
Taxonomic Description and Complete Genome Sequencing of Pseudomonas silvicola sp. nov. Isolated from Cunninghamia laceolata
 ,
,
Abstract
1. Introduction
2. Results and Discussion
2.1. The 16s rRNA and Multilocus Sequence Analysis (MLSA-)-Based Phylogenetic Analyses
2.2. Analyses of DNA G + C Content, DNA Relatedness, and Phylogenomic Analyses
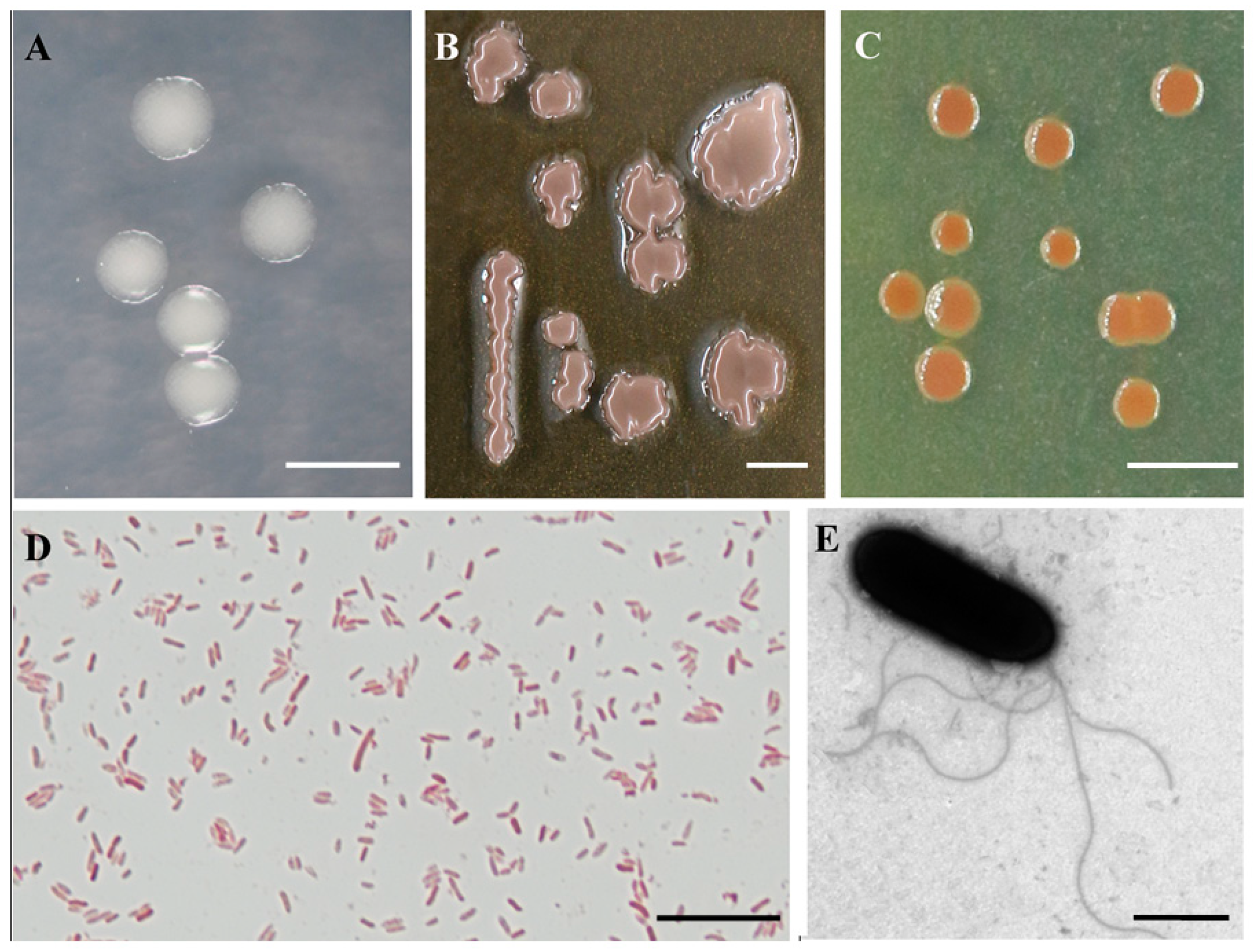
2.3. Morphological, Physiological, and Biochemical Characterization
2.4. Chemotaxonomic Characterization
2.5. Genomic Sequencing and the General Genomic Features of P. silvicola sp. nov.
2.6. Genomic Annotation Using the GO, KEGG, and eggNOG Databases
2.7. Further Annotation of the Features of P. silvicola sp. nov.
2.8. Genomic Identification of Biocontrol Determinants in P. silvicola T1-3-2T
3. Conclusions
4. Description of P. silvicola sp. nov.
5. Methods
5.1. Bacterial and Fungal Growth Conditions
5.2. PCR Amplification and DNA Sequencing
5.3. Phylogenetic Analysis
5.4. Genome Sequencing and Analysis
5.5. Morphological, Physiological, and Biochemical Tests
5.6. Chemotaxonomic Analysis
5.7. Statements
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Palleroni, N.J. Introduction to Family Pseudomonaceae; Springer: Berlin/Heidelberg, Germany, 1992. [Google Scholar]
- Lalucat, J.; Mulet, M.; Gomila, M.; García-Valdés, E. Genomics in Bacterial Taxonomy: Impact on the Genus Pseudomonas. Genes 2020, 11, 139. [Google Scholar] [CrossRef] [PubMed]
- Ikemoto, S.; Kuraishi, H.; Komagata, K.; Azuma, R.; Suto, T.; Murooka, H. Cellular fatty acid composition in Pseudomonas species. J. Gen. Appl. Microbiol. 1978, 24, 199–213. [Google Scholar] [CrossRef]
- Parte, A.C.; Sardà Carbasse, J.; Meier-Kolthoff, J.P.; Reimer, L.C.; Göker, M. List of Prokaryotic names with Standing in Nomenclature (LPSN) moves to the DSMZ. Int. J. Syst. Evol. Microbiol. 2020, 70, 5607–5612. [Google Scholar] [CrossRef] [PubMed]
- Zilber-Rosenberg, I.; Rosenberg, E. Role of microorganisms in the evolution of animals and plants: The hologenome theory of evolution. FEMS Microbiol. Rev. 2008, 32, 723–735. [Google Scholar] [CrossRef]
- Saati-Santamaría, Z.; Rivas, R.; Kolařik, M.; García-Fraile, P. A New Perspective of Pseudomonas—Host Interactions: Distribution and Potential Ecological Functions of the Genus Pseudomonas within the Bark Beetle Holobiont. Biology 2021, 10, 164. [Google Scholar] [CrossRef]
- Yang, R.; Li, S.; Li, Y.; Yan, Y.; Fang, Y.; Zou, L.; Chen, G. Bactericidal Effect of Pseudomonas oryziphila sp. nov., a Novel Pseudomonas Species Against Xanthomonas oryzae Reduces Disease Severity of Bacterial Leaf Streak of Rice. Front. Microbiol. 2021, 12, 759536. [Google Scholar] [CrossRef]
- Preston, G.M. Plant perceptions of plant growth-promoting Pseudomonas. Philos. Trans. R. Soc. B Biol. Sci. 2004, 359, 907–918. [Google Scholar] [CrossRef]
- Martínez-García, P.M.; Ruano-Rosa, D.; Schilirò, E.; Prieto, P.; Ramos, C.; Rodríguez-Palenzuela, P.; Mercado-Blanco, J. Complete genome sequence of Pseudomonas fluorescens strain PICF7, an indigenous root endophyte from olive (Olea europaea L.) and effective biocontrol agent against Verticillium dahliae. Stand. Genomic Sci. 2015, 10, 10. [Google Scholar] [CrossRef]
- Chlebek, D.; Pinski, A.; Żur, J.; Michalska, J.; Hupert-Kocurek, K. Genome Mining and Evaluation of the Biocontrol Potential of Pseudomonas fluorescens BRZ63, a New Endophyte of Oilseed Rape (Brassica napus L.) against Fungal Pathogens. Int. J. Mol. Sci. 2020, 21, 8740. [Google Scholar] [CrossRef]
- Shen, X.; Chen, M.; Hu, H.; Wang, W.; Peng, H.; Xu, P.; Zhang, X. Genome Sequence of Pseudomonas chlororaphis GP72, a Root-Colonizing Biocontrol Strain. J. Bacteriol. 2012, 194, 1269–1270. [Google Scholar] [CrossRef]
- Calderón, C.E.; Ramos, C.; de Vicente, A.; Cazorla, F.M. Comparative Genomic Analysis of Pseudomonas chlororaphis PCL1606 Reveals New Insight into Antifungal Compounds Involved in Biocontrol. Mol. Plant-Microbe Interact. 2015, 28, 249–260. [Google Scholar] [CrossRef] [PubMed]
- Glick, B.R. Modulation of plant ethylene levels by the bacterial enzyme ACC deaminase. FEMS Microbiol. Lett. 2005, 251, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Vacheron, J.; Desbrosses, G.; Bouffaud, M.-L.; Touraine, B.; Moënne-Loccoz, Y.; Muller, D.; Legendre, L.; Wisniewski-Dyé, F.; Prigent-Combaret, C. Plant growth-promoting rhizobacteria and root system functioning. Front. Plant Sci. 2013, 4, 356. [Google Scholar] [CrossRef]
- Arrebola, E.; Tienda, S.; Vida, C.; de Vicente, A.; Cazorla, F.M. Fitness Features Involved in the Biocontrol Interaction of Pseudomonas chlororaphis With Host Plants: The Case Study of PcPCL1606. Front. Microbiol. 2019, 10, 719. [Google Scholar] [CrossRef]
- Beskrovnaya, P.; Melnyk, R.A.; Liu, Z.; Liu, Y.; Higgins, M.A.; Song, Y.; Ryan, K.S.; Haney, C.H. Comparative Genomics Identified a Genetic Locus in Plant-Associated Pseudomonas spp. That Is Necessary for Induced Systemic Susceptibility. mBio 2020, 11, e00575-20. [Google Scholar] [CrossRef]
- Sue, B.; David, W. Chemical composition of decomposing stumps in successive rotation of Chinese fir (Cunninghamia lanceolata (Lamb.) Hook.) plantations. Chin. Sci. Bull. 2005, 50, 2581–2586. [Google Scholar]
- Shucheng, L.; Ddexun, Q.; Yuanxiang, L. Control of blight of Chinese fir by antibiotic strain F_051. For. Pest Dis. 1988, 2, 19–20. [Google Scholar]
- Li, B.; Zuo, Q.; Wang, Z.; Li, Y. Control of blight of Chinese fir by antibiotic strain Trichoderma harzianum F060 and Dexon. J. Sichuan Agric. Univ. 1992, 10, 301–306. [Google Scholar]
- Wang, S.; He, X.; Liu, H.; Zhou, L.; Zhang, M.; Li, P. Identification of an endophytic bacterium having antagonistic activity isolated from Cunninghamia lanceolata and optimization of fermentation condition. Genomics Appl. Biol. 2014, 33, 1275–1280. [Google Scholar]
- Li, D.; Tan, Y.; Lu, Z.; Zhou, G. Development and forest control of antagonistic bacteria AM53 against Colletotrichum gloesporioides on Chinise fir. J. Fujian Agric. For. Univ. 2015, 44, 154–158. [Google Scholar]
- Liu, Y.; Zeng, H.; Zhang, L.; Liang, W.; Song, Z.; Lu, M.; Zhu, W.; Zhang, Q. Endophytic bacteria for biocontrol of copper blight on Cunninghamia lanceolate. Fujian J. Agric. Sci. 2021, 36, 556–563. [Google Scholar]
- Tian, L.Y.; Huang, H.; Yang, H.; Qiu, H.L.; Zhang, C.H.; Xu, J.Z.; Qin, C.S. Pseudomonas sp. T1-3-2 with antimicrobial activities improves the diseases resistance and promotes the growth promotion of Cunninghamia lanceolata. Microbiol. China 2022, 49, 1–14. [Google Scholar]
- Meier-Kolthoff, J.P.; Klenk, H.-P.; Göker, M. Taxonomic use of DNA G + C content and DNA–DNA hybridization in the genomic age. Int. J. Syst. Evol. Microbiol. 2014, 64, 352–356. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.; Rosselló-Móra, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Göker, M. TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat. Commun. 2019, 10, 1–10. [Google Scholar] [CrossRef]
- Liu, Y.; Song, Z.; Zeng, H.; Lu, M.; Zhu, W.; Wang, X.; Lian, X.; Zhang, Q. Pseudomonas eucalypticola sp. nov., a producer of antifungal agents isolated from Eucalyptus dunnii leaves. Sci. Rep. 2021, 11, 3630. [Google Scholar] [CrossRef]
- Hiroshi, O.; Kazuo, K. Grouping of Pseudomonas species on the basis of cellular fatty acid composition and the quinone system with special reference to 3-hydroxy fatty acids. J. Gen. Appl. Microbiol. 1983, 29, 17–40. [Google Scholar]
- Bertelli, C.; Brinkman, F.S.L. Improved genomic island predictions with IslandPath-DIMOB. Bioinformatics 2018, 34, 2161–2167. [Google Scholar] [CrossRef]
- Lanteigne, C.; Gadkar, V.J.; Wallon, T.; Novinscak, A.; Filion, M. Production of DAPG and HCN by Pseudomonas sp. LBUM300 Contributes to the Biological Control of Bacterial Canker of Tomato. Phytopathology 2012, 102, 967–973. [Google Scholar] [CrossRef]
- Santoyo, G.; Orozco-Mosqueda, M.D.C.; Govindappa, M. Mechanisms of biocontrol and plant growth-promoting activity in soil bacterial species of Bacillus and Pseudomonas: A review. Biocontrol Sci. Technol. 2012, 22, 855–872. [Google Scholar] [CrossRef]
- Biessy, A.; Filion, M. Phenazines in plant-beneficial Pseudomonas spp.: Biosynthesis, regulation, function and genomics. Environ. Microbiol. 2018, 20, 3905–3917. [Google Scholar] [CrossRef] [PubMed]
- Coates, R.C.; Bowen, B.P.; Oberortner, E.; Thomashow, L.; Hadjithomas, M.; Zhao, Z.; Ke, J.; Silva, L.; Louie, K.; Wang, G.; et al. An integrated workflow for phenazine-modifying enzyme characterization. J. Ind. Microbiol. Biotechnol. 2018, 45, 567–577. [Google Scholar] [CrossRef] [PubMed]
- Cunrath, O.; Geoffroy, V.A.; Schalk, I.J. Metallome of Pseudomonas aeruginosa: A role for siderophores. Environ. Microbiol. 2015, 18, 3258–3267. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.B.; Sayyed, R.Z.; Trivedi, M.H.; Gobi, T.A. Phosphate solubilizing microbes: Sustainable approach for managing phosphorus deficiency in agricultural soils. SpringerPlus 2013, 2, 587. [Google Scholar] [CrossRef]
- Shen, Y.-Q.; Bonnot, F.; Imsand, E.M.; RoseFigura, J.M.; Sjölander, K.; Klinman, J.P. Distribution and Properties of the Genes Encoding the Biosynthesis of the Bacterial Cofactor, Pyrroloquinoline Quinone. Biochemistry 2012, 51, 2265–2275. [Google Scholar] [CrossRef]
- Wanner, B.L. Gene regulation by phosphate in enteric bacteria. J. Cell. Biochem. 1993, 51, 47–54. [Google Scholar] [CrossRef]
- Etesami, H.; Alikhani, H.A.; Hosseini, H.M. Indole-3-acetic acid (IAA) production trait, a useful screening to select endophytic and rhizosphere competent bacteria for rice growth promoting agents. Methodsx 2015, 2, 72–78. [Google Scholar] [CrossRef]
- Penrose, D.M.; Glick, B.R. Methods for isolating and characterizing ACC deaminase-containing plant growth-promoting rhizobacteria. Physiol. Plant. 2003, 118, 10–15. [Google Scholar] [CrossRef]
- Goswami, D.; Borah, S.N.; Lahkar, J.; Handique, P.J.; Deka, S. Antifungal properties of rhamnolipid produced byPseudomonas aeruginosaDS9 againstColletotrichum falcatum. J. Basic Microbiol. 2015, 55, 1265–1274. [Google Scholar] [CrossRef]
- Anzai, Y.; Kim, H.; Park, J.-Y.; Wakabayashi, H.; Oyaizu, H. Phylogenetic affiliation of the pseudomonads based on 16S rRNA sequence. Int. J. Syst. Evol. Microbiol. 2000, 50, 1563–1589. [Google Scholar] [CrossRef]
- Yamamoto, S.; Kasai, H.; Arnold, D.L.; Jackson, R.W.; Vivian, A.; Harayama, S. Phylogeny of the genus Pseudomonas: Intrageneric structure reconstructed from the nucleotide sequences of gyrB and rpoD genes. Microbiology 2000, 146, 2385–2394. [Google Scholar] [CrossRef] [PubMed]
- Tayeb, L.A.; Ageron, E.; Grimont, F.; Grimont, P. Molecular phylogeny of the genus Pseudomonas based on rpoB sequences and application for the identification of isolates. Res. Microbiol. 2005, 156, 763–773. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.-H.; Ha, S.-M.; Kwon, S.; Lim, J.; Kim, Y.; Seo, H.; Chun, J. Introducing EzBioCloud: A taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 2017, 67, 1613–1617. [Google Scholar] [CrossRef] [PubMed]
- Sudhir, K.; Glen, S.; Koichiro, T. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870. [Google Scholar]
- Felsenstein, J. Evolutionary trees from DNA sequences: A maximum likelihood approach. J. Mol. Evol. 1981, 17, 368–376. [Google Scholar] [CrossRef]
- Alexandros, S. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar]
- Hyatt, D.; Chen, G.-L.; Locascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef]
- Nawrocki, E.P.; Eddy, S.R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 2013, 29, 2933–2935. [Google Scholar] [CrossRef]
- She, R.; Chu, J.S.-C.; Wang, K.; Pei, J.; Chen, N. genBlastA: Enabling BLAST to identify homologous gene sequences. Genome Res. 2008, 19, 143–149. [Google Scholar] [CrossRef]
- Birney, E.; Clamp, M.; Durbin, R. GeneWise and Genomewise. Genome Res. 2004, 14, 988–995. [Google Scholar] [CrossRef]
- Akhter, S.; Aziz, R.; Edwards, R.A. PhiSpy: A novel algorithm for finding prophages in bacterial genomes that combines similarity- and composition-based strategies. Nucleic Acids Res. 2012, 40, e126. [Google Scholar] [CrossRef] [PubMed]
- Blin, K.; Shaw, S.; Steinke, K.; Villebro, R.; Ziemert, N.; Lee, S.Y.; Medema, M.H.; Weber, T. antiSMASH 5.0: Updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 2019, 47, W81–W87. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Bairoch, A. The SWISS-PROT protein sequence data bank and its new supplement TREMBL. Nucleic Acids Res. 1996, 24, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [PubMed]
- Powell, S.; Forslund, K.; Szklarczyk, D.; Trachana, K.; Roth, A.; Huerta-Cepas, J.; Gabaldón, T.; Rattei, T.; Creevey, C.; Kuhn, M.; et al. eggNOG v4.0: Nested orthology inference across 3686 organisms. Nucleic Acids Res. 2014, 42, D231–D239. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes database (CAZy): An expert resource for Glycogenomics. Nucleic Acids Res. 2009, 37, D233–D238. [Google Scholar] [CrossRef]
- Jia, B.; Raphenya, A.R.; Alcock, B.; Waglechner, N.; Guo, P.; Tsang, K.K.; Lago, B.A.; Dave, B.M.; Pereira, S.; Sharma, A.N.; et al. CARD 2017: Expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2017, 45, D566–D573. [Google Scholar] [CrossRef]
- Urban, M.; Cuzick, A.; Seager, J.; Wood, V.; Rutherford, K.; Venkatesh, S.Y.; De Silva, N.; Martinez, M.C.; Pedro, H.; Yates, A.D.; et al. PHI-base: The pathogen–host interactions database. Nucleic Acids Res. 2019, 48, D613–D620. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Yang, J.; Yu, J.; Yao, Z.; Sun, L.; Shen, Y.; Jin, Q. VFDB: A reference database for bacterial virulence factors. Nucleic Acids Res. 2005, 33, D325–D328. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.Z.; Cai, M.Y. Determination of Biochemical Properties Manual for the Systematic Identification of General Bacteria; Science Press: Beijing, China, 2001; pp. 370–398. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Summary for T1-3-2T | ANI Value and Aligned Percentage [%] b | Tetra [%] c | DDH [%] d | G + C mol% Difference e | ||
|---|---|---|---|---|---|---|
| Genome a | ANIb | ANIm | ||||
| Pseudomonas eucalypticola NP-1T GCF_013374995 | 90.3 [66.5] | 92.8 [64.5] | 99.3 | 48.2 | 1.48 | |
| Pseudomonas rhizosphaerae DSM 16299T GCA_000761155 | 76.9 [35.7] | 85.4 [17.8] | 95.6 | 23.8 | 0.35 | |
| Pseudomonas coleopterorum LMG 28558T GCA_900105555 | 76.5 [37.1] | 85.4 [17.6] | 95.5 | 23.6 | 0.38 | |
| Characteristic | T1-3-2T | NP-1T | Characteristic | T1-3-2T | NP-1T |
|---|---|---|---|---|---|
| Bacterial morphology | rod-shaped | rod-shaped | Gram staining | - | - |
| Size (μm) | 0.6–0.9 × 1.8–2.6 | 1.0 × 2.0 | Fluorescent pigments | + | - |
| Number of polar flagella | 3–5 | 1 | O-F test | O/- | O/- |
| Oxidase | - | - | Hydrolysis of gelatin | + | - |
| Catalase | + | + | Hydrolysis of starch | + | + |
| Nitrate reduction | + | - | Hydrolysis of DNA | - | - |
| Arginine dihydrolase | + | + | Citrate utilization | + | + |
| Acetamidase hydrolysis | - | D-fructose | w | + | |
| dextrin | - | + | D-galactose | + | + |
| D-mannose | + | + | gentiobiose | w | + |
| D-Mannose | w | + | α-D-glucose | + | + |
| L-Rhamnose | + | + | D-mannitol | w | + |
| Quinic Acid | + | + | L-Aspartic Acid | + | + |
| Bromo-Succinic Acid | w | + | D-trehalose | w | + |
| Glucuronamide | + | + | formic acid | w | + |
| D-galacturonic acid | + | + | D-Gluconic Acid | + | + |
| α-keto glutaric acid | - | w | D-saccharic acid | + | w |
| methyl pyruvate | - | w | D-turanose | - | w |
| Growth at temperature °C | 10–37 | 4–37 | Maltose | - | - |
| NaCl (%, w/v) | 0–2 | 0–2 | pH | 5–8 | 3–7 |
| Chemotaxonomic Characterization | Peak Name | T1-3-2T Percent Named% | NP-1T Percent Named% | |
|---|---|---|---|---|
| Fatty acid | Straight-chain fatty acids | 10:0 | 0.99 | 2.57 |
| 12:0 | 3.61 | 4.39 | ||
| 14:0 | 0.31 | 0.19 | ||
| 16:0 | 25.08 | 19.67 | ||
| 17:0 | 0.11 | 0.17 | ||
| 18:0 | 0.86 | 0.7 | ||
| 19:0 | 0.15 | 0.13 | ||
| Unsaturated fatty acids | 17:1 w7c | 0.11 | 0.15 | |
| 18:1 w7c 11-methyl | 0.16 | 0.19 | ||
| 20:2 w6,9c | - | 0.07 | ||
| Branched fatty acid | 19:0 iso | - | 0.06 | |
| Hydroxy fatty acid | 10:0 3OH | 5.96 | 11.15 | |
| 12:0 2OH | 5.39 | 5.13 | ||
| 12:1 3OH | 2.76 | 6.6 | ||
| 12:0 3OH | 5.64 | 7.16 | ||
| 14:0 2OH | - | 0.17 | ||
| 15:0 3OH | - | 0.05 | ||
| 16:0 3OH | 0.12 | 0.25 | ||
| Cyclopropane acids | 17:0 cyclo | 17.64 | 13.56 | |
| 19:0 cyclo w8c | 5.51 | 6.97 | ||
| Summed feature | Summed feature 2 | 0.44 | 0.72 | |
| Summed feature 3 | 10.13 | 7.58 | ||
| Summed feature 5 | 0.27 | 0.21 | ||
| Summed feature 7 | - | 0.11 | ||
| Summed feature 8 | 14.71 | 12.06 | ||
| Summed feature 9 | 0.07 | - | ||
| Quinone system | Ubiquinone | Q-8 | 15.99 | 34.08 |
| Q-9 | 84.02 | 65.92 | ||
| Methylnaphthoquinone | MK8 | 100.00 | - | |
| Attributes | T1-3-2T |
|---|---|
| Genome size (bp) | 8,650,947 bp |
| G + C content (%) | 61.7 |
| Total genes | 8116 |
| Protein coding genes | 8116 |
| RNAs | 197 |
| tRNAs | 95 |
| Ribosomal RNAs (5S, 16S, 23S) | 9, 8, 8 |
| ncRNAs | 77 |
| Pseudogenes | 38 |
| Genomic island | 20 |
| Prophage | 4 |
| Gene cluster | 15 |
| Genes with predicted functions | |
| Annotated genes databases | 7884 |
| Nonredundant protein database | 7870 |
| eggNOG_Annotation | 6862 |
| Gene Ontology_Annotation | 5779 |
| Kyoto encyclopedia of genes and genomes | 4055 |
| Pfam_Annotation | 6644 |
| Swissprot_Annotation | 4667 |
| TrEMBL_Annotation | 4667 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, L.; Zhang, Y.; Yang, H.; Zhao, Q.; Qiu, H.; Xu, J.; Qin, C. Taxonomic Description and Complete Genome Sequencing of Pseudomonas silvicola sp. nov. Isolated from Cunninghamia laceolata. Forests 2023, 14, 1089. https://doi.org/10.3390/f14061089
Tian L, Zhang Y, Yang H, Zhao Q, Qiu H, Xu J, Qin C. Taxonomic Description and Complete Genome Sequencing of Pseudomonas silvicola sp. nov. Isolated from Cunninghamia laceolata. Forests. 2023; 14(6):1089. https://doi.org/10.3390/f14061089
Chicago/Turabian StyleTian, Longyan, Yanfeng Zhang, Hua Yang, Qian Zhao, Hualong Qiu, Jinzhu Xu, and Changsheng Qin. 2023. "Taxonomic Description and Complete Genome Sequencing of Pseudomonas silvicola sp. nov. Isolated from Cunninghamia laceolata" Forests 14, no. 6: 1089. https://doi.org/10.3390/f14061089
APA StyleTian, L., Zhang, Y., Yang, H., Zhao, Q., Qiu, H., Xu, J., & Qin, C. (2023). Taxonomic Description and Complete Genome Sequencing of Pseudomonas silvicola sp. nov. Isolated from Cunninghamia laceolata. Forests, 14(6), 1089. https://doi.org/10.3390/f14061089