
Mutant Bisexual and Wild Male Flowers Were Compared by Integrated Proteome and Transcriptome Analyses to Provide Insight into the Sex Conversion of Idesia polycarpa Maxim

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Cytological Study
2.3. Protein Identification and Quantification
2.4. mRNA Library Construction, Sequencing and Quantification
2.5. Bioinformatic Analysis
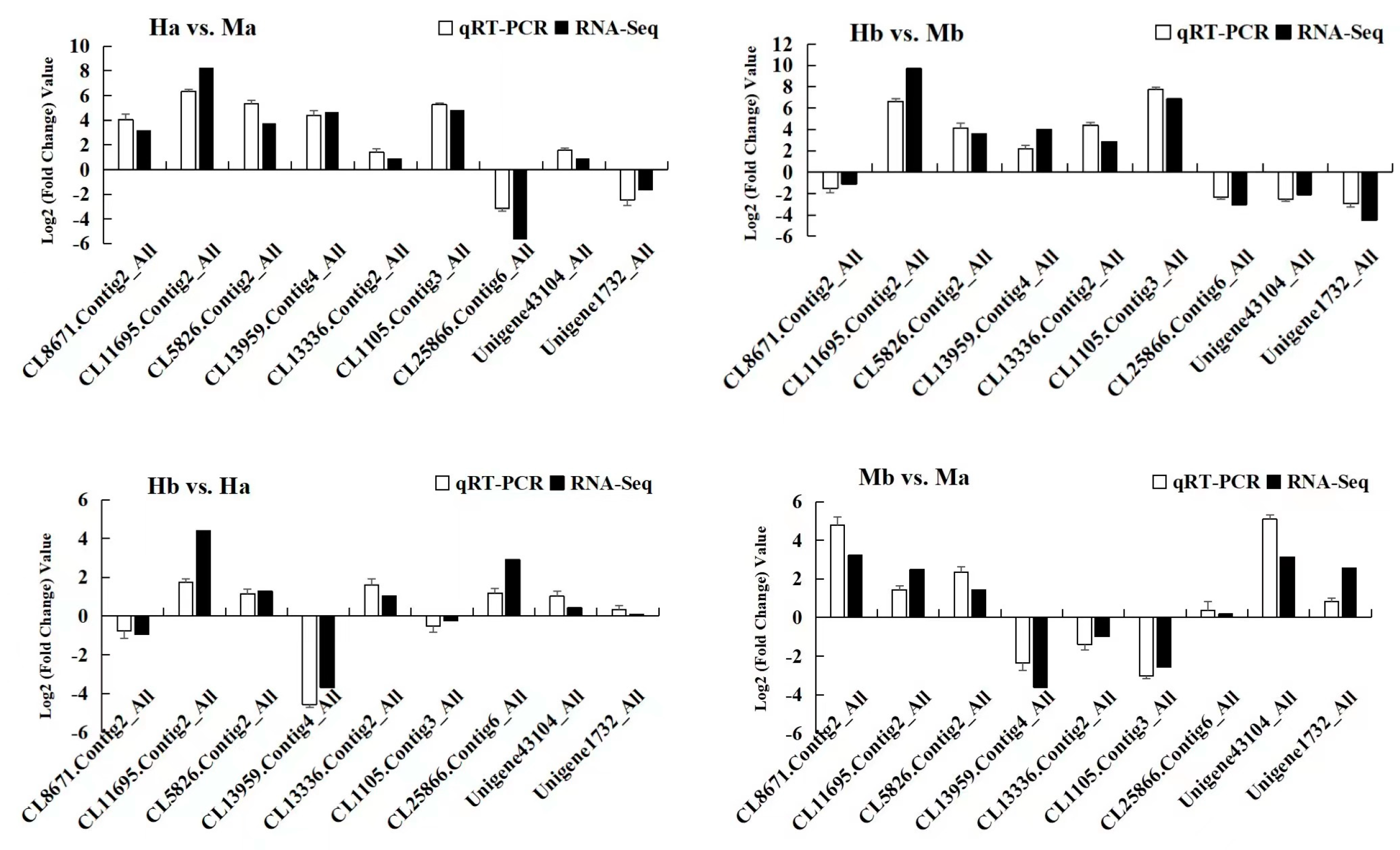
2.6. qRT-PCR Analysis
3. Results
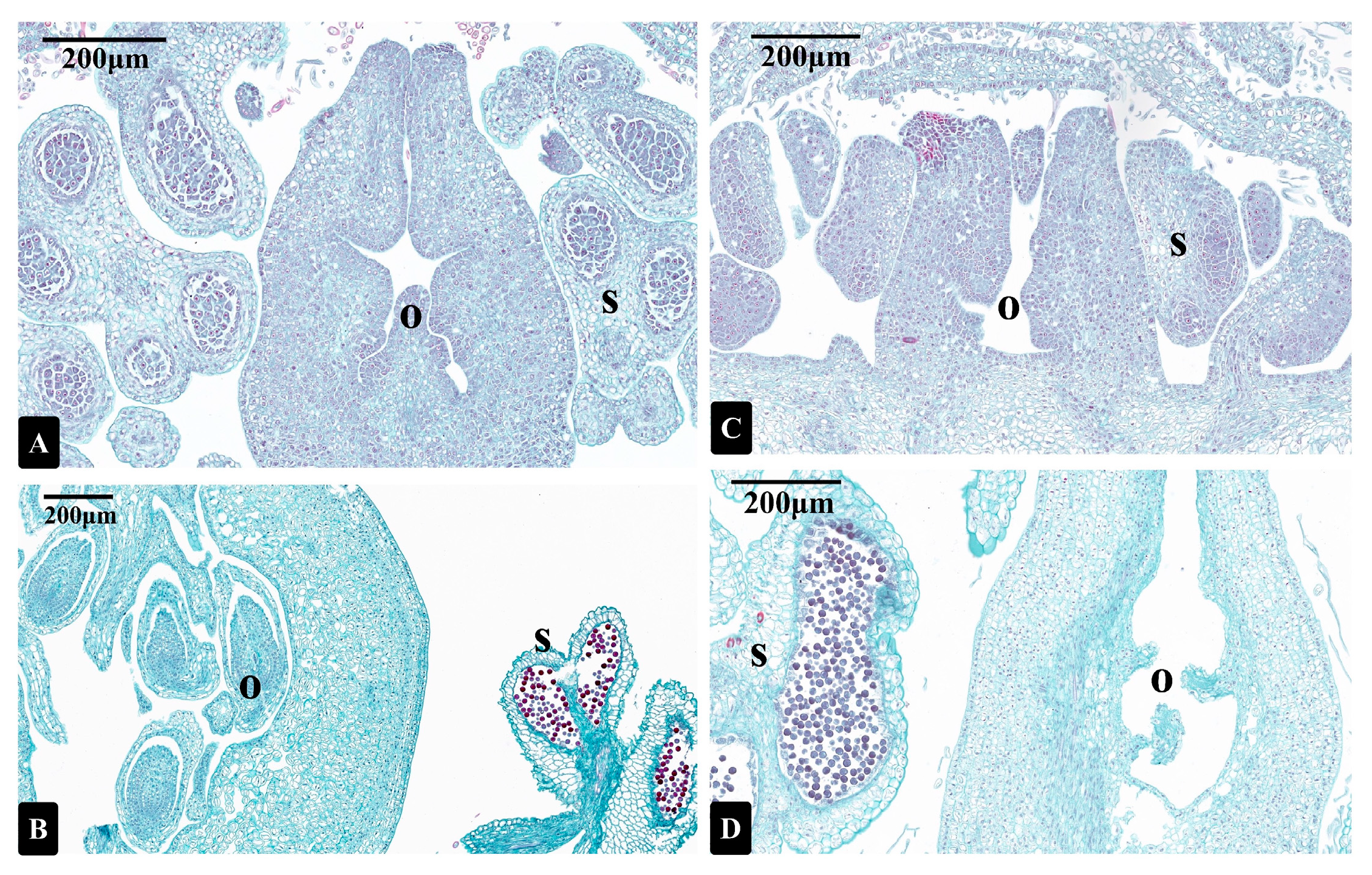
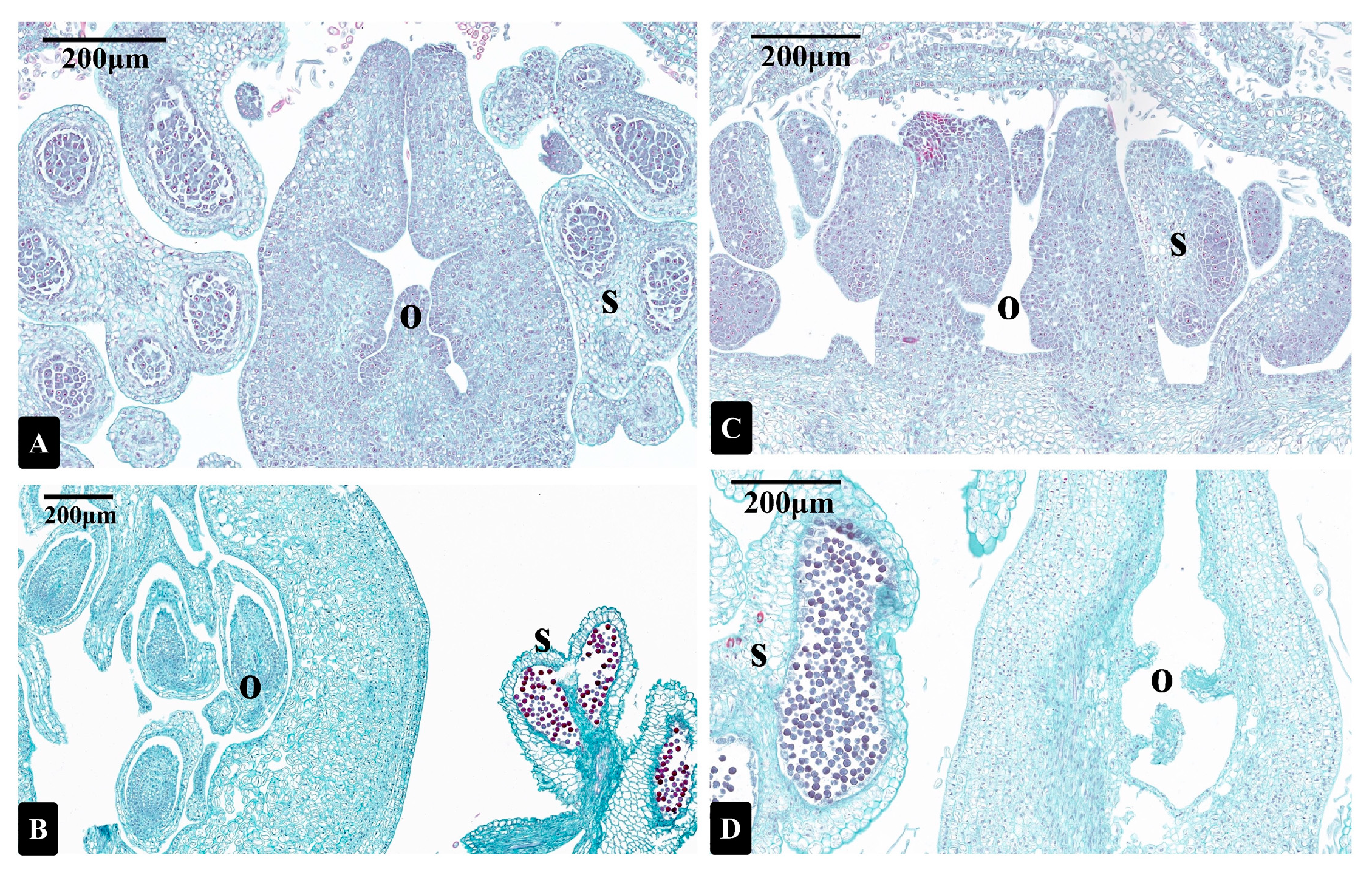
3.1. Photomicrographs of Longitudinal Sections of I. polycarpa Male and Bisexual Flowers
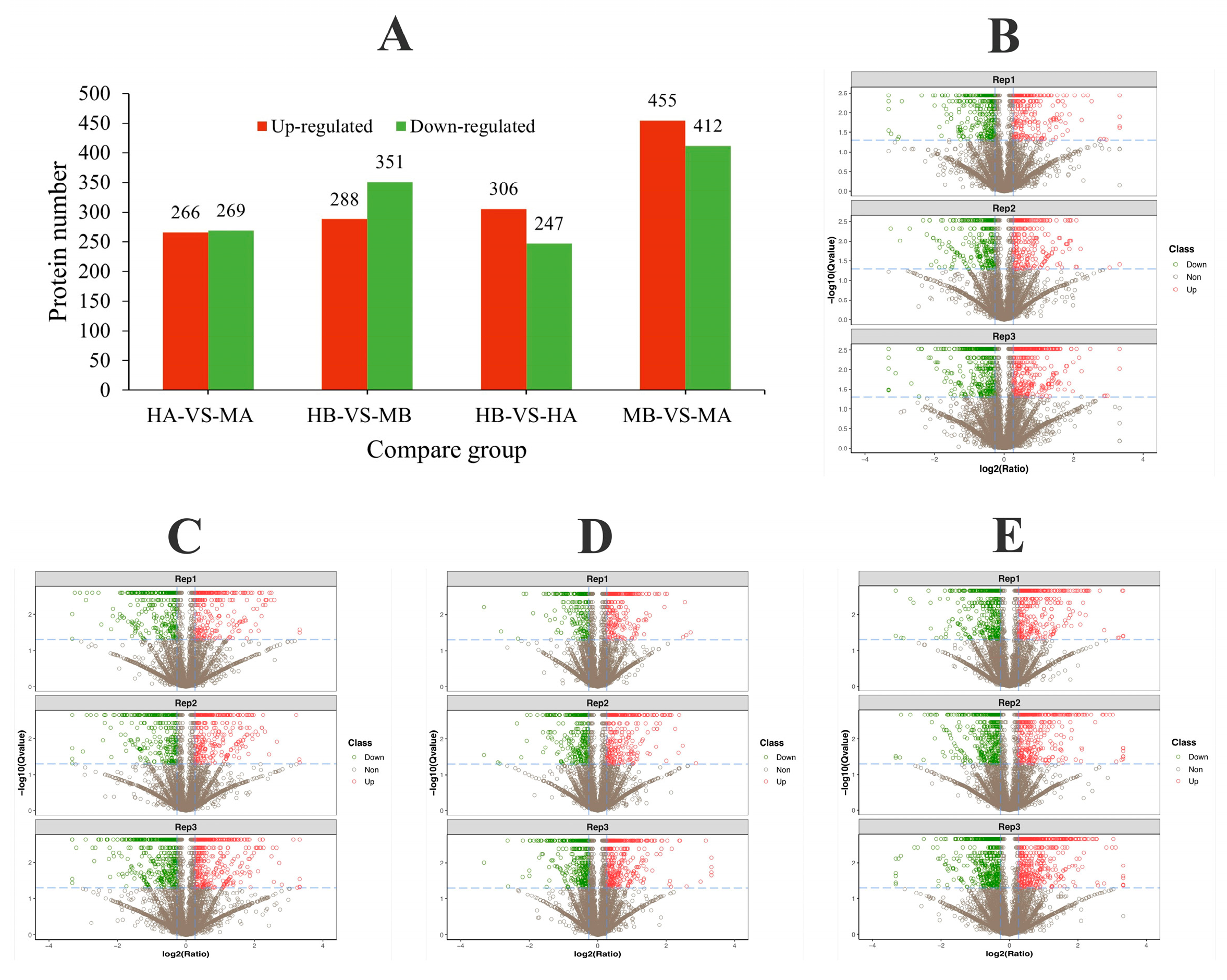
3.2. The ITRAQ Proteomic Analysis and Identification of DEPs
3.3. KEGG and GO Enrichment Analyses of DEPs
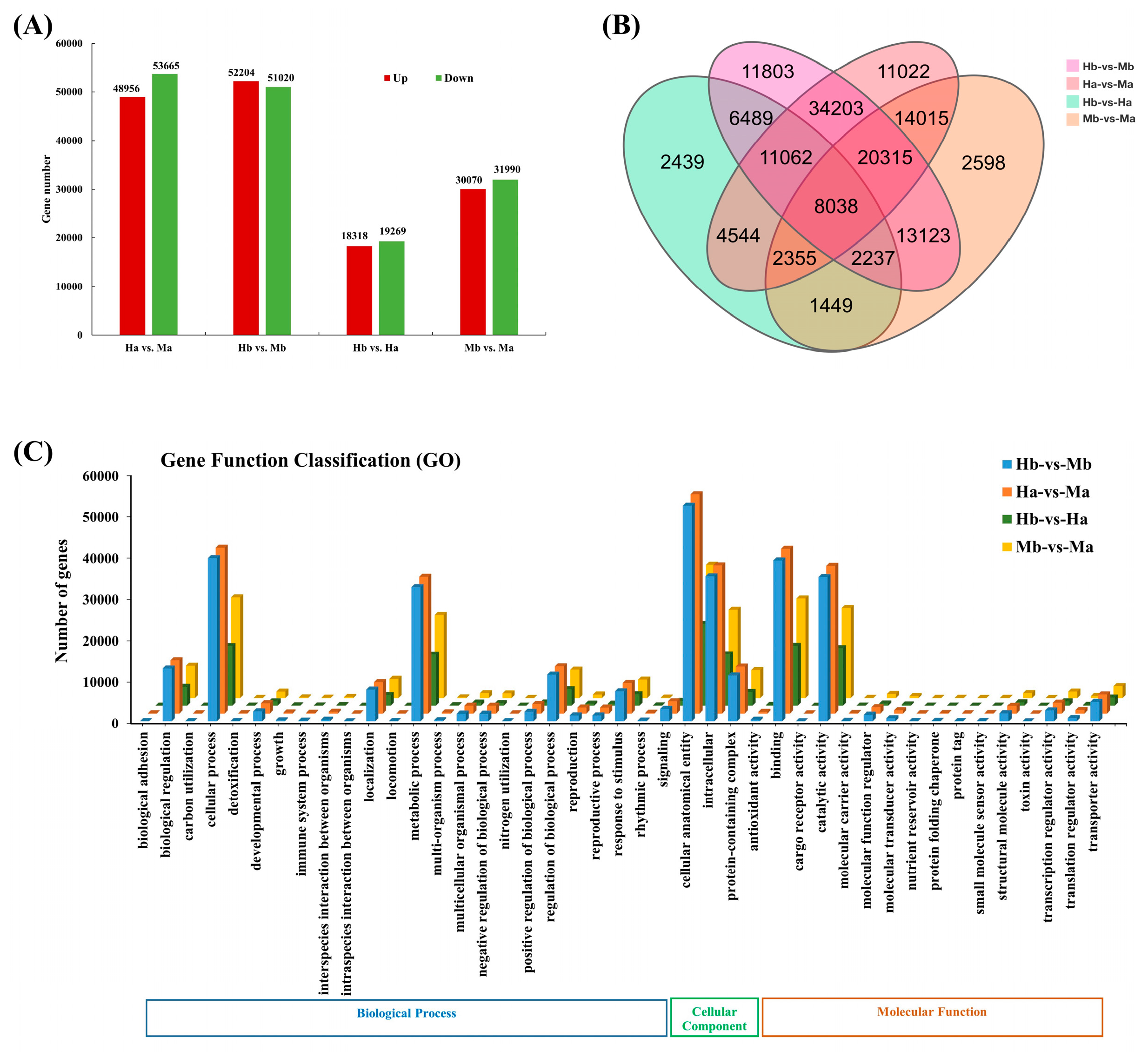
3.4. Results of RNA-seq Assembly and Unigenes Annotation
3.5. The Enriched GO Terms and KEGG Pathways of Unigenes
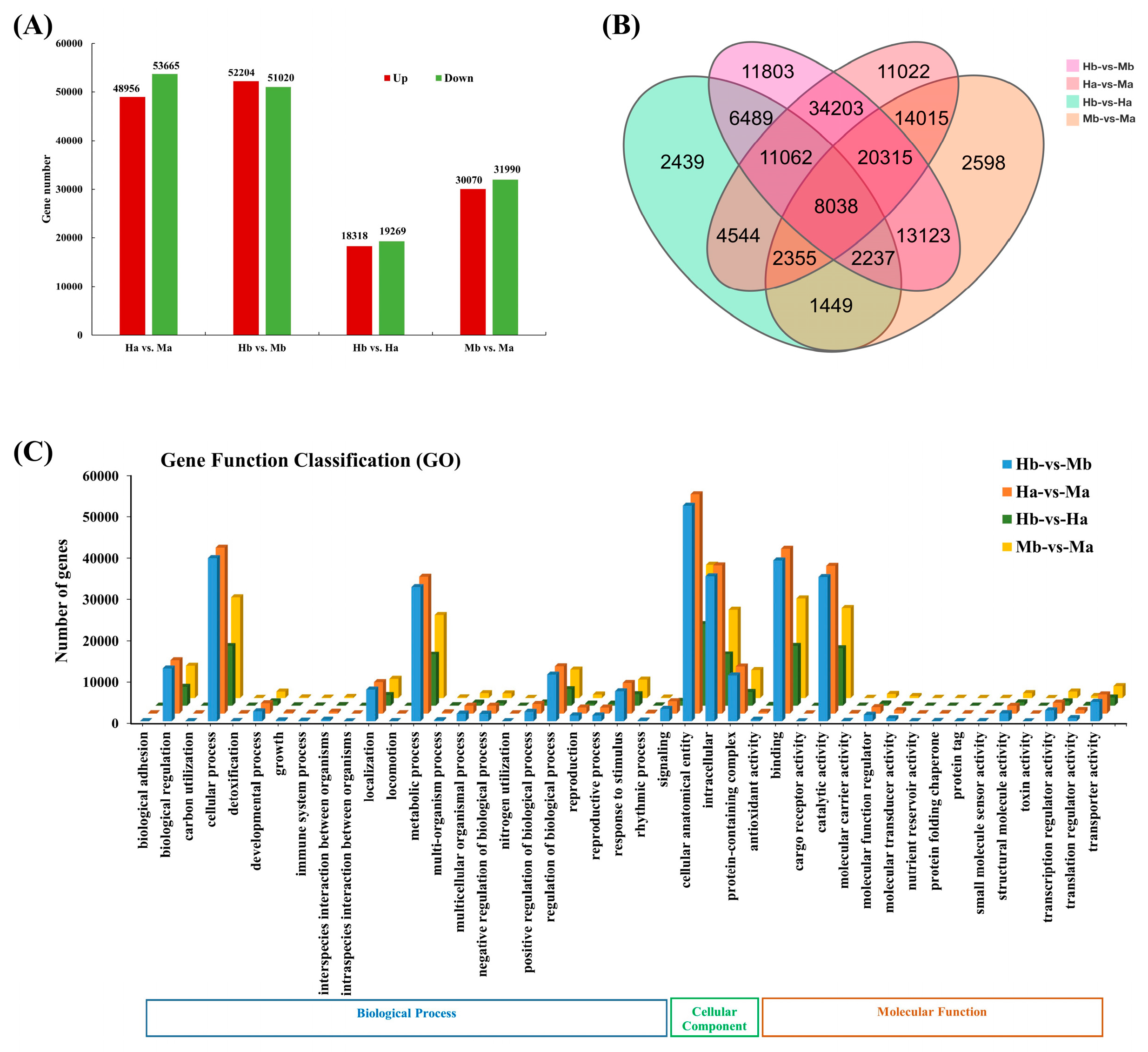
3.6. Identification and Enrichment Analysis of DEGs
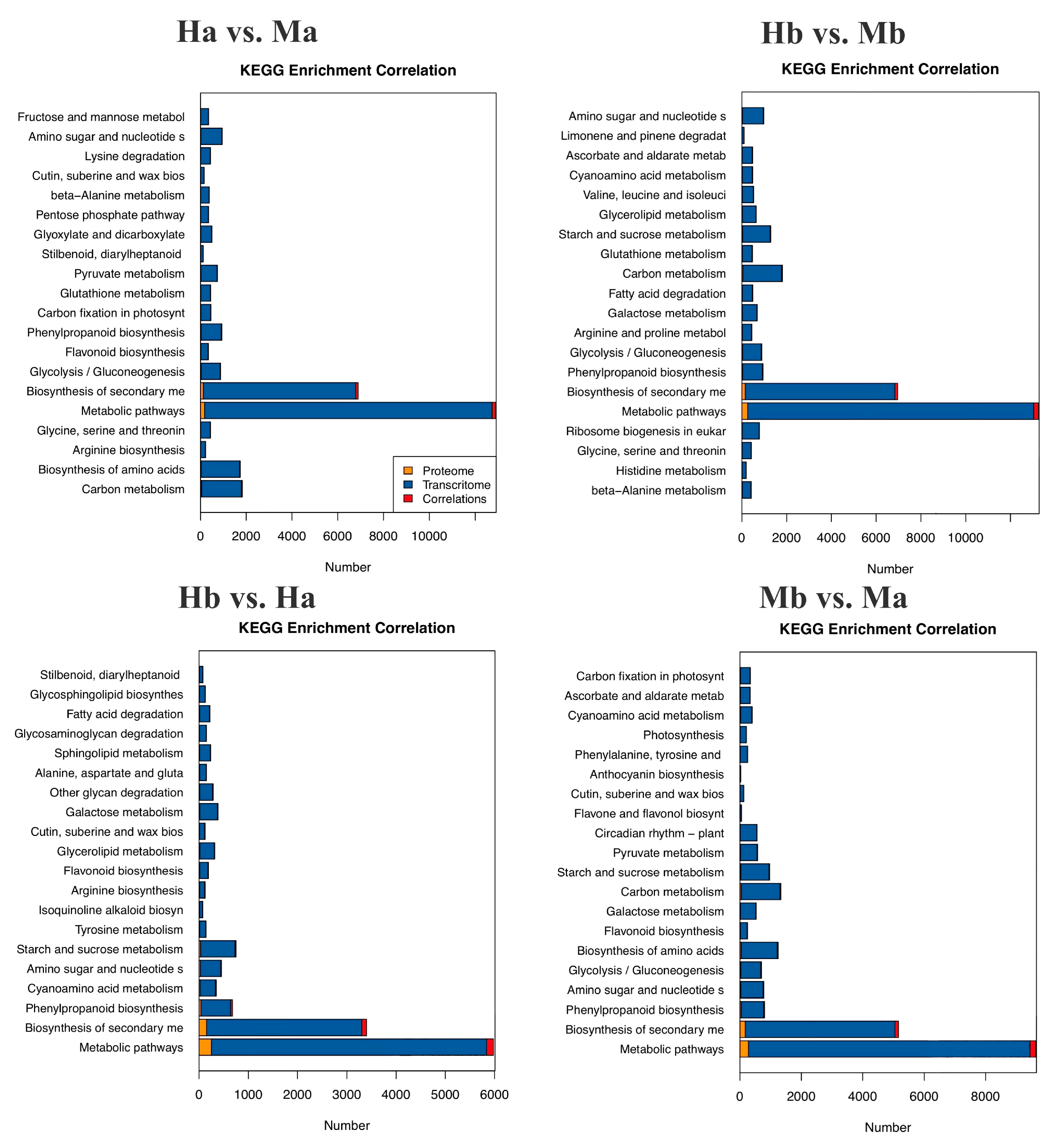
3.7. Correlation Analysis of the Proteome and Transcriptome Data
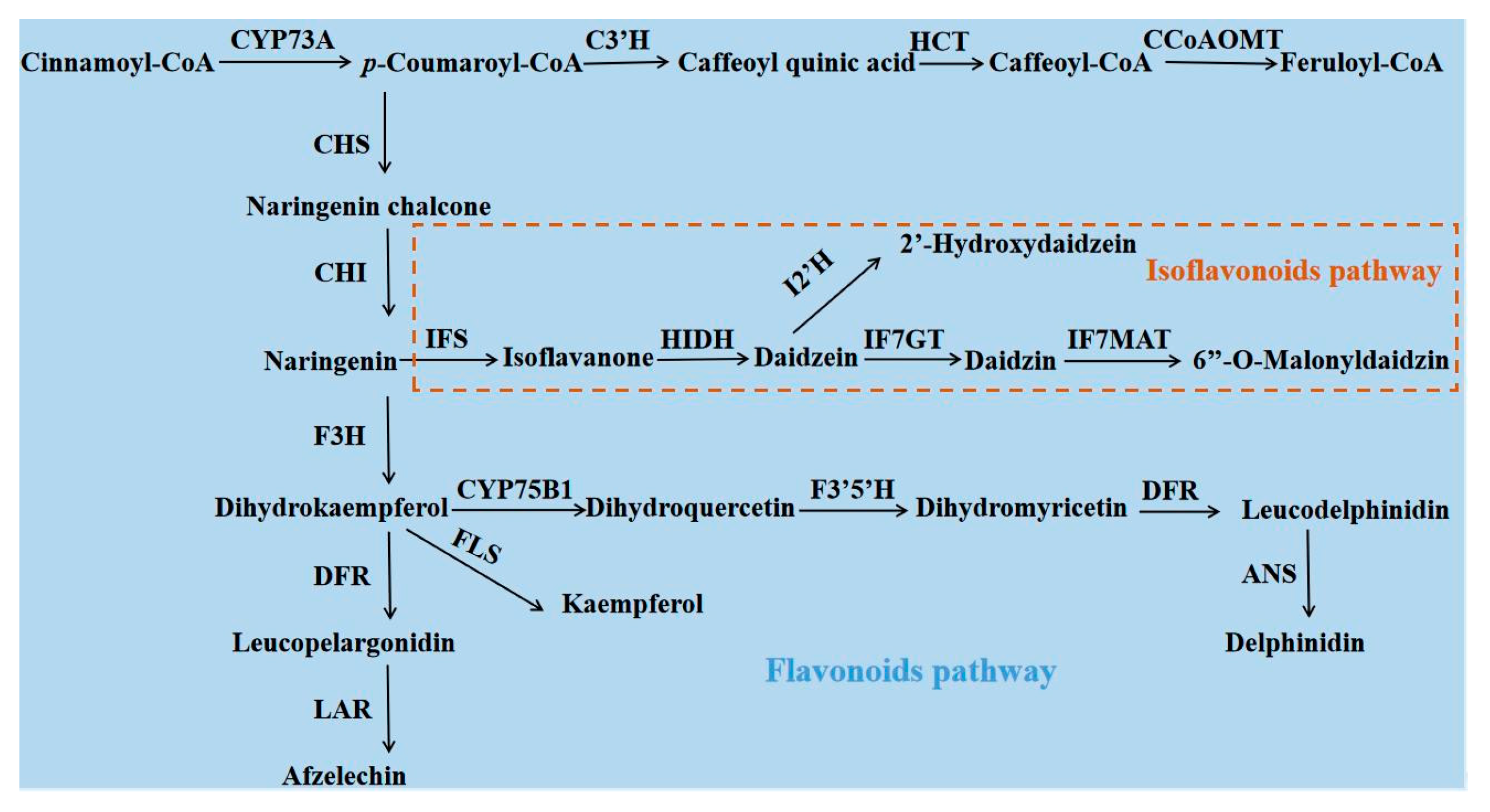
3.8. qRT-PCR Analysis of Flavonoids Biosynthesis-Related Genes
4. Discussion
4.1. Evolutionary Factors Associated with I. polycarpa Sex Conversion
4.2. Integrated Proteome and Transcriptome Analysis of Male and Bisexual Flowers
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rana, S.; Liu, Z. Study on the pattern of vegetative growth in young dioecious trees of Idesia polycarpa maxim. Trees 2021, 35, 69–80. [Google Scholar] [CrossRef]
- Wang, H.; Rana, S.; Li, Z.; Geng, X.; Wang, Y.; Cai, Q.; Li, S.; Sun, J.; Liu, Z. Morphological and anatomical changes during floral bud development of the trioecious Idesia polycarpa Maxim. Rev. Bras. Bot. 2022, 45, 679–688. [Google Scholar] [CrossRef]
- Yang, F.; Su, Y.; Li, X.; Zhang, Q.; Sun, R. Preparation of biodiesel from Idesia polycarpa var. vestita fruit oil. Ind. Crop. Prod. 2009, 29, 622–628. [Google Scholar] [CrossRef]
- Dai, L. Study on Geographic Variation of Fruit and Seed of Idesia polycarpa Maxim. Ph.D. Thesis, Henan Agricultural University, Hanan, Zhengzhou, 2014. (In Chinese) [Google Scholar] [CrossRef]
- Xiang, X.; Wen, L.; Wang, Z.; Yang, G.; Mao, J.; An, X.; Kan, J. A comprehensive study on physicochemical properties, bioactive compounds, and emulsified lipid digestion characteristics of Idesia polycarpa var. Vestita Diels fruits oil. Food Chem. 2023, 404, 134634. [Google Scholar] [CrossRef]
- Jung, M.; Yoo, J.; Kang, Y.; Lee, H.; Kim, S.; Sung, S.; Lee, Y.; Choi, I.; Kim, T. Idesolide, an isolate of Idesia polycarpa, inhibits apoptosis through induction of intracellular heat shock protein 70 in C2C12 muscle cells. Biol. Pharm. Bull. 2010, 33, 1063–1066. [Google Scholar] [CrossRef]
- Lee, M.; Lee, H.; Lee, J.; Ye, S.; Kim, S.; Sung, S. Anti-adipogenic activity of compounds isolated from Idesia polycarpa on 3T3-L1 cells. Bioorganic Med. Chem. Lett. 2013, 23, 3170–3174. [Google Scholar] [CrossRef] [PubMed]
- Charlesworth, B.; Charlesworth, D. A model for the evolution of dioecy and gynodioecy. Am. Nat. 1978, 112, 975–997. [Google Scholar] [CrossRef]
- Baránková, S.; Pascual-Díaz, J.P.; Sultana, N.; Alonso-Lifante, M.P.; Balant, M.; Barros, K.; D’Ambrosio, U.; Malinská, H.; Peska, V.; Pérez Lorenzo, I.; et al. Sex-chrom, a database on plant sex chromosomes. New Phytol. 2020, 227, 1594–1604. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.; Holzinger, E.; Li, R.; Pendergrass, S.; Kim, D. Methods of integrating data to uncover genotype-phenotype interactions. Nat. Rev. Genet. 2015, 16, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Deng, W.; Gunasekara, C.; Chiang, V.; Chen, H.; Ma, H.; Davis, X.; Wei, H. Bottom-up GGM algorithm for constructing multilayered hierarchical gene regulatory networks that govern biological pathways or processes. BMC Bioinf. 2016, 17, 132. [Google Scholar] [CrossRef]
- Iwasaki, M.; Tabata, T.; Kawahara, Y.; Ishihama, Y.; Nakagawa, M. Removal of interference MS/MS spectra for accurate quantification in isobaric tag-based proteomics. J. Proteome Res. 2019, 18, 2535–2544. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.; Marcotte, E.M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 2012, 13, 227–232. [Google Scholar] [CrossRef]
- Zhang, C.; Yu, D.; Ke, F.; Zhu, M.; Xu, J.; Zhang, M. Seedless mutant ‘Wuzi Ougan’ (Citrus suavissima Hort. ex Tanaka ‘seedless’) and the wild type were compared by iTRAQ-based quantitative proteomics and integratedly analyzed with transcriptome to improve understanding of male sterility. BMC Genet. 2018, 19, 106. [Google Scholar] [CrossRef]
- Dai, F.; Wang, Z.; Li, Z.; Luo, G.; Wang, Y.; Tang, C. Transcriptomic and proteomic analyses of mulberry (Morus atropurpurea) fruit response to Ciboria carunculoides. J. Proteom. 2018, 193, 142–153. [Google Scholar] [CrossRef]
- Wu, L.; Fang, Z.; Lin, J.; Sun, Y.; Du, Z.; Guo, Y.; Liu, J.; Liang, Y.; Ye, J. Complementary iTRAQ proteomic and transcriptomic analyses of leaves in tea plant (Camellia sinensis L.) with different maturity and regulatory network of flavonoid biosynthesis. J. Proteome Res. 2018, 18, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, S.; Song, S.; Xu, F.; Pan, Y.; Wang, H. Transcriptomic and proteomic analyses reveal new insight into chlorophyll synthesis and chloroplast structure of maize leaves under zinc deficiency stress. J. Proteom. 2019, 199, 123–134. [Google Scholar] [CrossRef]
- Li, Z.; Liu, N.; Zhang, W.; Wu, C.; Jiang, Y.; Ma, J.; Li, M.; Sui, S. Integrated transcriptome and proteome analysis provides insight into chilling-induced dormancy breaking in Chimonanthus praecox. Hortic. Res. 2020, 7, 198. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Yu, J.; Yan, W.; Zhang, J.; Yang, D.; Yao, G.; Liu, Z.; Wu, Y.; Hou, X. Integrative iTRAQ-based proteomic and transcriptomic analysis reveals the accumulation patterns of key metabolites associated with oil quality during seed ripening of Camellia oleifera. Hortic. Res. 2021, 8, 157. [Google Scholar] [CrossRef]
- Chen, T.; Sheng, Y.; Hao, Z.; Long, X.; Fu, F.; Liu, Y.; Tang, Z.; Ali, A.; Peng, Y.; Liu, Y.; et al. Transcriptome and proteome analysis suggest enhanced photosynthesis in tetraploid Liriodendron sino-americanum. Tree Physiol. 2021, 41, 1953–1971. [Google Scholar] [CrossRef]
- Dong, N.; Lin, H. Contribution of phenylpropanoid metabolism to plant development and plant-environment interactions. J. Integr. Plant Biol. 2020, 63, 180–209. [Google Scholar] [CrossRef]
- Brenda, W.S. Biosynthesis of flavonoids and effects of stress. Curr. Opin. Plant Biol. 2002, 5, 218–223. [Google Scholar] [CrossRef]
- Tohge, T.; Watanabe, M.; Hoefgen, R.; Fernie, A. The evolution of phenylpropanoid metabolism in the green lineage. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 123–152. [Google Scholar] [CrossRef] [PubMed]
- Panche, A.; Diwan, A.; Chandra, S. Flavonoids: An overview. J. Nutr. Sci. 2016, 5, 1–15. [Google Scholar] [CrossRef]
- Shimada, N.; Akashi, T.; Aoki, T.; Ayabe, S. Induction of isoflavonoid pathway in the model legume Lotus japonicus: Molecular characterization of enzymes involved in phytoalexin biosynthesis. Plant Sci. 2000, 160, 37–47. [Google Scholar] [CrossRef]
- Payyavula, R.S.; Singh, R.K.; Navarre, D.A. Transcription factors, sucrose, and sucrose metabolic genes interact to regulate potato phenylpropanoid metabolism. J. Exp. Bot. 2013, 64, 5115–5131. [Google Scholar] [CrossRef]
- Ban, Z.; Qin, H.; Mitchell, A.J.; Liu, B.; Zhang, F.; Weng, J.K.; Dixon, R.A.; Wang, G. Noncatalytic Chalcone Isomerase-fold Proteins in Humulus lupulus are Auxiliary Components in Prenylated Flavonoid Biosynthesis. Proc. Natl. Acad. Sci. USA 2018, 115, E5223–E5232. [Google Scholar] [CrossRef]
- Zhang, S.; Yang, J.; Li, H.; Chiang, V.L.; Fu, Y. Cooperative Regulation of Flavonoid and Lignin Biosynthesis in Plants. Crit. Rev. Plant Sci. 2021, 40, 109–126. [Google Scholar] [CrossRef]
- Van der Meer, I.; Stam, M.; van Tunen, A.; Mol, J.; Stuitje, A. Antisense inhibition of flavonoid biosynthesis in petunia anthers results in male sterility. Plant Cell. 1992, 4, 253–262. [Google Scholar] [CrossRef]
- Napoli, C.; Deirdre, F.; Wang, H.; Taylor, L. White anther: A petunia mutant that abolishes pollen flavonol accumulation, induces male sterility, and is complemented by a chalcone synthase transgene1. Plant Physiol. 1999, 120, 615–622. [Google Scholar] [CrossRef]
- Mahajan, M.; Ahuja, P.; Yadav, S. Post-transcriptional silencing of flavonol synthase mRNA in tobacco leads to fruits with arrested seed set. PLoS ONE 2011, 6, e28315. [Google Scholar] [CrossRef]
- Shan, X.; Li, Y.; Yang, S.; Yang, Z.; Qiu, M.; Gao, R.; Han, T.; Meng, X.; Xu, Z.; Wang, L.; et al. The spatio-temporal biosynthesis of floral flavonols is controlled by differential phylogenetic MYB regulators in Freesia hybrida. New Phytol. 2020, 228, 1864–1879. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Zhang, B.; Zhan, H.; Lv, Y.; Jia, X.; Wang, T.; Yang, N.; Lou, Y.; Zhang, Z.; Hu, W.; et al. Phenylpropanoid Derivatives Are Essential Components of Sporopollenin in Vascular Plants. Mol. Plant 2020, 13, 1–10. [Google Scholar] [CrossRef]
- Diggle, P.K.; Di Stilio, V.S.; Gschwend, A.R.; Golenberg, E.M.; Moore, R.C.; Russell, J.R.W.; Sinclair, J. Multiple developmental processes underlie sex differentitation in angiosperms. Trends Genet. 2011, 27, 368–376. [Google Scholar] [CrossRef]
- Aggarwal, S.; Yadav, A.K. Dissecting the iTRAQ Data Analysis. Methods Mol. Biol. 2016, 1362, 277–291. [Google Scholar] [CrossRef]
- Unwin, R.D.; Griffiths, J.R.; Whetton, A.D. Simultaneous analysis of relative protein expression levels across multiple samples using ITRAQ isobaric tags with 2D nano LC-MS/MS. Nat. Protoc. 2010, 5, 1574–1582. [Google Scholar] [CrossRef] [PubMed]
- Brosch, M.; Yu, L.; Hubbard, T.; Choudhary, J. Accurate and Sensitive Peptide Identification with Mascot Percolator. J. Proteome Res. 2009, 8, 3176–3181. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Statist. Soc. B. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Storey, J.D.; Tibshirani, R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA 2003, 100, 9440–9445. [Google Scholar] [CrossRef]
- Pertea, G.; Huang, X.; Liang, F.; Antonescu, V.; Sultana, R.; Karamycheva, S.; Lee, Y.; White, J.; Cheung, F.; Parvizi, B.; et al. TIGR Gene Indices clustering tools (TGICL): A software system for fast clustering of large EST datasets. Bioinformatics 2003, 19, 651–652. [Google Scholar] [CrossRef]
- Savitski, M.; Wilhelm, M.; Hahne, H.; Kuster, B.; Bantscheff, M. A Scalable Approach for Protein False Discovery Rate Estimation in Large Proteomic Data Sets. Mol. Cell. Proteom. 2015, 14, 2394–2404. [Google Scholar] [CrossRef]
- Wen, B.; Zhou, R.; Feng, Q.; Wang, Q.; Wang, J.; Liu, S. IQuant: An automated pipeline for quantitative proteomics based upon isobaric tags. Proteomics 2014, 14, 2280–2285. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Liang, X.; Xuan, Y.; Geng, C.; Li, Y.; Lu, H.; Qu, S.; Mei, X.; Chen, H.; Yu, T.; et al. A reference human genome dataset of the BGISEQ-500 sequencer. Gigascience 2017, 6, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhao, X.; Zhang, W.; Wang, L.; Wang, J.; Xu, D.; Mei, Z.; Liu, Q.; Du, S.; Li, Z.; et al. Reliable multiplex sequencing with rare index mis-assignment on DNB-based NGS platform. BMC Genom. 2019, 20, 215. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Li, Y.; Kristiansen, K.; Wang, J. SOAP: Short oligonucleotide alignment program. Bioinformatics 2008, 24, 713–714. [Google Scholar] [CrossRef]
- Zhang, F.; Gao, Q.; Khan, G.; Luo, K.; Chen, S. Comparative transcriptome analysis of aboveground and underground tissues of Rhodiola algida, an important ethno-medicinal herb endemic to the Qinghai-Tibetan Plateau. Gene 2014, 553, 90–97. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinf. 2011, 12, 323. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Deutsch, E.W.; Bandeira, N.; Perez-Riverol, Y.; Sharma, V.; Carver, J.J.; Mendoza, L.; Kundu, D.; Wang, S.; Bandla, C.; Kamatchinathan, S.; et al. The ProteomeXchange Consortium at 10 years: 2023 update. Nucleic Acids Res. 2023, 51, D1539–D1548. [Google Scholar] [CrossRef]
- Miller, W.; Myers, E.W.; Lipman, D.J. Blast (basic local alignment search tool). In Encyclopedia of Genetics, Genomics, Proteomics and Informatics; Springer: Dordrecht, The Netherlands, 2008. [Google Scholar] [CrossRef]
- Harris, M.A.; Clark, J.; Ireland, A.; Lomax, J.; Ashburner, M.; Foulger, R.; Eilbeck, K.; Lewis, S.; Marshall, B.; Mungall, C.; et al. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004, 32, 258–261. [Google Scholar] [CrossRef]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Li, W.V.; Li, J.J. Modeling and analysis of RNA-seq data: A review from a statistical perspective. Quant. Biol. 2018, 6, 195–209. [Google Scholar] [CrossRef] [PubMed]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef]
- Spearman, C.E. “General Intelligence,” Objectively Determined and Measured. Am. J. Psychol. 1904, 15, 201–293. [Google Scholar] [CrossRef]
- Hauke, J.; Kossowski, T. Comparison of values of Pearson’s and Spearman’s correlation coefficients on the same sets of data. Quaest. Geogr. 2011, 30, 87–93. [Google Scholar] [CrossRef]
- Fan, R.; Cai, G.; Zhou, X.; Qiao, Y.; Wang, J.; Zhong, H.; Bo, J.; Miao, F.; Tu, W.; Long, F.; et al. Characterization of diacylglycerol acyltransferase 2 from Idesia polycarpa and function analysis. Chem. Phys. Lipids. 2021, 234, 105023. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCt Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Cruden, R.W.; Lloyd, R.M. Embryophytes have equivalent sexual phenotypes and breeding systems: Why not a common terminology to describe them? Am. J. Bot. 1995, 82, 816–825. [Google Scholar] [CrossRef]
- Rottenberg, A. Sex ratio and gender stability in the dioecious plants of Israel. Bot. J. Linn. Soc. 1998, 128, 137–148. [Google Scholar] [CrossRef]
- Fechter, I.; Hausmann, L.; Daum, M.; Rosleff Sörensen, T.; Viehöver, P.; Weisshaar, B.; Töpfer, R. Candidate genes within a 143 kb region of the flower sex locus in Vitis. Mol. Genet. Genom. 2012, 287, 247–259. [Google Scholar] [CrossRef]
- Wang, J.; Na, J.; Yu, Q.; Gschwend, A.; Han, J.; Zeng, F.; Aryal, R.; Van Buren, R.; Murray, J.; Zhang, W.; et al. Sequencing Papaya X and Yh Chromosomes Reveals Molecular Basis of Incipient Sex Chromosome Evolution. Proc. Natl. Acad. Sci. USA 2012, 109, 13710–13715. [Google Scholar] [CrossRef]
- Van Buren, R.; Zeng, F.; Chen, C.; Zhang, J.; Wai, C.M.; Han, J.; Aryal, R.; Gschwend, A.; Wang, J.; Na, J.; et al. Origin and domestication of papaya Yh chromosome. Genome Res. 2015, 25, 524–533. [Google Scholar] [CrossRef]
- Nagata, T.; Hasebe, M.; Toriba, T.; Crane, P. Sex conversion in Ginkgo biloba (Ginkgoaceae). J. Jpn. Bot. 2016, 91, 120–127. Available online: https://www.researchgate.net/publication/311859168 (accessed on 19 January 2016).
- Venkatasamy, S.; Khittoo, G.; Keeley, K.S. Leaky dioecy in Diospyros (Ebenaceae) endemic to the Island of Mauritius. Plant Ecol. 2007, 189, 139–146. [Google Scholar] [CrossRef]
- Krizek, B.A.; Fletcher, J.C. Molecular Mechanisms of Flower Development: An Armchair Guide. Nat. Rev. Genet. 2005, 6, 688–698. [Google Scholar] [CrossRef]
- Ming, R.; Moore, P.H. Genomics of sex chromosomes. Curr. Opin. Plant Biol. 2007, 10, 123–130. [Google Scholar] [CrossRef]
- Renner, S.S. The Relative and Absolute Frequencies of Angiosperm Sexual Systems: Dioecy, Monoecy, Gynodioecy, and An Updated Online Database. Am. J. Bot. 2014, 101, 1588–1596. [Google Scholar] [CrossRef]
- Charlesworth, D. Plant contributions to our understanding of sex chromosome evolution. New Phytol. 2015, 208, 52–65. [Google Scholar] [CrossRef]
- Renner, S.S.; Ricklefs, R.E. Dioecy and its correlates in the flowering plants. Am. J. Bot. 1995, 82, 596–606. [Google Scholar] [CrossRef]
- Charlesworth, D. Mogens Westergaard’s Contributions to Understanding Sex Chromosomes. Genetics 2018, 210, 1143–1149. [Google Scholar] [CrossRef]
- Ross, M.D. The Evolution of Gynodioecy and Subdioecy. Evolution 1978, 32, 174–188. [Google Scholar] [CrossRef]
- Harder, L.D.; Barrett, S.C.H. Ecology and Evolution of Flowers; Oxford University Press: New York, NY, USA, 2007. [Google Scholar]
- Barrett, S.C.H. The evolution of plant sexual diversity. Nat. Rev. Genet. 2002, 3, 274–284. [Google Scholar] [CrossRef]
- Charlesworth, D. Plant sex Chromosome Evolution. J. Exp. Bot. 2012, 64, 405–420. [Google Scholar] [CrossRef]
- Lloyd, D.G. Breeding systems in Cotula L. (Compositae, Anthemideae). I. The array of monoclinous and diclinous systems. New Phytol. 1972, 71, 1181–1194. [Google Scholar] [CrossRef]
- Opler, P.A.; Baker, H.G.; Frankie, G.W. Reproductive Biology of Some Costa Rican Cordia Species (Boraginaceae). Biotropica 1975, 7, 234–247. [Google Scholar] [CrossRef]
- Aryal, R.; Ming, R. Sex determination in flowering plants: Papaya as a model system. Plant Sci. 2014, 217–218, 56–62. [Google Scholar] [CrossRef]
- Akagi, T.; Henry, I.; Ohtani, H.; Morimoto, T.; Beppu, K.; Kataoka, I.; Tao, R. A Y-Encoded Suppressor of Feminization Arose via Lineage-Specific Duplication of a Cytokinin Response Regulator in Kiwifruit. Plant Cell. 2018, 30, 780–795. [Google Scholar] [CrossRef]
- Ideker, T.; Thorsson, V.; Ranish, J.A.; Christmas, R.; Buhler, J.; Eng, J.K.; Bumgarner, R.; Goodlett, D.; Aebersold, R.; Hood, L. Integrated genomic and proteomic analyses of a systematically perturbed metabolic network. Science 2001, 292, 929–934. [Google Scholar] [CrossRef]
- Muers, M. Gene expression: Transcriptome to proteome and back to genome. Nat. Rev. Genet. 2011, 12, 518. [Google Scholar] [CrossRef] [PubMed]
- Maier, T.; Güell, M.; Serrano, L. Correlation of mRNA and protein in complex biological samples. FEBS Lett. 2009, 583, 3966–3973. [Google Scholar] [CrossRef]
- Du, S.; Sang, Y.; Liu, X.; Xing, S.; Li, J.; Tang, H.; Sun, L. Transcriptome Profile Analysis from Different Sex Types of Ginkgo biloba L. Front. Plant Sci. 2016, 7, 871. [Google Scholar] [CrossRef] [PubMed]
- Wils, C.; Brandt, W.; Manke, K.; Vogt, T. A single amino acid determines position specificity of an Arabidopsis thaliana CCoAOMT-like O-methyltransferase. FEBS Lett. 2013, 587, 683–689. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhao, C.; Gong, Q.; Wang, Y.; Cao, J.; Li, X.; Grierson, D.; Sun, C. Characterization of a caffeoyl-CoA O-methyltransferase-like enzyme involved in biosynthesis of polymethoxylated flavones in Citrus reticulata. J. Exp. Bot. 2020, 71, 3066–3079. [Google Scholar] [CrossRef]
- Hugueney, P.; Provenzano, S.; Verriès, C.; Ferrandino, A.; Meudec, E.; Batelli, G.; Merdinoglu, D.; Cheynier, V.; Schubert, A.; Ageorges, A. A novel cation-dependent O-methyltransferase involved in anthocyanin methylation in grapevine. Plant Physiol. 2009, 150, 2057–2070. [Google Scholar] [CrossRef]
- Gomez Roldan, M.V.; Outchkourov, N.; van Houwelingen, A.; Lammers, M.; Romero de la Fuente, I.; Ziklo, N.; Aharoni, A.; Hall, R.; Beekwilder, J. An O-methyltransferase modifies accumulation of methylated anthocyanins in seedlings of tomato. Plant J. 2014, 80, 695–708. [Google Scholar] [CrossRef]
- Du, H.; Wu, J.; Ji, K.; Zeng, Q.Y.; Bhuiya, M.W.; Su, S.; Shu, Q.; Ren, H.; Liu, Z.; Wang, L. Methylation mediated by an anthocyanin, O-methyltransferase, is involved in purple flower coloration in Paeonia. J. Exp. Bot. 2015, 66, 6563–6577. [Google Scholar] [CrossRef]
- Chowdhury, E.M.; Bo, S.C.; Sang, U.P.; Lim, H.S.; Bae, H. Transcriptional analysis of hydroxycinnamoyl transferase (HCT) in various tissues of Hibiscus cannabinus in response to abiotic stress conditions. Plant Omics 2012, 5, 305–313. [Google Scholar] [CrossRef]
- Vanholme, B.; Cesarino, I.; Goeminne, G.; Kim, H.; Marroni, F.; Van Acker, R.; Vanholme, R.; Morreel, K.; Ivens, B.; Pinosio, S.; et al. Breeding with rare defective alleles (BRDA): A natural Populus nigra HCT mutant with modified lignin as a case study. New Phytol. 2013, 198, 765–776. [Google Scholar] [CrossRef]
- Teng, L.; Zheng, K.; Qu, Y.T.; Cheng, Q. Cloning and Expression Characteristics Analysis of GbHCT13 Gene in Gossypium barbadense. Chin. J. Agric. Biotechnol. 2021, 29, 1050–1060. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Item | Value |
|---|---|
| Type of search | MS/MS ion search |
| Enzyme | Trypsin |
| Fragment mass tolerance | 0.05 Da |
| Peptide mass tolerance | 20 ppm |
| Fixed modifications | Carbamidomethyl (C), ITRAQ8plex (N-term), ITRAQ8plex (K) |
| Variable modifications | Oxidation (M), deamidated (NQ), ITRAQ8plex (Y) |
| Peptide FDR | ≤0.01 |
| Sample Name | Total Spectra | Spectra | Peptide | Unique Peptide | Protein |
|---|---|---|---|---|---|
| Replicate 1 | 928,921 | 111,497 | 47,195 | 31,514 | 10,122 |
| Replicate 2 | 953,111 | 115,434 | 48,006 | 31,904 | 10,158 |
| Replicate 3 | 938,500 | 111,138 | 47,990 | 32,052 | 10,243 |
| Total | 2,820,532 | 338,069 | 64,942 | 47,580 | 15,003 |
| Sample | Total Raw Reads (M) | Total Clean Reads (M) | Total Clean Bases (Gb) | Total Number of Unigenes | Clean Reads Q20 (%) | Clean Reads Q30 (%) | GC (%) |
|---|---|---|---|---|---|---|---|
| Ha1 | 47.33 | 43.71 | 6.56 | 87,618 | 97.05 | 88.87 | 40.44 |
| Ha2 | 47.33 | 43.1 | 6.46 | 87,263 | 96.89 | 88.38 | 40.45 |
| Ha3 | 47.33 | 43.05 | 6.46 | 86,168 | 97.2 | 89.12 | 40.48 |
| Hb1 | 47.33 | 43.24 | 6.49 | 93,899 | 97.02 | 88.76 | 40.08 |
| Hb2 | 49.08 | 44.96 | 6.74 | 114,098 | 97.08 | 88.99 | 39.5 |
| Hb3 | 47.33 | 43.5 | 6.53 | 90,575 | 96.98 | 88.63 | 40.18 |
| Ma1 | 47.33 | 43.39 | 6.51 | 98,947 | 97.08 | 88.97 | 40.03 |
| Ma2 | 49.08 | 45.05 | 6.76 | 112,130 | 96.9 | 88.39 | 39.44 |
| Ma3 | 47.33 | 43.4 | 6.51 | 70,122 | 97 | 88.7 | 40.88 |
| Mb1 | 47.33 | 43.76 | 6.56 | 48,720 | 96.95 | 88.61 | 41.41 |
| Mb2 | 47.33 | 43.44 | 6.52 | 102,848 | 96.9 | 88.41 | 39.71 |
| Mb3 | 49.08 | 44.9 | 6.74 | 115,564 | 96.91 | 88.44 | 39.18 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.; Li, Z.; Cai, Q.; Wang, Y.; Geng, X.; Li, S.; Fang, L.; Yao, S.; Li, H.; Liu, Z. Mutant Bisexual and Wild Male Flowers Were Compared by Integrated Proteome and Transcriptome Analyses to Provide Insight into the Sex Conversion of Idesia polycarpa Maxim. Forests 2023, 14, 1737. https://doi.org/10.3390/f14091737
Wang H, Li Z, Cai Q, Wang Y, Geng X, Li S, Fang L, Yao S, Li H, Liu Z. Mutant Bisexual and Wild Male Flowers Were Compared by Integrated Proteome and Transcriptome Analyses to Provide Insight into the Sex Conversion of Idesia polycarpa Maxim. Forests. 2023; 14(9):1737. https://doi.org/10.3390/f14091737
Chicago/Turabian StyleWang, Huimin, Zhi Li, Qifei Cai, Yanmei Wang, Xiaodong Geng, Shunfu Li, Lisha Fang, Shunyang Yao, Huiyun Li, and Zhen Liu. 2023. "Mutant Bisexual and Wild Male Flowers Were Compared by Integrated Proteome and Transcriptome Analyses to Provide Insight into the Sex Conversion of Idesia polycarpa Maxim" Forests 14, no. 9: 1737. https://doi.org/10.3390/f14091737
APA StyleWang, H., Li, Z., Cai, Q., Wang, Y., Geng, X., Li, S., Fang, L., Yao, S., Li, H., & Liu, Z. (2023). Mutant Bisexual and Wild Male Flowers Were Compared by Integrated Proteome and Transcriptome Analyses to Provide Insight into the Sex Conversion of Idesia polycarpa Maxim. Forests, 14(9), 1737. https://doi.org/10.3390/f14091737