, P.; Niu, S.; Yuan, H.; Li, Z.; Zhang, Y.; Yuan, L.; Li, W. Development and characterization of 25 EST-SSR markers in Pinus sylvestris var. mongolica (Pinaceae). Appl. Plant Sci. 2014, 2, 1300057. [Google Scholar] [CrossRef]
- Yang, B.; Niu, S.; El-Kassaby, Y.A.; Li, W. Monitoring genetic diversity across Pinus tabuliformis seed orchard generations using SSR markers. Can. J. For. Res. 2021, 51, 1534–1540. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, Y.; Shi, C.; Huang, Z.; Zhang, Y.; Li, S.; Li, Y.; Ye, J.; Yu, C.; Li, Z.; et al. SOAPnuke: A MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience 2018, 7, 1–6. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Pertea, G.; Huang, X.; Liang, F.; Antonescu, V.; Sultana, R.; Karamycheva, S.; Lee, Y.; White, J.; Cheung, F.; Parvizi, B.; et al. TIGR Gene Indices clustering tools (TGICL): A software system for fast clustering of large EST datasets. Bioinformatics 2003, 19, 651–652. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Audic, S.; Claverie, J.M. The significance of digital gene expression profiles. Genome Res. 1997, 7, 986–995. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Koressaar, T.; Remm, M. Enhancements and modifications of primer design program Primer3. Bioinformatics 2007, 23, 1289–1291. [Google Scholar] [CrossRef]
- Holland, M.M.; Parson, W. GeneMarker(R) HID: A reliable software tool for the analysis of forensic STR data. J. Forensic. Sci. 2011, 56, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Muse, S.V. PowerMarker: An integrated analysis environment for genetic marker analysis. Bioinformatics 2005, 21, 2128–2129. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Blazquez, M.A.; Nelson, D.C.; Weijers, D. Evolution of plant hormone response pathways. Annu. Rev. Plant Biol. 2020, 71, 327–353. [Google Scholar] [CrossRef]
- Qi, J.; Wang, Y.; Yu, T.; Cunha, A.; Wu, B.; Vernoux, T.; Meyerowitz, E.; Jiao, Y. Auxin depletion from leaf primordia contributes to organ patterning. Proc. Natl. Acad. Sci. USA 2014, 111, 18769–18774. [Google Scholar] [CrossRef]
- Dai, X.; Liu, Z.; Qiao, M.; Li, J.; Li, S.; Xiang, F. ARR12 promotes de novo shoot regeneration in Arabidopsis thaliana via activation of WUSCHEL expression. J. Integr. Plant Biol. 2017, 59, 747–758. [Google Scholar] [CrossRef]
- Wang, J.; Tian, C.; Zhang, C.; Shi, B.; Cao, X.; Zhang, T.Q.; Zhao, Z.; Wang, J.W.; Jiao, Y. Cytokinin signaling activates WUSCHEL expression during axillary meristem Initiation. Plant Cell 2017, 29, 1373–1387. [Google Scholar] [CrossRef]
- Sun, T.P. The molecular mechanism and evolution of the GA-GID1-DELLA signaling module in plants. Curr. Biol. 2011, 21, R338–R345. [Google Scholar] [CrossRef]
- Wang, Y.; Deng, D. Molecular basis and evolutionary pattern of GA-GID1-DELLA regulatory module. Mol. Genet. Genom. 2014, 289, 1–9. [Google Scholar] [CrossRef]
- Ma, Z.; Hu, X.; Cai, W.; Huang, W.; Zhou, X.; Luo, Q.; Yang, H.; Wang, J.; Huang, J. Arabidopsis miR 171-targeted scarecrow-like proteins bind to GT cis-elements and mediate gibberellin-regulated chlorophyll biosynthesis under light conditions. PLoS Genet. 2014, 10, e1004519. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.; Wang, Z.; He, Q.; Zhao, J.; Fan, Z.; Zhang, J. Development of EST-SSR markers in Larix principis-rupprechtii Mayr and evaluation of their polymorphism and cross-species amplification. Trees 2018, 32, 1559–1571. [Google Scholar] [CrossRef]
- Dutta, S.; Kumawat, G.; Singh, B.P.; Gupta, D.K.; Singh, S.; Dogra, V.; Gaikwad, K.; Sharma, T.R.; Raje, R.S.; Bandhopadhya, T.K.; et al. Development of genic-SSR markers by deep transcriptome sequencing in pigeonpea [Cajanus cajan (L.) Millspaugh]. BMC Plant Biol. 2011, 11, 17. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, X.; Wang, Y.H.; Shen, S.K. De Novo Assembly of Transcriptome and development of novel EST-SSR markers in Rhododendron rex Levl. through Illumina sequencing. Front. Plant Sci. 2017, 8, 1664. [Google Scholar] [CrossRef]
- Li, X.; Liu, X.; Wei, J.; Li, Y.; Tigabu, M.; Zhao, X. Development and transferability of EST-SSR markers for Pinus koraiensis from cold-stressed transcriptome through Illumina sequencing. Genes 2020, 11, 500. [Google Scholar] [CrossRef]
- Raza, Q.; Riaz, A.; Saher, H.; Bibi, A.; Raza, M.A.; Ali, S.S.; Sabar, M. Grain Fe and Zn contents linked SSR markers based genetic diversity in rice. PLoS ONE 2020, 15, e0239739. [Google Scholar] [CrossRef]
- Chen, S.; Dong, M.; Zhang, Y.; Qi, S.; Liu, X.; Zhang, J.; Zhao, J. Development and characterization of simple sequence repeat markers for, and genetic diversity analysis of Liquidambar formosana. Forests 2020, 11, 203. [Google Scholar] [CrossRef]
- Biao, Z.; Zhang, Z.; Li, Y.; Ma, Y.; Zhang, S.; Niu, S.; Li, Y. Genetic diversity, genetic structure, and germplasm source of Chinese pine in North China. Eur. J. For. Res. 2023, 142, 183–195. [Google Scholar] [CrossRef]
- Botstein, D.; White, R.L.; Skolnick, M.; Davis, R.W. Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am. J. Hum. Genet. 1980, 32, 314–331. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SSR Motif | Number of Motifs | Number of Repeats | Total | Percentage | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | ≥15 | ||||
| Mononucleotides | 2 | - | - | - | - | - | 707 | 290 | 153 | 102 | 69 | 208 | 1529 | 54.39% |
| Dinucleotides | 4 | - | 208 | 105 | 67 | 41 | 49 | 26 | 20 | 18 | 8 | 7 | 549 | 19.53% |
| Trinucleotides | 10 | 443 | 149 | 46 | 21 | 6 | 3 | 5 | 1 | 0 | 0 | 1 | 675 | 24.01% |
| Tetranucleotide | 14 | 21 | 5 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 27 | 0.96% |
| Pentanucleotide | 9 | 12 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 13 | 0.46% |
| Hexanucleotide | 16 | 12 | 6 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 18 | 0.64% |
| Total | 55 | 488 | 369 | 152 | 88 | 47 | 759 | 321 | 174 | 120 | 77 | 216 | 2811 | 100.00% |
| SSR Loci | Na | Ne | I | Ho | He | PIC |
|---|---|---|---|---|---|---|
| g3601_i0 | 7 | 2.769 | 1.282 | 0.655 | 0.639 | 0.5876 |
| g10617_i0 | 5 | 3.037 | 1.262 | 0.673 | 0.671 | 0.6118 |
| g7466_i0 | 6 | 3.673 | 1.396 | 0.727 | 0.728 | 0.6863 |
| g5114_i0 | 3 | 2.219 | 0.923 | 0.673 | 0.549 | 0.4857 |
| g24756_i0 | 4 | 2.265 | 0.912 | 0.618 | 0.559 | 0.4582 |
| g6422_i0 | 10 | 3.881 | 1.689 | 0.764 | 0.742 | 0.7073 |
| g6405_i0 | 12 | 5.623 | 1.962 | 0.855 | 0.822 | 0.8072 |
| g334_i0 | 5 | 3.779 | 1.427 | 0.600 | 0.735 | 0.6955 |
| g798_i0 | 5 | 3.479 | 1.339 | 0.764 | 0.713 | 0.6571 |
| g10239_i0 | 3 | 2.561 | 1.018 | 0.509 | 0.610 | 0.5407 |
| Mean | 6 | 3.329 | 1.321 | 0.684 | 0.677 | 0.624 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Guo, S.; Zhang, Y.; Zhang, F.; Yun, Y.; Zhang, G. Transcriptome Analysis and Novel EST-SSR Marker Development for Pinus tabuliformis Seedlings from Four Provenances. Forests 2023, 14, 1810. https://doi.org/10.3390/f14091810
Wang J, Guo S, Zhang Y, Zhang F, Yun Y, Zhang G. Transcriptome Analysis and Novel EST-SSR Marker Development for Pinus tabuliformis Seedlings from Four Provenances. Forests. 2023; 14(9):1810. https://doi.org/10.3390/f14091810
Chicago/Turabian StyleWang, Ju, Shuai Guo, Yongxin Zhang, Feng Zhang, Yufei Yun, and Guosheng Zhang. 2023. "Transcriptome Analysis and Novel EST-SSR Marker Development for Pinus tabuliformis Seedlings from Four Provenances" Forests 14, no. 9: 1810. https://doi.org/10.3390/f14091810
APA StyleWang, J., Guo, S., Zhang, Y., Zhang, F., Yun, Y., & Zhang, G. (2023). Transcriptome Analysis and Novel EST-SSR Marker Development for Pinus tabuliformis Seedlings from Four Provenances. Forests, 14(9), 1810. https://doi.org/10.3390/f14091810