Abstract
The alpine grasslands account for approximately 54.5% of the total carbon in China’s grasslands, and carbohydrate-active enzymes (CAZymes) play key roles in the turnover of carbon. However, the variation and factors influencing gene-encoding enzymes for plant- and microbial-derived carbon decomposition in alpine steppes and alpine meadows remain unclear. Here, the trends in microbial carbohydrate-active enzymes (CAZymes) and their responses to the decomposition of biomass of different origins were studied using metagenomics in the alpine steppes and alpine meadows on the Tibetan Plateau. Our results revealed the abundance of GTs and CBMs was higher in the alpine steppes than in the alpine meadows, whereas AAs were higher in the alpine steppes than in the alpine meadows. Soil properties (i.e., soil water content, soil ammonium nitrogen, and nitrate nitrogen) highly related to CAZyme genes (GTs, CBMs, and AAs) showed an abundant pattern between the alpine steppes and alpine meadows. Moreover, our results indicated that the relative abundance of genes encoding CAZymes involved in the decomposition of plant- (indicated by cellulose, hemicellulose, and lignin) and fungal-derived carbon (indicated by chitin and glucans) was higher by 8.7% and 10.1%, respectively, in the alpine steppes than in the alpine meadows, whereas bacterial-derived carbon (indicated by peptidoglycan) was lower by 7.9% in the alpine steppes than in the alpine meadows. Soil water content (SWC), nitrate nitrogen (NO3−), and pH influenced on the abundance of CAZyme genes involved in the decomposition of plant-, fungal-, bacterial-derived carbon. In addition, the dominant microbial phyla (Actinobacteria, Protebacteria, and Acidobacteria) mineralized carbon sources from plant- and microbial-derived carbon through their corresponding CAZyme families. In conclusion, our study compared plant- and microbial-derived carbon decomposition potentials and influencing factors to illustrate the contribution of dead biomass to carbon accumulation in alpine grasslands.
1. Introduction
Grassland ecosystems, which are significant components of the terrestrial biosphere, cover approximately one-third of the global land surface and play a crucial role in the global carbon cycle [1,2,3]. The Tibetan Plateau, also known as the third pole, is mainly occupied by alpine steppes and meadows [4,5]. The alpine grasslands store approximately 27.2 Pg C in the top 1 m of soil, accounting for approximately 54.5% of the total carbon in China’s grasslands [6,7]. Alpine steppes and meadows store about 4.32 Pg C SOC and 7.68 Pg C SOC, respectively [8]. The carbon decomposition rate is 51.76 mg CO2-C m−2 h−1 and 53.76 mg CO2-C m−2 h−1 in alpine steppes and meadows and is influenced by many factors [8]. Thus, a slight change in carbon stocks in alpine steppes and meadows may influence atmospheric CO2 concentrations and further global change [7].
Plants (i.e., leaf litter and root litter) and microbes are the main carbon sources in alpine steppes and meadows [9]. Dead plant biomass is a vital source of energy stored in the form of complex polysaccharides, primarily cellulose, hemicellulose, and lignin, which are relative recalcitrant matrices [9,10]. Dead fungal biomass is mostly made up of polysaccharides, which constitute 80–90% of the total cell wall, and it also contains lipids and mannose proteins [11,12], primarily chitin and glucan, the decomposition of which is correlated with the soil texture [13]. Dead bacterial biomass is the main peptidoglycan, which is abundant in soils and shows higher turnover rates than fungal biomass under conditions of high microbial activity [14]. The turnover of plants and microbial biomass represents a key step in the carbohydrate decomposition and can be tracked by analyzing microbial enzymes and carbohydrate-active enzymes (CAZymes) [15,16]. CAZymes can be classified as glycoside hydrolases (GHs), glycosyl transferases (GTs), polysaccharide lyases (PLs), carbohydrate esterases (CEs), auxiliary activities (AAs), and carbohydrate-binding modules (CBMs) based on the similarity of amino acid sequences in the protein domains [17]. Specifically, genes encoding several GH families in CAZymes are the degradation of plant biomass and microbial biomass including chitinases and peptidoglycan lytic transglycosylase and GT families are primarily involved in biosynthesis of disaccharides, oligosaccharides, and polysaccharides [16,18]. PLs are involved in the degradation of glycosaminoglycans and pectin by cleaving the glycosidic bonds of uronic acid-containing polysaccharides and CEs dislodge ester-based modifications present in mono-, oligo-, and polysaccharides [17,19,20]. Some genes encoding AA families in CAZymes are associated with lignin decomposition, and CBMs can associate CAZymes with carbohydrate-related substrates [21,22].
In addition, the abundance of the CAZyme genes are affected by several factors, including SOC, Total nitrogen (TN), Na+, and K+, which influence the decomposition potential [9,17]. Previous studies have shown that the carbon–nitrogen ratio, temperature, soil water content, soil pH, and normalized difference vegetation index (NDVI) are the main factors influencing the enrichment of carbohydrate enzyme genes [16,18,23]. For example, GH activities are very pH-dependent because pH variation boosts changes in folding, conformation, and protonation of enzymes [24]. PLs were temperature-sensitive, which means increased temperature can improve the potentials of cleaving complex polysaccharide chains to simple structural saccharides [18]. Previous studies indicated the types of grassland influence on soil organic matter decomposition and showed a higher soil organic matter decomposition rate in alpine meadows than in alpine steppes [25]. However, the microbial decomposition potential of and environmental factors affecting the decomposition of microbial- and plant-derived carbon in alpine steppes and meadows remain unknown. Therefore, it is fundamental to identify the environmental factors influencing the potential microbial genes involved in the decomposition of organic compounds, depending on the ecosystem properties.
In this study, we performed metagenomic analysis of soil microbial genes and investigated their responses to soil environmental factors in alpine steppes and meadows on the Tibetan Plateau. We hypothesized that (i) plant- and microbial-derived carbon decomposition potentials will be higher in alpine meadows than in alpine steppes, and (ii) the different carbon decomposition potentials may be due to differences in environmental factors, such as SWC, soil nutrients, and soil organic matter, between alpine steppes and meadows. Our aims were (i) to determine the trends of microbial CAZyme genes and the degradation potential of soil microbes between alpine steppes and meadows, and (ii) to investigate the major factors affecting microbial CAZyme genes in alpine steppes and meadows.
2. Material and Methods
2.1. Study Area and Soil Properties
Our study selected two types of alpine grasslands, alpine steppes and alpine meadows, on the Tibetan Plateau (Figure S1). For each grassland type, four sites were selected for multiple sampling. The alpine steppe at Site 1 was situated in Datong (latitude 37°08.93′ N, longitude 101°47.30′ E; altitude 2968 m above sea level), and the alpine meadow at Site 1 was situated in Datong (latitude 37°07.62′ N, longitude 101°47.83′ E; altitude 2882 m above sea level). The alpine steppe at Site 2 was situated in Gonghe (latitude 36°25.99′ N, longitude 100°53.64′ E; altitude 3244 m above sea level), and the alpine steppe at Site 2 was situated in Gonghe (latitude 36°26.08′ N, longitude 100°53.70′ E; altitude 3239 m above sea level). The alpine steppe at Site 3 was situated in Maqin (latitude 34°28.96′ N, longitude 100°12.59′ E; altitude 3738 m above sea level), and the alpine steppe at Site 3 was situated in Maqin (latitude 34°27.63′ N, longitude 100°14.30′ E; altitude 3727 m above sea level). The alpine steppe at Site 4 was situated in Maduo (latitude 34°52.41′ N, longitude 98°14.94′ E; altitude 4255 m above sea level), and the alpine steppe at Site 4 was situated in Maduo (latitude 34°51.73′ N, longitude 98°14.6159′ E; altitude 4212 m above sea level). The study area is characterized by an alpine, semi-humid, and semi-arid climate. The annual mean temperature (MAT) is −4~2 °C and annual mean precipitation (MAP) is 235~974.6 mm. Alpine grassland is the main vegetation type in the Tibetan alpine region, with the dominant species being Stipa purpurea and Carex moorcroftii for alpine steppes and Kobresia pygmaea and Kobresia humilis for alpine meadows; the major soil types are Xerosols for the alpine steppes and Cambisols for the alpine meadows [26].
2.2. Soil Sampling and Processing
To prevent a single season from being representative of the sample, we selected three seasons for the analysis: April, June, and August 2021. However, we did not find a significant difference in the CAZyme genes among the three seasons (Figure S2). Therefore, we considered the data from the three seasons as replicates (n = 9). The ecological factors (slope) were kept as consistent as possible when setting up the experimental plots. At each site, three experimental plots (10 × 10 m) were randomly selected for each grassland type. In each experimental plot, three 1 × 1 m quadrats were used to measure soil properties. Soil samples were collected through drills on the soil surface (0–5 cm) at each experimental plot. Rocks, roots, plants, animal residues, and other sundries were immediately removed, and the samples were rolled through a 2 mm sieve [18]. A portion of each freshly collected soil sample was stored at 4 °C and another portion was stored on dry ice at −80 °C and immediately transported from the field to the laboratory for carrying out soil physicochemical property analysis and subsequent DNA analysis, respectively [9].
Among the soil basic physicochemical properties, SWC (%) was determined by weighing the soil sample after drying it at 105 °C for 24 h (until the weight remained constant). The pH of each soil sample was measured in aqueous solution (soil:water, 1:2.5 w/v) using a pH meter. Soil organic carbon concentration (SOC, g C kg−1) was determined using the K2Cr2O7 oxidation method, total nitrogen content (TN, g kg−1) was determined using the Kjeldahl method, and total phosphorus content (TP, g kg−1) was determined using the Mo-Sb Anti spectrophotometric method, having initially digested the soil samples with H2SO4–H2O2 [27]. For determination of soil ammonium nitrogen (NH4+, mg kg−1) and nitrate nitrogen (NO3−, mg kg−1) concentrations, samples were extracted with a 2 mol L−1 KCl solution, and, following filtration, extracts were analyzed using a Dionex ICS 1500 ion chromatograph, as described in a previous study [28]. We selected physicochemical properties from the growing season in June.
2.3. DNA Extraction and Sequencing
Total microbial genomic DNA was extracted using the OMEGA Soil DNA Kit (Omega Bio-tek, Norcross, GA, USA) in accordance with the manufacturer’s instructions. The purity and concentration of the extracted DNA were measured using NanoDrop2000 and TBS-380, respectively [29]. The quality of the extracted DNA was examined using 1% agarose gel. The extracted microbial DNA was fragmented to an average size of approximately 400 bp using a Covaris M220 (Gene Company Limited, Shanghai, China) for paired-end library construction. A paired-end library was established using the NEXTFLEX® Rapid DNA Seq (Bioo Scientific, Austin, TX, USA). Adapters containing the full complement of the sequencing primer hybridization sites were ligated into the blunt ends of the fragments. Paired-end sequencing was performed on an Illumina Novaseq 6000 (Illumina Inc., San Diego, CA, USA) at Majorbio Bio-Pharm Technology Co., Ltd. (Shanghai, China), using NovaSeq Reagent Kits in accordance with the manufacturer’s instructions (www.illumina.com, accessed on 20 June 2023). The sequences were obtained from the National Center for Biotechnology Information website (PRJNA916885).
2.4. Metagenomics Analysis
The raw sequencing reads were processed to obtain quality-filtered reads for further analysis. Briefly, paired-end Illumina reads were trimmed of adaptors, and low-quality reads (length < 50 bp, quality value < 20, or having N bases) were removed using fastp [30] (https://github.com/OpenGene/fastp, version 0.20.0, accessed on 20 June 2023). The metagenomic data were constructed using MEGAHIT [31] (https://github.com/voutcn/megahit, version 1.1.2, accessed on 20 June 2023), which uses succinct de Bruijn graphs. Contigs were selected when the length was ≥300 bp as the final assembling result, and then the contigs were used for further gene prediction and annotation. Open reading frames (ORFs) from each assembled contig were predicted using Prodigal/MetaGene [32] (http://metagene.cb.k.u-tokyo.ac.jp/, accessed on 20 June 2023). Predicted ORFs with a length ≥ 100 bp were searched and translated into amino acid sequences using the NCBI translation table (http://www.ncbi.nlm.nih.gov/Taxonomy/taxonomyhome.html/index.cgi?chapter=tgencodes#SG1, accessed on 20 June 2023). A non-redundant gene catalog was constructed using CD-HIT [33] (http://www.bioinformatics.org/cd-hit/, version 4.6.1, accessed on 20 June 2023) with 90% coverage and 90% sequence identity. High-quality reads were aligned to non-redundant gene catalogs to calculate gene abundance with 95% identity using SOAPaligner [34] (http://soap.genomics.org.cn/, version 2.21, accessed on 20 June 2023).
2.5. CAZyme Annotation
Carbohydrate-active enzyme annotation was established using hmmscan (http://hmmer.janelia.org/search/hmmscan, accessed on 20 June 2023) against the CAZy database (http://www.cazy.org/, accessed on 20 June 2023) with an e-value cutoff of 1 × 10−5, followed by obtaining carbohydrate-active enzyme annotation information for a gene. The abundance of carbohydrate-active enzymes was calculated by applying the sum of gene abundances corresponding to carbohydrate-active enzymes, and transcripts per kilobase per million mapped reads (TPM) were employed to normalize the abundance value in the metagenome.
2.6. Statistical Analysis
Before analysis, all data were tested for normal distribution. Non-metric multidimensional scaling (NMDS) based on Bray–Curtis distance was used to reveal changes in the soil microbial diversity and CAZyme gene structure. Significant differences in the relative abundance of genes coding for CAZyme between alpine steppes and meadows were determined by Wilcoxon test by ‘stats’ package in R v4.2.2. Spearman analysis was conducted to identify the relationship between the abundance of CAZymes involved in the degradation of plant- and microbial-derived components and environmental factors. Mantel tests were conducted to identify the relationship between the microorganism involved in the degradation of plant- and microbial-derived components and environmental factors, which was analyzed by ‘linkET’ package in R v4.2.2. Significant differences in abundance of CAZyme genes in season were determined by one way ANOVA. In microbial composition, we selected the abundance and accounted for the top eleven, which was calculated as the percentage of microbial group; the other represented the rest of the microbes. All graphs were drawn using the ‘ggplot2′ package in R v4.2.2. All plots were drawn using Adobe Illustrator 2020.
3. Results
3.1. Characteristics of CAZyme Encoding Genes in the Alpine Steppes and Meadows
At the family level, 599 CAZyme encoding genes were detected in all normalized metagenomes, including 24 auxiliary enzymes (AAs), 97 polysaccharide lyases (PLs), 16 carbohydrate esterases (CEs), 106 glycosyltransferases (GTs), 65 carbohydrate-binding modules (CBMs), and 291 glycoside hydrolases (GHs). GTs were the most abundant enzymes in the alpine steppes and meadows, followed by GHs, CEs, AAs, CBMs, and PLs. NMDS showed that the structure of CAZyme-encoding genes in the alpine steppes was significantly different from that in the alpine meadows (Figure 1). The abundance of AAs, CBMs, and GTs were significantly different between the alpine steppes and meadows (Figure 2A, p < 0.001), and all four sites showed the same trends (Figure S3). The abundance of AAs genes was higher in the alpine steppes than in the alpine meadows (4404.01 vs. 3524.48 TPM) (Figure 2A, p < 0.001). The gene abundance of AAs had a negative relationship with NO3− (p < 0.01) in the alpine steppes and a positive relationship with SON (p < 0.01) and a negative relationship with SWC (p < 0.05) in the alpine meadows (Figure 2B). The abundances of CBMs (1062.66 vs. 1203.08 TPM) and GTs (10,033.99 vs. 11,682.77 TPM) were lower in the alpine steppes than in the alpine meadows (Figure 2A, p < 0.001). The gene abundance of CBMs was positively correlated with NH4+ (p < 0.01), SON (p < 0.05), SOC (p < 0.001), and TN (p < 0.01); it was negatively correlated with pH (r = −0.60, p < 0.001) in the alpine steppes and positively correlated with SWC (p < 0.05) in the alpine meadows (Figure 2B). The gene abundance for GTs had a positive relationship with NO3− (p < 0.01) in the alpine steppes. There were no significant differences between the CEs, GHs, or PLs in the alpine meadows and steppes (Figure 2A). The gene abundance of CEs was negatively correlated with pH (p < 0.01) and positively correlated with SOC (p < 0.01) and TN (p < 0.05) in the alpine steppes and positively correlated with SON (p < 0.05) in the alpine meadows (Figure 2B). The gene abundance of GHs had a negative relationship with NO3− (p < 0.05) and a positive relationship with pH (p < 0.05) in the alpine steppes and a negative relationship with TP (p < 0.05) in the alpine meadows (Figure 2B). The gene abundance of PLs was negatively correlated with SWC (p < 0.01), NH4+ (r = −0.63, p < 0.001), SON (p < 0.05), SOC (r = −0.61, p < 0.001), and TN (r = −0.65, p < 0.001) and positively correlated with pH and NO3− (p < 0.05) in the alpine steppes (Figure 2B).
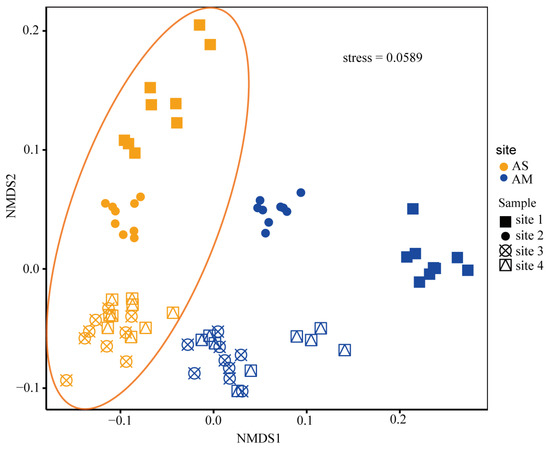
Figure 1.
Nonmetric multidimensional scaling (NMDS) based on Bray–Curtis dissimilarity displaying samples in the alpine steppes and meadows. Different shapes show different soil sampling points, orange and blue represent alpine steppes and alpine meadows, respectively. AS and AM represent alpine steppes and alpine meadows in all figures. Site 1, site 2, site 3, and site 4 represent sampling sites situated in Datong, Gonghe, Maqin, and Maduo, respectively.
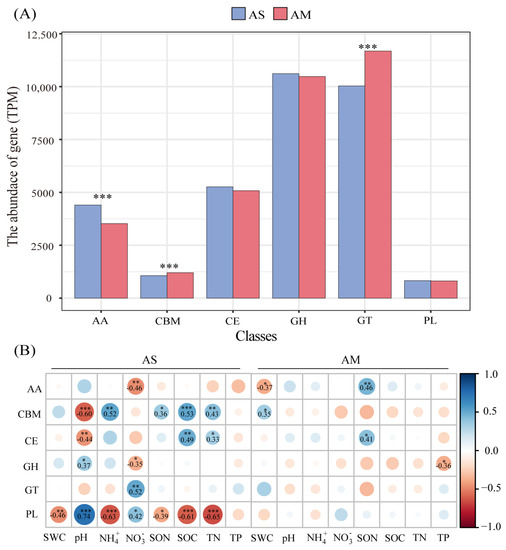
Figure 2.
The abundance of CAZyme genes at class level in the alpine steppes and meadows (A). AAs: Auxiliary activities, CBMs: Carbohydrate-binding module, CEs: Carbohydrate esterase, GHs: Glycoside hydrolase, GTs: Glycosyl transferase, PLs: Polysaccharidelyase, CEs: Carbohydrate esterase. Significant p values as tested by Mann–Wilcoxon test are indicated (*, p < 0.05; **, p < 0.01; ***, p < 0.001). Relationships between the abundance of CAZyme families and environmental factors based on Spearman (significance: *, p < 0.05; **, p < 0.01; ***, p < 0.001) (B).
3.2. Characteristics of CAZyme Encoding Genes Involved in the Degradation of Plant- and Microbial-Derived Components in the Alpine Steppes and Meadows
The main CAZyme families encoding the enzymatic activities were divided into the degradation of plant- and microbial-derived components according to CAZy (Table S1). Among them, 37 CAZymes encoding genes were involved in the degradation of plant-derived components, 10 were involved in the degradation of fungal-derived components, and 9 were involved in the degradation of bacterial-derived components. In addition, we found different trends in gene abundance for the decomposition of plant- and microbial-derived carbon in the alpine steppes and meadows, and the four sites showed the same trends (Figure S4A). The most abundant CAZyme-encoding genes were AA3, GH16, and GH23, which were involved in the degradation of plant-, fungal-, and bacterial-derived components, respectively (Figure S4B–D). Specifically, gene abundances for the decomposition of plant- (4742.97 vs. 4363.52 TPM) and fungal-derived carbon (342.42 vs. 310.93 TPM) were higher in the alpine steppes than in the alpine meadows, respectively (Figure 3A, p < 0.001 and p < 0.05). Gene abundance for the decomposition of plant-derived carbon (cellulose, hemicellulose, and lignin) showed a positive relationship with pH (p < 0.01) in the alpine steppes and a negative relationship with SWC (p < 0.01) in the alpine meadows (Figure S5). Gene abundance for the decomposition of fungal-derived carbon (chitin and glucans) was positively correlated with pH (p < 0.01) in the alpine steppes and positively correlated with NO3− (p < 0.01) and negatively correlated with pH (p < 0.05) in the alpine meadows (Figure S5). Gene abundance for the decomposition of bacterial-derived carbon (756.40 vs. 696.86 TPM) was lower in the alpine steppes than in the alpine meadows (Figure 3A, p < 0.001). Gene abundance for the decomposition of bacterial-derived carbon (peptidoglycan) was negatively correlated with NO3− (p < 0.01) in the alpine steppes (Figure S5). Moreover, the cumulative abundance of CAZyme-encoding genes involved in the degradation of plant-derived carbon was higher than that of microbial-derived carbon (Figure 3A).
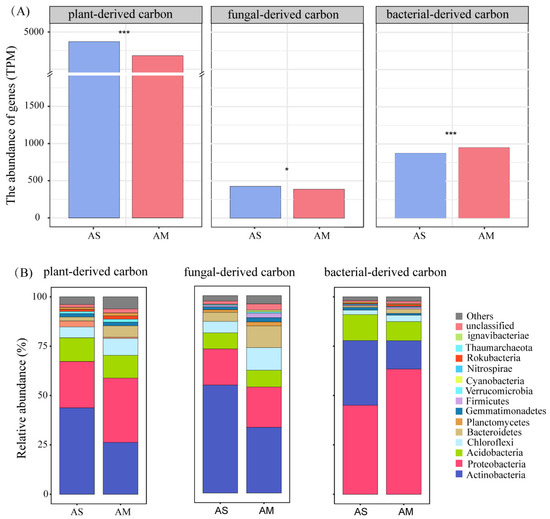
Figure 3.
The abundance of genes encoding enzymes degrading plant-, fungal-, and bacterial-derived carbon between alpine steppes and meadows (A). Significant p values as tested by the Mann–Wilcoxon test are indicated (*, p < 0.05; ***, p < 0.001). Changes of microbial composition with decomposing plant-, fungal-, and bacterial-derived carbon in the alpine steppes and meadows (B). Microbial composition shown at the phylum level; the abundance of each taxon (accounted for the top eleven) was calculated as the percentage of microbial group. Different colors represent different phylum.
3.3. Changes of Microbial Composition and Diversity Involved in the Degradation of Plant- and Microbial-Derived Components in the Alpine Steppes and Meadows
Our results indicated that Actinobacteria, Proteobacteria, Acidobacteria, and Chloroflexi were the dominant bacterial phyla decomposing plant-, fungal-, and bacterial-derived carbon (Figure 3B). Among them, Actinobacteria and Proteobacteria accounted for more than 60% in all groups. The relative abundances of Actinobacteria in decomposing plant-, fungal-, and bacterial-derived carbon accounted for 43.7%, 54.6%, and 32.8% in the alpine steppes and 26.2%, 33.2%, and 14.4% in the alpine meadows, respectively. The relative abundance of Proteobacteria in decomposing plant-, fungal-, and bacterial-derived carbon accounted for 23.5%, 18.3%, and 45.0% in the alpine steppes and 32.6%, 20.5%, and 63.4% in the alpine meadows, respectively (Figure 3B). In addition, Acidobacteria and Chloroflexi accounted for more than 10% in all groups. Specifically, the relative abundances of Acidobacteria in decomposing plant-, fungal-, and bacterial-derived carbon accounted for 12.0%, 8.2%, and 13.1% in the alpine steppes and 11.6%, 8.4%, and 9.6% in the alpine meadows, respectively. The relative abundance of Chloroflexi in decomposing plant-, fungal-, and bacterial-derived carbon accounted for 5.6%, 5.8%, and 2.3% in the alpine steppes and 8.6%, 11.4%, and 3.3% in the alpine meadows, respectively (Figure 3B).
As shown in Figure S6A, the Shannon index values indicated the diversity of microbial communities in decomposing plant-, fungal-, and bacterial-derived carbon in the alpine steppes and meadows. The results showed that the Shannon index of the microbial communities was higher in decomposing bacterial-derived carbon than in plant- and fungal-derived carbon. In addition, the Shannon index of the microbial communities’ decomposing plant-derived carbon was not significantly different between the alpine steppes and meadows, but decomposing fungal-derived carbon (p < 0.001) and bacterial-derived carbon (p < 0.05) were significantly higher in the alpine steppes than in the alpine meadows. NMDS analysis further illustrated that the soil microbial communities in decomposing plant-, fungal-, and bacterial-derived carbon in the alpine steppes were separated from those in the alpine meadows (Figure S6B–D).
3.4. Relationships between Soil Microbial Communities and Soil Properties
In the alpine steppes, Mantel test analysis showed that SWC, pH, SOC, and TN (Mantel’s r < 0.2, Mantel’s p < 0.05) were significantly related to the microorganism degradation of plant-derived carbon. In addition, pH, SON, SOC, and TN (Mantel’s r > 0.2, Mantel’s p < 0.05) had a stronger correlation with the microorganism degradation of bacterial-derived carbon (Figure 4A). Soil pH was an important factor influencing plant-derived carbon decomposition and had a strong correlation with microorganism degradation of fungal-derived carbon decomposition (Mantel’s r > 0.2, Mantel’s p < 0.01). In the alpine meadows, SWC, pH, SOC, and TN (Mantel’s r < 0.2, Mantel’s p < 0.05) were strongly correlated with the microbial degradation of plant-derived carbon. Only SWC (Mantel’s r < 0.2, Mantel’s p < 0.05) was significantly related to microbial degradation during bacterial-derived carbon decomposition. SWC, pH, NH4+, SOC, and TN (Mantel’s r < 0.2, Mantel’s p < 0.05) were correlated with the microbial degradation of fungal-derived carbon (Figure 4B).
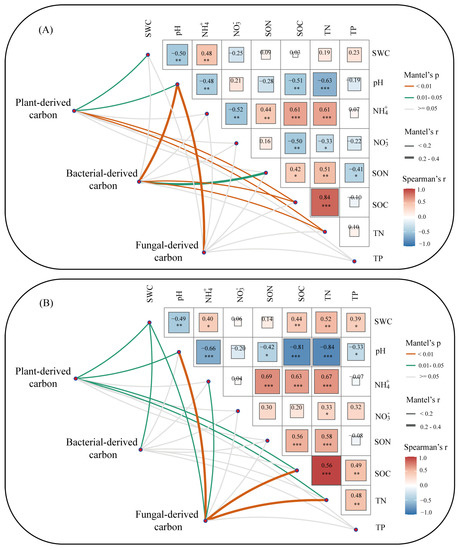
Figure 4.
Microbial community composition was related to each environmental factor by partial Mantel test in the alpine steppes (A) and meadows (B). Edge width corresponds to the Mantel’s r statistic for the corresponding distance correlations, and edge color denotes the statistical significance. Spearman’s correlation based on pairwise comparisons of environmental factors (*, p < 0.05; **, p < 0.01; ***, p < 0.001).
4. Discussion
4.1. Characteristics of CAZyme-Encoding Genes in Alpine Steppes and Meadows
Our results showed that the abundance of genes encoding GTs and CBMs in the alpine steppes was significantly lower than that in the alpine meadows on the Tibetan Plateau, whereas the abundance of genes encoding AAs was higher in the alpine steppes (Figure 2A, p < 0.001). Higher abundance of GTs and CBMs indicated higher biosynthesis (e.g., coding for the biosynthesis of disaccharides, oligosaccharides, and polysaccharides) [18] and more carbohydrate enzymes in alpine steppes [20]. AAs are associated with lignin, which manifested higher decomposition potential in alpine meadows [21,22]. Our results can be attributed to three factors. Firstly, compared with alpine steppes, alpine meadows had better vegetation conditions (more litter and roots), which can lead to higher soil substrates and improve microbial activities by releasing a wide variety of compounds, including ethylene, sugars, vitamins, amino acids, organic acids, and polysaccharides [35,36,37,38], thereby enhancing biosynthesis potential. This is consistent with a previous study’s findings that GTs are abundant in the organic layer of litter-rich soil [17]. Secondly, soil nutrients are necessary for microbial metabolism, boost soil microbial growth and activities [37], and promote the secretion of CAZymes by soil microbes. Compared to alpine steppes, soils in alpine meadows were nutrient-rich (NH4+ and NO3−) [39] (Table S2), and our results showed that CAZymes (CBMs and GTs) were significantly positively correlated with NH4+ and NO3− (Figure 1B). Thirdly, higher soil water content may have resulted in a lower abundance of genes encoding AAs in the alpine meadows. For example, high SWC may cause anaerobic fungi to lack the enzymatic machinery to catabolize or mineralize lignin. The enzymatic reaction cleaves the aromatic ring requiring oxygen or its partially reduced species, resulting in a lower microbial decomposition potential [21]. Confirming this expectation, our results showed that AA levels were significantly and negatively correlated with SWC in alpine meadows (Figure 2B).
4.2. Characteristics of CAZyme-Encoding Genes Involved Plant- and Microbial-Derived Carbon Decomposition and Soil Microbial Communities in the Alpine Steppes and Meadows
Our results showed that the abundance of CAZyme genes encoding plant-derived carbon degradation was significantly higher in the alpine steppes than in the alpine meadows (Figure 3A, p < 0.001). This is inconsistent with previous evidence showing that higher soil organic matter and favorable moisture and nutrient conditions in alpine meadows favor microbes, which would result in faster decomposition of plant litter [39,40]. One possible reason is the influence of the extreme environment of the Tibetan Plateau [8]. A previous study also found that sufficient plant C inputs can drive microorganisms to utilize more recalcitrant C, but this study did not include ecosystems under extreme climates [18]. Previous studies have shown that temperature is the primary factor affecting CAZyme genes that target recalcitrant plant-derived carbon and fungal biomass turnover and that the microbial decomposition potential of organic C increases with increasing temperature [16,18,41]. Another possible reason was higher pH (8.07 vs. 7.03) in the alpine steppes than in the alpine meadows. GHs are mostly involved in the decomposition of plant-derived carbon (Table S1), and GH activities are very pH-dependent because pH variation boosts changes in folding, conformation, and protonation of enzymes [24]. The CAZyme-encoding genes involved in plant-derived carbon decomposition are positively related with pH in alpine steppes, and a previous study indicated soil pH increases may accelerate the microbial decomposition of recalcitrant organic C [18].
In addition, our results showed that the abundance of CAZyme genes encoding fungal-derived carbon was significantly higher in the alpine steppes than in the alpine meadows (Figure 3A, p < 0.05), whereas the abundance of CAZyme genes encoding bacterial-derived carbon was significantly lower in the alpine steppes than in the alpine meadows (Figure 3A, p < 0.001). In line with our results, previous evidence indicates that the turnover mechanisms of fungal and bacterial cell walls are different [14], thereby affecting fungal-derived carbon and bacterial-derived carbon decomposition potential. First, peptidoglycan, a major and universal component of bacterial cell walls that decomposes, was correlated with soil microbial biomass (the microbial biomass in alpine steppes was lower than that in alpine meadows) and was primarily decomposed to muropeptides in soils, which can be directly utilized by microbes without further depolymerization to free amino compounds [13,42,43]. However, fungal biomass fractions are highly recalcitrant compared to peptidoglycan decomposition [44]. Additionally, some β-glucans may indirectly increase the decomposability of dead fungal biomass by affecting water availability. β-glucans can increase the water holding capacity of the fungal cell wall, which is incredibly important when water availability is a limiting factor (e.g., alpine steppes) [45,46]. Therefore, bacterial-derived carbon may have a significantly higher decomposition potential in alpine meadows, whereas fungal-derived carbon has a higher decomposition potential in the alpine steppes. A previous study conducted an in situ decomposition experiment that demonstrated that bacterial- and fungal-derived carbon decomposition correlated with microbial biomass and soil texture, respectively [13]. Alpine meadows have higher microbial biomass than alpine steppes [39], which may result in higher bacteria-derived carbon decomposition in alpine meadows.
Our results showed that most gene-encoding enzymes for decomposing plant- and microbial-derived carbon were attributed to Actinobacteria, Proteobacteria, Acidobacteria, and Chloroflexi (Figure 3B), which can produce CAZymes of decomposing recalcitrant organic compounds such as lignin, chitin, cellulose, and so on [16,47]. Actinobacteria is a dominant phylum of degrading plant- and fungal-derived carbon through secreting β-glucosidase and xylanases, which usually lives well in lower soil water content and aerobic conditions [17,48]. Specifically, cellulose degradation in Actinobacteria has concentrated on two model organisms, Thermobifida fusca and Cellulomonas fimi. The system of T. fusca consists of three non-processive endocellulases (E1/Cel9B, E2/Cel6A, E5/Cel5A), two exocellulases (E3/Cel6B and E6/Cel48A), and one processive endocellulase (E4/Cel9A), which cleave cellulose at random sites along cellulose chains [49]. Another system combines features of both endo- and exo-type enzymes that made an initial endocellulolytic cleavage, followed by the release of cellotetraose units from the cleaved substrate [50]. Therefore, a lower soil water content could significantly facilitate the growth of Actinobacteria and indirectly affect the abundance of genes encoding the degradation of plant- and fungal-derived carbon in alpine steppes. Moreover, Proteobacteria was the most abundant phylum degrading bacterial-derived carbon (higher relative abundance in the alpine meadows than in the alpine steppes), with rapid turnover and favorable growth in soils richer in carbon because it is a symbiotic bacterium that can grow rapidly [51], thus improving bacterial-derived carbon (indicated by peptidoglycan) decomposition in the alpine meadows. Chloroflexi predominantly contributes to the hydrolysis of the internal glycosidic linkages of the heteroxylan backbone (endoxylanase) [48].
5. Conclusions
This study investigated the differences in functional genes relevant to C decomposition between alpine steppes and meadows on the Tibetan Plateau using metagenomic analyses. The abundance of AA genes was higher in the alpine steppes than in the alpine meadows, whereas CBMs and GTs were lower in the alpine steppes. Additionally, specific microbial CAZyme families with plant- and fungal-derived carbon decomposition were significantly higher in the alpine steppes than in the alpine meadows, whereas bacterial-derived carbon decomposition was lower in the alpine steppes. Actinobacteria and Proteobacteria were dominant in the decomposing plant and microbial biomass, which accounted for more than 60% of all groups. Soil properties, especially soil pH, SWC, and NO3−, are related to the composition of soil microbial communities and influence on the biodegradation potential of plants and dead microbial biomass. In future studies, to better study the number of substances specifically decomposed or synthesized by enzyme family genes, we need to combine the data of carbon mineralization with CAZyme genes by using metagenomics and macro-transcriptomics. Nonetheless, this paper was the first study that compared plant- and microbial-derived carbon decomposition potentials and influencing factors to illustrate the contribution of dead biomass to carbon accumulation in alpine grasslands on the Tibetan Plateau. Additionally, these findings promote our understanding of functional genes in association with ecosystem functions between alpine grasslands.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/f14081580/s1: Figure S1: Sampling sites of alpine grasslands on the Tibetan Plateau; Figure S2: The abundance of CAZyme genes at class level on seasonal variations in April, June and August; Figure S3: The abundance of CAZyme genes at class level between in the alpine steppes and meadows; Figure S4: The abundance of gene encoding enzymes for plant-, bacterial- and fungal-derived carbon decomposition of soil sampling points between in the alpine steppes and meadows (A). Abundance of selected GHs and AAs encoding the decomposition of the plant- (B), bacterial- (C) and fungal-derived carbon (D) between in the alpine steppes and meadows; Figure S5: Relationships between the abundance of genes encoding enzymes degrading plant-, bacterial-, fungal-derived carbon and environmental factors based on Spearman (significance: *, p < 0.05; **, p < 0.01; ***, p < 0.001); Figure S6: Change of microbial diversity with decomposing plant-, bacterial- and fungal-derived carbon in the alpine steppes and meadows; Table S1: The main CAZyme families involved in the decomposition of plant-derived and microbial-derived carbon in this study; Table S2: Mean ± SE (standard error) values soil properties between in the alpine steppes and meadows.
Author Contributions
F.Z., W.W. and N.W. conceived the project; Y.Y. and Y.L. performed the data compilation and analysis; C.R., Y.G. and J.W. (Jun Wang) interpreted the results; Y.Y., L.C. and J.W. (Jieying Wang) wrote the manuscript with assistance of all other coauthors. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. 42277284); the 2021 first funds for central government to guide local science and technology development in Qinghai Province (No. 2021ZY002); and the Second Tibetan Plateau Scientific Expedition and Research Program and Research (2019QZKK020102; 2019QZKK0302).
Data Availability Statement
All the sequencing data have been deposited on the National Center for Biotechnology Information website (PRJNA916885). The data and scripts used are saved in GitHub https://github.com/yyhhyj/code.git (accessed on 20 June 2023).
Conflicts of Interest
The authors have declared no competing interest.
References
- Liang, W.; Lü, Y.; Zhang, W.; Li, S.; Jin, Z.; Ciais, P.; Fu, B.; Wang, S.; Yan, J.; Li, J.; et al. Grassland gross carbon dioxide uptake based on an improved model tree ensemble approach considering human interventions: Global estimation and covariation with climate. Glob. Chang. Biol. 2017, 23, 2720–2742. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xiao, J.; Ma, Y.; Luo, Y.; Hu, Z.; Li, F.; Li, Y.; Gu, L.; Li, Z.; Yuan, L. Carbon fluxes and environmental controls across different alpine grassland types on the Tibetan Plateau. Agric. For. Meteorol. 2021, 311, 108694. [Google Scholar] [CrossRef]
- Zhao, Y.F.; Wang, X.; Jiang, S.L.; Zhou, X.H.; Liu, H.Y.; Xiao, J.J.; Hao, Z.G.; Wang, K.C. Climate and geochemistry interactions at different altitudes influence soil organic carbon turnover times in alpine grasslands. Agric. Ecosyst. Environ. 2021, 320, 107591. [Google Scholar] [CrossRef]
- Chen, H.; Zhu, Q.A.; Peng, C.H.; Wu, N.; Wang, Y.F.; Fang, X.; Gao, Y.H.; Zhu, D.; Yang, G.; Tian, J.Q.; et al. The Impacts of Climate Change and Human Activities on Biogeochemical Cycles on the Qinghai-Tibetan Plateau. Glob. Chang. Biol. 2013, 19, 2940–2955. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Feng, J.; Yuan, X.; Zhu, B. Effects of warming on carbon and nitrogen cycling in alpine grassland ecosystems on the Tibetan Plateau: A meta-analysis. Geoderma 2020, 370, 114363. [Google Scholar] [CrossRef]
- Xie, Z.; Zhu, J.; Liu, G.; Cadisch, G.; Hasegawa, T.; Chen, C.; Sun, H.; Tang, H.; Zeng, Q. Soil organic carbon stocks in China and changes from 1980s to 2000s. Glob. Chang. Biol. 2007, 13, 1989–2007. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhan, T.; Li, Y.; Wang, Y.; Yu, T.; Sun, J. Soil organic carbon stock responded more sensitively to degradation in alpine meadows than in alpine steppes on the Qinghai-Tibetan Plateau. Land Degrad. Dev. 2022, 34, 353–361. [Google Scholar] [CrossRef]
- Chen, H.; Ju, P.; Zhu, Q.; Xu, X.; Wu, N.; Gao, Y.; Feng, X.; Tian, J.; Niu, S.; Zhang, Y.; et al. Carbon and nitrogen cycling on the Qinghai–Tibetan Plateau. Nat. Rev. Earth Environ. 2022, 3, 701–716. [Google Scholar] [CrossRef]
- Ren, C.; Zhang, X.; Zhang, S.; Wang, J.; Xu, M.; Guo, Y.; Wang, J.; Han, X.; Zhao, F.; Yang, G.; et al. Altered microbial CAZyme families indicated dead biomass decomposition following afforestation. Soil Biol. Biochem. 2021, 160, 108362. [Google Scholar] [CrossRef]
- Jiménez, D.J.; de Lima Brossi, M.J.; Schückel, J.; Kračun, S.K.; Willats, W.G.T.; van Elsas, J.D. Characterization of three plant biomass-degrading microbial consortia by metagenomics- and metasecretomics-based approaches. Appl. Microbiol. Biotechnol. 2016, 100, 10463–10477. [Google Scholar] [CrossRef]
- Fesel, P.H.; Zuccaro, A. Beta-Glucan: Crucial Component of the Fungal Cell Wall and Elusive Mamp in Plants. Fungal Genet. Biol. 2016, 90, 53–60. [Google Scholar] [CrossRef]
- Free, S.J. Fungal Cell Wall Organization and Biosynthesis. In Advances in Genetics; Friedmann, T., Dunlap, J.C., Goodwin, S.F., Eds.; Elsevier: Amsterdam, The Netherlands, 2013; Volume 81, pp. 33–82. [Google Scholar]
- Hu, Y.; Zheng, Q.; Noll, L.; Zhang, S.; Wanek, W. Direct measurement of the in situ decomposition of microbial-derived soil organic matter. Soil Biol. Biochem. 2020, 141, 107660. [Google Scholar] [CrossRef]
- Gunina, A.; Dippold, M.; Glaser, B.; Kuzyakov, Y. Turnover of Microbial Groups and Cell Components in Soil: 13C Analysis of Cellular Biomarkers. Biogeosciences 2017, 14, 271–283. [Google Scholar] [CrossRef]
- Eichorst, S.A.; Kuske, C.R. Identification of Cellulose-Responsive Bacterial and Fungal Communities in Geographically and Edaphically Different Soils by Using Stable Isotope Probing. Appl. Environ. Microbiol. 2012, 78, 2316–2327. [Google Scholar] [CrossRef] [PubMed]
- Žifčáková, L.; Větrovský, T.; Lombard, V.; Henrissat, B.; Howe, A.; Baldrian, P. Feed in summer, rest in winter: Microbial carbon utilization in forest topsoil. Microbiome 2017, 5, 122. [Google Scholar] [CrossRef]
- Yin, F.; Zhang, F. Reclamation of Abandoned Saline-Alkali Soil Increased Soil Microbial Diversity and Degradation Potential. Plant Soil 2022, 477, 521–538. [Google Scholar] [CrossRef]
- Dai, Z.; Zang, H.; Chen, J.; Fu, Y.; Wang, X.; Liu, H.; Shen, C.; Wang, J.; Kuzyakov, Y.; Becker, J.N.; et al. Metagenomic Insights into Soil Microbial Communities Involved in Carbon Cycling Along an Elevation Climosequences. Environ. Microbiol. 2021, 23, 4631–4645. [Google Scholar] [CrossRef]
- Yip, V.L.; Withers, S.G. Breakdown of oligosaccharides by the process of elimination. Curr. Opin. Chem. Biol. 2006, 10, 147–155. [Google Scholar] [CrossRef]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes database (CAZy): An expert resource for Glycogenomics. Nucleic Acids Res. 2009, 37, D233–D238. [Google Scholar] [CrossRef]
- Oh, H.N.; Park, D.; Seong, H.J.; Kim, D.; Sul, W.J. Antarctic tundra soil metagenome as useful natural resources of cold-active lignocelluolytic enzymes. J. Microbiol. 2019, 57, 865–873. [Google Scholar] [CrossRef]
- Lombard, V.; Golaconda Ramulu, H.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, D490–D495. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, L.; Yang, J.F.; Duan, Y.M.; Luo, Y.; Taherzadeh, M.J.; Li, Y.F.; Li, H.K.; Awasthi, M.K.; Zhao, Z.Y. The diversity of microbial community and function varied in response to different agricultural residues composting. Sci. Total. Environ. 2020, 715, 136983. [Google Scholar] [CrossRef] [PubMed]
- Bernardes, A.; Pellegrini, V.O.A.; Curtolo, F.; Camilo, C.M.; Mello, B.L.; Johns, M.A.; Scott, J.L.; Guimaraes, F.E.C.; Polikarpov, I. Carbohydrate binding modules enhance cellulose enzymatic hydrolysis by increasing access of cellulases to the substrate. Carbohydr. Polym. 2019, 211, 57–68. [Google Scholar] [CrossRef]
- Li, J.; He, N.; Wei, X.; Gao, Y.; Zuo, Y. Changes in Temperature Sensitivity and Activation Energy of Soil Organic Matter Decomposition in Different Qinghai-Tibet Plateau Grasslands. PLoS ONE 2015, 10, e0132795. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Chen, L.; Ji, C.; Hugelius, G.; Li, Y.; Liu, L.; Qin, S.; Zhang, B.; Yang, G.; Li, F.; et al. Decadal soil carbon accumulation across Tibetan permafrost regions. Nat. Geosci. 2017, 10, 420–424. [Google Scholar] [CrossRef]
- Wang, J.Y.; Ren, C.J.; Feng, X.X.; Zhang, L.; Doughty, R.; Zhao, F.Z. Temperature sensitivity of soil carbon decomposition due to shifts in soil extracellular enzymes after afforestation. Geoderma 2020, 374, 114426. [Google Scholar] [CrossRef]
- Zhang, J.J.; Li, Y.F.; Chang, S.X.; Jiang, P.K.; Zhou, G.M.; Liu, J.; Wu, J.S.; Shen, Z.M. Understory vegetation management affected greenhouse gas emissions and labile organic carbon pools in an intensively managed Chinese chestnut plantation. Plant Soil 2014, 376, 363–375. [Google Scholar] [CrossRef]
- Wu, F.; Ding, X.; Zhang, Y.; Gu, J.D.; Liu, X.; Guo, Q.; Li, J.; Feng, H. Metagenomic and Metaproteomic Insights into the Mi-crobiome and the Key Geobiochemical Potentials on the Sandstone of Rock-Hewn Beishiku Temple in Northwest China. Sci. Total Environ. 2023, 893, 164616. [Google Scholar] [CrossRef]
- Chen, S.F.; Zhou, Y.Q.; Chen, Y.R.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Li, D.H.; Liu, C.-M.; Luo, R.B.; Sadakane, K.; Lam, T.-W. Megahit: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Noguchi, H.; Park, J.; Takagi, T. MetaGene: Prokaryotic gene finding from environmental genome shotgun sequences. Nucleic Acids Res. 2006, 34, 5623–5630. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Li, R.Q.; Li, Y.R.; Kristiansen, K.; Wang, J. SOAP: Short oligonucleotide alignment program. Bioinformatics 2008, 24, 713–714. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yi, S.; Wu, Q.; Yang, K.; Ding, Y. The role of permafrost and soil water in distribution of alpine grassland and its NDVI dynamics on the Qinghai-Tibetan Plateau. Glob. Planet. Change 2016, 147, 40–53. [Google Scholar] [CrossRef]
- Ren, C.J.; Chen, J.; Deng, J.; Zhao, F.Z.; Han, X.H.; Yang, G.; Tong, X.G.; Feng, Y.Z.; Shelton, S.; Ren, G.X. Response of microbial diversity to C:N:P stoichiometry in fine root and microbial biomass following afforestation. Biol. Fertil. Soils 2017, 53, 457–468. [Google Scholar] [CrossRef]
- Xu, M.; Gao, D.; Fu, S.; Lu, X.; Wu, S.; Han, X.; Yang, G.; Feng, Y. Long-term effects of vegetation and soil on the microbial communities following afforestation of farmland with Robinia pseudoacacia plantations. Geoderma 2020, 367, 114263. [Google Scholar] [CrossRef]
- Garbeva, P.; van Veen, J.A.; van Elsas, J. Microbial Diversity in Soil: Selection of Microbial Populations by Plant and Soil Type and Implications for Disease Suppressiveness. Annu. Rev. Phytopathol. 2004, 42, 243–270. [Google Scholar] [CrossRef]
- Wang, S.; Jiao, C.; Zhao, D.; Zeng, J.; Xing, P.; Liu, Y.; Wu, Q.L. Disentangling the Assembly Mechanisms of Bacterial Communities in a Transition Zone between the Alpine Steppe and Alpine Meadow Ecosystems on the Tibetan Plateau. Sci. Total. Environ. 2022, 847, 157446. [Google Scholar] [CrossRef]
- Xu, S.; Li, P.; Sayer, E.J.; Zhang, B.; Wang, J.; Qiao, C.; Peng, Z.; Diao, L.; Chi, Y.; Liu, W.; et al. Initial Soil Organic Matter Content Influences the Storage and Turnover of Litter, Root and Soil Carbon in Grasslands. Ecosystems 2018, 21, 1377–1389. [Google Scholar] [CrossRef]
- Stone, M.M.; Weiss, M.S.; Goodale, C.L.; Adams, M.B.; Fernandez, I.J.; German, D.P.; Allison, S.D. Temperature sensitivity of soil enzyme kinetics underN-fertilization in two temperate forests. Glob. Change Biol. 2011, 18, 1173–1184. [Google Scholar] [CrossRef]
- Chen, X.; Tian, Y.; Zhang, Y.; Cui, Y.; Zhao, Y.; Sun, W. Plant community dynamics during the growing season of typical ecosystems on the Tibetan Plateau. Geogr. Sustain. 2020, 1, 266–274. [Google Scholar] [CrossRef]
- Egan, A.J.F.; Cleverley, R.M.; Peters, K.; Lewis, R.J.; Vollmer, W. Regulation of bacterial cell wall growth. FEBS J. 2017, 284, 851–867. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Xu, Y.-Z.; Han, X.-Z.; He, H.-B.; Zhang, X.-D.; Zhang, B. Fungi contribute more than bacteria to soil organic matter through necromass accumulation under different agricultural practices during the early pedogenesis of a Mollisol. Eur. J. Soil Biol. 2015, 67, 51–58. [Google Scholar] [CrossRef]
- Kiianko, M.V.; Canel, R.S.; Ludemann, V.; Pose, G.; Wagner, J.R. Beta-Glucan Content and Hydration Properties of Fila-mentous Fungi. Prikl. Biokhimiia Mikrobiol. 2013, 49, 48–52. [Google Scholar]
- Fernandez, C.W.; Langley, J.A.; Chapman, S.; McCormack, M.L.; Koide, R.T. The decomposition of ectomycorrhizal fungal necromass. Soil Biol. Biochem. 2016, 93, 38–49. [Google Scholar] [CrossRef]
- Ivanova, A.A.; Wegner, C.-E.; Kim, Y.; Liesack, W.; Dedysh, S.N. Identification of microbial populations driving biopolymer degradation in acidic peatlands by metatranscriptomic analysis. Mol. Ecol. 2016, 25, 4818–4835. [Google Scholar] [CrossRef]
- Wang, C.; Dong, D.; Wang, H.; Müller, K.; Qin, Y.; Wang, H.; Wu, W. Metagenomic analysis of microbial consortia enriched from compost: New insights into the role of Actinobacteria in lignocellulose decomposition. Biotechnol. Biofuels 2016, 9, 22. [Google Scholar] [CrossRef]
- Anderson, I.; Abt, B.; Lykidis, A.; Klenk, H.-P.; Kyrpides, N.; Ivanova, N. Genomics of Aerobic Cellulose Utilization Systems in Actinobacteria. PLoS ONE 2012, 7, e39331. [Google Scholar] [CrossRef]
- Wilson, D.B. Studies of Thermobifida fusca plant cell wall degrading enzymes. Chem. Rec. 2004, 4, 72–82. [Google Scholar] [CrossRef]
- Zhao, F.; Wang, J.; Li, Y.; Xu, X.; He, L.; Wang, J.; Ren, C.; Guo, Y. Microbial functional genes driving the positive priming effect in forest soils along an elevation gradient. Soil Biol. Biochem. 2022, 165, 108498. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).