Abstract
Plant tissue regeneration is a key process for genetic transformation and genome editing. The exploration of regulatory mechanisms in plant regeneration would improve regeneration efficiency. In comparison to some model plants, the genomic heterozygosity is much higher in forest trees, increasing the complexity of transcriptional regulation. Here, we report the allele-specific transcriptional analysis in hybrid poplar 84K (Populus alba × P. tremula var. glandulosa cv. 84K) during the shoot regeneration process. Firstly, 180 regeneration-related genes (REGs) and 2446 REG-homologous genes (REGHs) were identified in hybrid poplar. The expression patterns of REGs exhibited that about half of them were positively correlated between poplar and Arabidopsis at the locus level. The expression levels of REGHs vary among the gene family at different stages during callus and shoot induction. Among the gene clusters with similar expression patterns, the distribution of gene families in poplar and Arabidopsis also exhibits large variations. At the allele level, most of the allele pairs of REGs were positively correlated in expression. The expression patterns of genes in auxin synthesis, transport, and signaling pathways agree with the general patterns. Due to the presence/absence of variations between two subgenomes, two YUC alleles and two IAA alleles are present only in one subgenome, and the expression patterns of the two alleles are greatly different. Our analysis indicates the conservativeness and diversity of transcriptional regulation during shoot regeneration in poplar and Arabidopsis. The complexity in allele expression contributed by heterozygosity suggests the importance of genotyping in the screening of explants for plant regeneration.
1. Introduction
Plant regeneration is the process of repair, renewal, and organogenesis of tissues [1], which is indispensable for plant growth and has great importance for plant survival and reproduction [2]. Plants have evolved adaptive mechanisms that allow somatic cells near the damaged sites to regain their ability to divide and produce new cells [3,4]. These mechanisms result in different types of regeneration, including wounding-triggered callus formation [5], de novo organ regeneration (including de novo root regeneration and de novo shoot regeneration) [6], and somatic embryogenesis (SE) [7]. In tissue culture, different combinations of plant hormones were used to induce plant regeneration [8]. Typically, the explants were first cultured on an auxin-rich callus-inducing medium (CIM) and then on a cytokinin-rich shoot-inducing medium (SIM) to stimulate the formation of shoots [9]. Plant tissue culture techniques have evolved to be foundational for genetic improvement, micropropagation, genetic engineering, and genome editing [10].
More and more studies have revealed biological pathways involved in regeneration regulation, including wound and stress signals [11], hormones, and transcriptional and epigenetic regulation processes. Under wounding and stress conditions, plants can generate physical or chemical signals, triggering downstream hormone signals [5,12]. Transcription factors and epigenetic regulators are usually involved in these signaling pathways [9,13], generating gene networks through cross-talks at diverse gene nodes [10]. With the development of single-cell RNA sequencing (scRNA-seq) technologies, the transcriptional dynamics during plant regeneration can be explored at the single-cell level to reveal new cell types and states and continuous developmental trajectories [14,15]. An understanding of regulatory mechanisms in shoot regeneration would provide more efficient biotechnical tools to improve regeneration rates [16].
Previous studies have shown that there are significant differences in regeneration abilities among plants of diverse categories [17]. As a dicotyledonous model plant, Arabidopsis thaliana is relatively convenient for tissue regeneration [18,19]. In Bryophytes, such as Marchantia polymorpha, the injured tissues can regenerate stem cells within about two days, indicating high regenerative capacity [20,21]. In monocotyledonous plants, rice and maize have low regeneration efficiency because regenerative cells are only present at the base of immature leaves [22,23]. At the same time, regeneration efficiency varies significantly in populations with individuals of different genotypes [24]. Many efforts have been made to overexpress key transcription factors to elevate the regeneration and transformation efficiency of crop plants and forest trees, such as growth-regulating factors (GRF), baby boom (BBM), WUSCHEL-related homeobox (WOX), and other transcription factors or chimeric constructions [24,25].
The development of tissue regeneration and transgenic techniques for forest tree breeding is particularly important because of the long cycle of traditional breeding [26]. At the same time, transgenic approaches were not yet developed for most forest trees. In comparison to crop and model plants, most forest trees are large in genome size and high in genetic heterozygosity. The genomic variations between two parental alleles would contribute to the transcriptional diversities during shoot regeneration, similar to the heterosis effects in plant growth and development [27]. Poplar, as a model tree species for forest molecular biology research, has advanced in genetic transformation. The transgenic systems have been developed for multiple poplar species or clones, especially inter-species and intra-species hybrids, such as hybrid poplar 84K (Populus alba × P. tremula var. glandulosa clone 84K) [28], hybrid aspen T89 (P. tremula × P. tremuloides), and hybrid poplar INRA 7171-B4 (P. tremula × P. alba) [29]. These hybrids are ideal materials for transcriptional studies at the allele level.
To meet the demands for highly efficient genome editing techniques in poplar and other forest trees, the molecular mechanisms in regeneration need to be further explored to identify key factors for perturbation. There are some outstanding questions about the regulation of regeneration in forests, such as: (1) Are the key drivers in regeneration conserved between model herbaceous plants and woody trees? (2) Do the functions and regulations of these drivers remain the same in trees as in herbaceous plants? and (3) How does genome heterozygosity affect transcriptional regulation in regeneration? In this study, we summarized the regeneration-related genes (REGs) in Arabidopsis and identified their orthologs in hybrid poplar. The alleles of REGs in hybrid poplar were also recognized to check their expression patterns at allele levels. Our analysis would provide clues for the genetic engineering of poplar clones to elevate regeneration efficiency.
2. Materials and Methods
2.1. Plant Materials
In our analysis, we compared the transcriptional dynamics during shoot regeneration in hybrid poplar 84K (Populus alba × P. tremula var. glandulosa cv. 84K) and Arabidopsis. In hybrid poplar, the leaf explants were derived from tissue-cultured seedlings of 6–7 weeks, and the induction of shoot organogenesis was performed directly from leaf explants and indirectly via callus. During the indirect inducing process, plant samples were harvested at 0 DAC (S2_Pag_CIM0), 7 DAC (S2_Pag_CIM7), and 14 DAC (S2_Pag_CIM14) on callus induction medium (CIM), and 7 DAC (S2_Pag_SIM7), 14 DAC (S2_Pag_SIM14), 21 DAC (S2_Pag_SIM21), and 28 DAC (S2_Pag_SIM28) on SIM. During the direct inducing process, plant samples were collected at 15 (S2_Pag_Leaf15) and 30 (S2_Pag_Leaf30) days after culture (DAC) on shoot induction medium (SIM), respectively. The collected samples are stored at −80 °C before transcriptional sequencing. Besides the dataset described above, a publicly available dataset from an indirect inducing approach with fewer samples of hybrid poplar 84K was also included in our analysis (S1_Pag_CIM0, S1_Pag_CIM8, S1_Pag_SIM4, and S1_Pag_SIM8) [30].
Similar to the experiments in hybrid poplar, two datasets from indirect shoot organogenesis induction were downloaded for comparative analysis [31,32]. Based on the cellular status of cell fate transition in the phases of shoot regeneration, the samples of Arabidopsis were selected in alignment with the samples in hybrid poplar. In the smaller dataset (S1), four stages of samples were compared, including S1_Pag_CIM0, S1_Pag_CIM8, S1_Pag_SIM4, and S1_Pag_SIM8 in poplar and S1_At_CIM0, S1_At_CIM4, S1_At_SIM4, and S1_At_SIM6 in Arabidopsis. In the larger dataset (S2), six stages of samples were compared, including S2_Pag_CIM0, S2_Pag_CIM7, S2_Pag_CIM14, S2_Pag_SIM7, S2_Pag_SIM14, and S2_Pag_SIM21 in hybrid poplar, and S2_At_CIM0, S2_At_CIM3, S2_At_CIM7, S2_At_SIM3, S2_At_SIM6, and S2_At_SIM8 in Arabidopsis.
2.2. Transcriptome Sequencing and RNA-Seq Data Analysis
Total RNA was extracted from plant samples using the RNA Prep Pure Plant Kit (Tiangen, Beijing, China), and the sequencing libraries were further constructed using the TruSeq RNA Library Prep Kit v2. The Illumina HiSeq 2500 platform was used to sequence the libraries to obtain reads of 150 nt in paired-end. Raw RNAseq reads were trimmed and filtered to remove adaptor sequences and lower-quality reads using Trimmomatic (v0.39). The clean reads were mapped to two subgenomes using STAR (v.2.7.9a) [33]. Then, featureCounts (v2.0.3) [34] was applied to calculate RNAseq reads associated with alleles. The read counts were applied for allelic expression analysis. To calculate the gene expression level at the locus level, the read numbers of two alleles were summed to obtain the total read number. The DESeq2 package in R was used to calculate gene expression values in TPMs (Transcripts Per Million) at locus and allele levels. A differential expression test was also performed using a one-way ANOVA-like model in a likelihood ratio test (LRT) based on read counts [35]. The cutoff for differentially expressed genes (DEGs) was set as Q value ≤ 0.05 and the absolute value of log2 (fold change) ≥ 1, in which the fold change was calculated between two samples with maximum and minimum averaged gene expression values. The differential expression analysis of REGs and REGHs followed the same processes, and the same cutoffs were applied to screen significantly differentially expressed genes.
Gene expression data were visualized in heatmaps using TBtools (v1.098745) [36]. The expression pattern of DEGs was also applied for STEM (Short Time-series Expression Miner) analysis. The numbers of gene families with diverse trends were summed by two plant species. Pearson Correlation Coefficients (PCCs) were calculated using expression values of orthologous gene pairs between hybrid poplar 84K and Arabidopsis and also two alleles of each gene locus in hybrid poplar 84K. The PCC calculation and significance test were applied using cor() and cor.test() functions in R, respectively. The threshold value of the |PCC| ≥ 0.6 and p-value < 0.05 indicates a significant correlation.
2.3. Identification of Regeneration-Related Genes and Gene Families in Hybrid Poplar
The identification of regeneration-related genes (REGs) was based on the literature and homolog search. Genes of diverse pathways related to plant regeneration in Arabidopsis were obtained from research and review papers. The protein sequences of REGs were downloaded from the Arabidopsis Information Resource (TAIR) database (https://www.Arabidopsis.org, accessed on 3 December 2022). The protein sequences of poplar 84K, including sequences from two haplotypes, were used to compare with the Arabidopsis genes using blast search (E-value < 1 × 10−5). The best hits from two haplotypes were selected as the orthologous genes in two subgenomes of the assembly [37].
To identify genes in each gene family, models of protein domains were downloaded from the Pfam database (https://pfam.xfam.org/, accessed on 21 December 2022), and candidate genes were obtained through the Hidden Markov Model (HMM) search. Genes identified from BLAST and HMM searches were further validated using the Simple Modular Architecture Research Tool (SMART) (https://smart.embl.de/, accessed on 21 December 2022) database. The gene members of transcription factor (TF) families were identified using the iTAK tool [38].
2.4. Identification of Allele Pairs in Two Subgenomes in Hybrid Poplar
The protein sequences of two subgenomes were compared using blast search, and the results were applied to identify synteny blocks using WGDI [39]. The list of alleles was screened from synteny blocks for REG orthologous genes in poplar. The loci of REGs in poplar were named after the homologous genes in Arabidopsis. The genes from two subgenomes (alba and glandulosa) were distinguished using prefixes A and G, respectively. For genes with multiple copies in poplar, the genes were distinguished with suffixes using the alphabet.
3. Results
3.1. Regeneration-Related Genes and Gene Families in Poplar 84K
Based on the literature search, 72 regeneration-related genes (REGs) were obtained in Arabidopsis (Table S1). These genes were mainly involved in the development processes of callus, shoot, root, and somatic embryogenesis (SE). Some of these genes are components of hormone-signaling transduction pathways (Figure 1). Homologous genes of these REGs were also identified in Arabidopsis, including 895 genes from 30 families (Table S2); these genes were termed REG-homologous genes (REGHs). The orthologous genes of REGs and REGHs in poplar were further identified in two subgenomes of poplar 84K (Table S3). Through blast and HMM searches, 93 and 87 REG alleles were identified for the P. alba (A) subgenome and the P. glandulosa (G) subgenome, respectively. And 1217 REGH alleles were identified for the A subgenome and 1229 REGH alleles for the G subgenome (Table S4). These gene sets serve as a start for further transcriptional analysis.
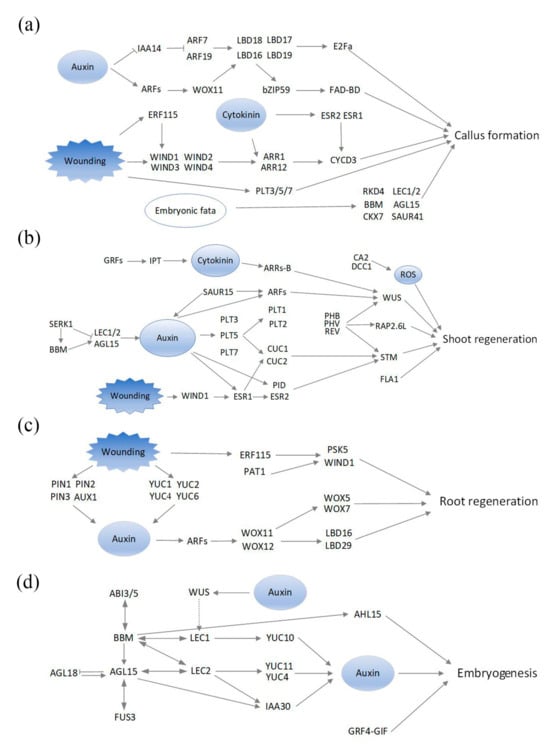
Figure 1.
Signal transduction pathways in plant regeneration are summarized based on [2]. The molecular components of signaling pathways are visualized in four processes, including callus formation (a), shoot regeneration (b), root regeneration (c), and embryogenesis (d).
3.2. Expression Patterns of REGs at Locus Level in Poplar 84K and Arabidopsis
To compare the transcriptional regulation in shoot regeneration between poplar 84K and Arabidopsis, we collected RNAseq data from both CIM and SIM stages with similar cellular status in two plant species [30,32]. Based on the annotation of orthologous pairs between two plant species, we first calculated the correlation coefficients of expression values at the locus level (Figure 2a and Table S5). The results indicated that about half (51) of the gene pairs exhibited positive correlations in expression values; for nine of them, the expression levels were significantly positively correlated (PCC > 0.6), including YUC6, GRF5A, LBD16B, GRF5B, LBD29, CYCD3.3A, WOX12, REVC, and DCC1. Meanwhile, the expression levels of 47 genes exhibited negative correlations, including PIN2, AHL15A, ARR10C, GIF1A, PSK5A, YUC1, YUC2, and GIF1B. It is worth noting that in gene families like YUC, the expression patterns of some gene pairs are positively correlated in two plant species, whereas the others are negatively correlated.
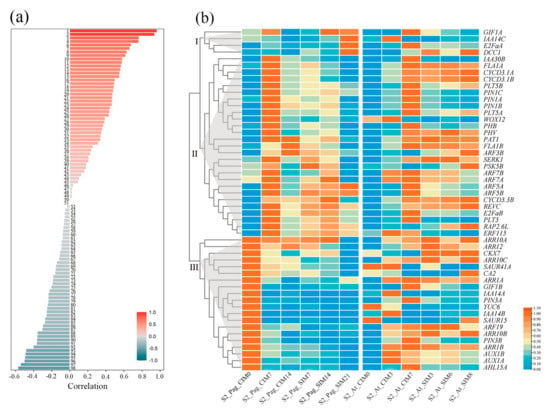
Figure 2.
Expression patterns of regeneration-related genes in poplar 84K and Arabidopsis. (a) Correlations of expression levels of regeneration-related genes (REGs) between poplar 84K and Arabidopsis. The calculation was performed using expression levels in TPM (Transcripts Per Million). The PCC values range from −1 to 1. The names of 98 genes are listed in Table S5. (b) Expression patterns of differentially expressed REGs on poplar 84K and their orthologs in Arabidopsis. The genes were clustered into three clades based on expression patterns. The expression data were normalized using the ZeroToOne method for heatmap visualization. The labels in each column indicate the stages during shoot regeneration. The columns on the left side were from hybrid poplar (shown as Pag). The columns on the right side were from Arabidopsis (shown as At). S2 in the labels indicates dataset 2.
Differentially expressed genes were further identified, followed by clustering analysis in poplar 84K. In the differential expression test, a one-way ANOVA-like model in likelihood ratio testing (LRT) was performed, which resulted in the identification of 51 differentially expressed REGs in hybrid poplar. The expression patterns of these REGs can be grouped into mainly three clades (Figure 2b). In clade I, the four genes were highly expressed in the third stage of the SIM process of poplar 84K. Three of these REGs were highly expressed in the CIM process of Arabidopsis, and one REG (DCC1) was highly expressed in all three stages in the SIM process of Arabidopsis. In clade II, almost all the REGs were highly expressed in the second stage of the CIM process of poplar 84K and gradually downregulated in the third stage of CIM and the following SIM stages. In Arabidopsis, most of these genes were highly expressed in the third stage of CIM. About half of these REGs stayed at a highly expressed level in the SIM process, and the remaining half of the genes depressed the expression level gradually in the SIM process. In clade III, all the genes were highly expressed in the first stage of the CIM process of poplar 84K, whereas most of these genes were expressed in the first stage of CIM in Arabidopsis at low levels, indicating reversed expression patterns in two species. The detailed expression patterns of REGs agree with the correlation of gene expression levels of gene pairs in two species.
3.3. The Expression Patterns of REGH Genes in Poplar 84K
To explore the expression patterns of REGH genes in poplar 84K, we first examined their expression levels at different regeneration stages (Figure 3). Among the 30 gene families in poplar, nearly all the gene members in CYCD3, GIF, and DCC families were expressed (TPM ≥ 1) at all the developmental stages from two experiments, whereas less than 40% of genes in the YUCCA and MADS families are expressed during the regeneration process. The portion of expressed genes varies greatly in the process for the WOX, CKX, E2F, and GRF families. For gene families LOB, CKX, E2F, AHL, SERK, and NAC, the portions of expressed genes were relatively lower at the CIM0 stages of the two experiments, with higher values at other stages. These data indicated the transcriptional dynamics during shoot regeneration poplar.
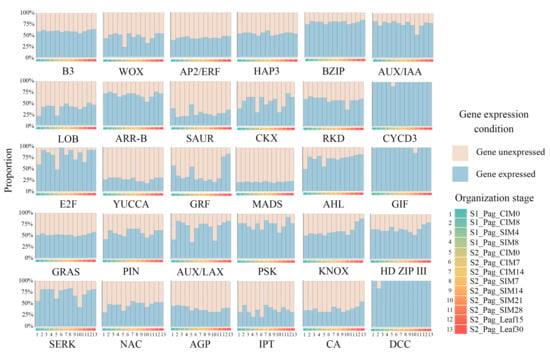
Figure 3.
Expression levels of REG-homologous genes in poplar 84K. The proportions of expressed (TPM ≥ 1) and unexpressed (TPM < 1) genes of each REGH family are shown in light-blue and gray bars, respectively. The order of samples is indicated with colored blocks. S1 and S2 indicate datasets 1 and 2, respectively; Pag indicates poplar 84K, CIM indicates days on callus induction media, and SIM indicates days on shoot induction media.
Differential expression analysis was applied for REGH genes using the same approaches as REGs. The expression trends of REGH genes were further analyzed to identify the major expression patterns of these differentially expressed genes. In the S1 dataset, 455 and 572 DEGs were identified for Arabidopsis and poplar 84K, respectively. These genes were then clustered into six major clusters using expression trend analysis (Figure 4a, Figure S1). In cluster 1, 98 Arabidopsis and 119 poplar genes were downregulated during the callus induction stage and displayed stable expression levels during subsequent shoot induction stages. In this cluster, most of the genes were from the AP2/ERF, BZIP, and AGP families in Arabidopsis and the AP2/ERF, SAUR, and HD ZIP III families in poplar 84K (Figure 4). Cluster 2 includes 103 Arabidopsis and 284 poplar genes. These genes were induced in the callus induction process and were also highly expressed in the following stages. Many genes in the AP2/ERF and NAC families of Arabidopsis and the AP2/ERF, AHL, GRAS, and NAC families of poplar are grouped in this cluster (Figure 4a).
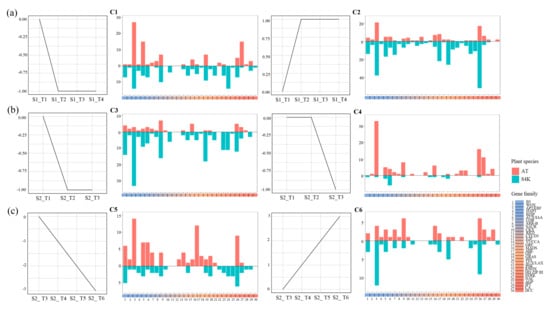
Figure 4.
Expression trends of REG-homologous genes in poplar 84K and Arabidopsis. (a) Clusters from data set S1, (b) Clusters from samples T1 to T3 of data set S2, and (c) Clusters from T3 to T6 of data set S2. Six clusters (C1–C6) were identified through STEM (Short Time-series Expression Miner) trend analysis. In each cluster, the trend line plot on the left side was used to show the gene expression patterns of poplar and Arabidopsis genes. The bar plot of each cluster on the right side shows the gene number of each gene family. The coral bars above the x-axis indicated gene numbers in Arabidopsis (AT), and the cyan bars below the x-axis indicated gene numbers in hybrid poplar (84K). The names of gene families are shown in colored bars on the bottom right. In the trend line plot, the stages of shoot regeneration are indicated at the x-axis. S1 and S2 indicate the two datasets of poplar and Arabidopsis. S1_T1, S1_T2, S1_T3, and S1_T4 were from dataset S1, corresponding to S1_Pag_CIM0, S1_Pag_CIM8, S1_Pag_SIM4, and S1_Pag_SIM8 in poplar, and S1_At_CIM0, S1_At_CIM4, S1_At_SIM4, and S1_At_SIM6 in Arabidopsis. S2_T1, S2_T2, S2_T3, S2_T4, S2_T5, and S2_T6 were from dataset S2, corresponding to S2_Pag_CIM0, S2_Pag_CIM7, S2_Pag_CIM14, S2_Pag_SIM7, S2_Pag_SIM14, and S2_Pag_SIM21 in poplar, and S2_At_CIM0, S2_At_CIM3, S2_At_CIM7, S2_At_SIM3, S2_At_SIM6, and S2_At_SIM8 in Arabidopsis.
The S2 dataset was further analyzed in the CIM and SIM groups. In the CIM group, 280 and 610 DEGs were identified in Arabidopsis and poplar, respectively. And in the SIM group, 434 and 340 DEGs were identified in Arabidopsis and poplar, respectively (Figure 4 and Figure S2). These genes were also clustered, and the results indicated that in some clusters like C3 and C6, the gene numbers of gene families in poplar are higher than in Arabidopsis. In clusters C4 and C5, the gene numbers of gene families in Arabidopsis are higher than in poplar. These data indicated the diversity of transcriptional regulations between poplar and Arabidopsis.
3.4. Allele-Specific Expression of REGs in Hybrid Poplar
As an inter-species hybrid clone, poplar 84K is an ideal system to study allele-specific expression during tissue regeneration. Among the 180 REGs in poplar, 78 pairs of alleles were identified based on the locations of alleles on the chromosomes. The expression levels of the alleles were analyzed using the S2 dataset in poplar. After filtering non-expressed alleles, the correlations of the expression values of 65 allele pairs were calculated. The result indicated that in most (59) of the allele pairs, the expression patterns were positively correlated, and only six pairs showed a negative correlation in expression level, including WUSA, LBD18A, CUC2B, DCC1, LBD18B, and YUC4B (Figure 5, Table S6). The negative correlation of allele-specific expression in the identified alleles may be due to the variation in cis-elements between the two alleles.
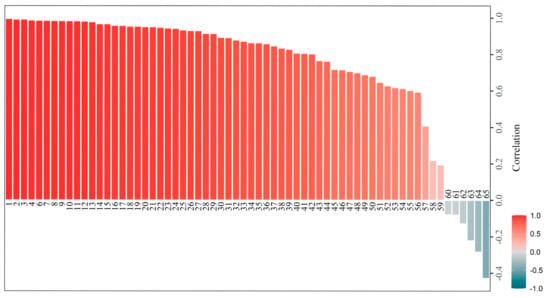
Figure 5.
Correlations of expression levels between two alleles of regeneration-related genes in poplar 84K. The calculation was performed using expression levels in TPM (Transcripts Per Million). The PCC values range from −1 to 1. The names of gene loci and the IDs of two alleles are shown in Table S6.
3.5. Expression Patterns of Genes in Auxin Signaling-Related Pathways
Genes in the auxin signaling pathway play significant roles in plant tissue regeneration [40]. In our analysis, the expression patterns of genes in pathways of auxin synthesis, transport, and signaling were examined in detail (Figure 6). Among the five expressed YUC genes in the auxin synthesis pathway, four YUC genes were highly expressed before callus induction in hybrid poplar. The expression patterns of YUC2 and YUC6 were similar between hybrid poplar and Arabidopsis, whereas YUC1 and YUC11 were highly expressed at the early SIM stages in Arabidopsis. In the auxin transport pathway, the expression patterns of PIN1 genes were similar between hybrid poplar and Arabidopsis, which were induced after treatment with CIM media. PIN2, PIN3, and AUX1 were highly expressed in leaf tissues before callus induction in hybrid poplar but lowly expressed in Arabidopsis at the same stage. Among the gene families in the auxin signaling pathway, including ARF, SAUR, and IAA, the expression patterns of genes between hybrid poplar and Arabidopsis exhibited diverse patterns, indicating the diversity in transcriptional dynamics (Figure 6). It is worth noting that the expressions of two alleles in auxin signaling-related pathways were similar in hybrid poplar. Among these alleles, A.YUC11A and A.IAA14B were only present in the alba subgenome, and G.YUC11B and G.IAA14C were only present in the glandulosa subgenome. The expression of the two allele pairs was greatly different, suggesting their subfunctionalization was caused by genomic variation. These data indicated an example of the similarity and differential expression of transcriptional regulators during the process of shoot regeneration.
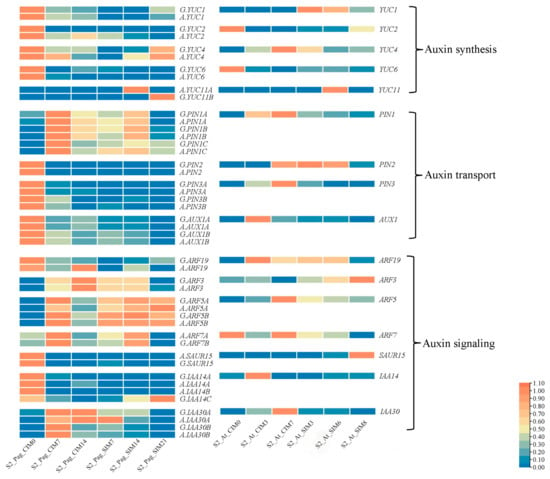
Figure 6.
Expression patterns of poplar alleles in the auxin signaling pathway and their orthologs in Arabidopsis. The expression patterns of alleles in hybrid poplar were visualized in heatmaps on the left side. In most cases, both alleles from two subgenomes were shown. In some cases where one allele was missing in one subgenome due to genomic variation, only the retained allele was shown. The expression patterns of orthologs in Arabidopsis were shown on the right to compare with their orthologs in poplar. The expression data were normalized using the ZeroToOne method for heatmap visualization.
4. Discussion
Shoot regeneration is widely employed in tissue culture techniques for plant breeding, which is crucial for transgenic or genome-editing to improve plant traits [41]. Currently, the study of the molecular mechanisms involved in the transition of cell fate during regeneration is primarily focused on the model plant Arabidopsis [42,43,44]. The characterization of transcriptional dynamics during shoot regeneration provides candidate genes for perturbation to improve transgenic efficiency. In our study, 93 and 87 REG alleles were identified for the P. alba (A) subgenome and the P. glandulosa (G) subgenome, respectively. These genes can be used as a starting point to study the regulation of genes in different pathways and their interactions at the transcription level. Further analysis of the differential expression of genes during shoot regeneration between poplar trees with different regeneration efficiency would help to identify the determining driver genes in poplar. These genes can also be used to design molecular markers to distinguish poplar clones with high regeneration efficiency.
The comparative analysis of transcriptional data between Arabidopsis and poplar at the locus level indicated that about half and half of REGs exhibited positive and negative correlations in expression levels between these two plant species (Figure 2a). The gene expression of key regulators, such as AUX/LAX, bZIP, and GRAS families, showed positive correlations during shoot regeneration, suggesting the core regulatory elements are conserved in plant species. Different genes in the same pathway may also exhibit different patterns. For example, the expression patterns of PIN1 genes were similar between poplar and Arabidopsis, which were induced after treatment on CIM media (Figure 6). In contrast, the expression patterns of PIN2, PIN3, and AUX1 were different in two plant species, which were highly expressed in leaf tissues before callus induction in hybrid poplar but lowly expressed in Arabidopsis at the same stage. The variations in gene expression during shoot regeneration among different plants agree with the diverse regeneration efficiency of plant species [45]. This also suggests that the top candidates for perturbation to improve regeneration efficiency could vary among plants. Multiple transcription factors (TFs) such as Baby boom, Wuschel2, GROWTH-REGULATING FACTOR5(GRF5), GRF4, and Wuschel-homeobox 5 related genes were reported to enhance regeneration and transformation efficiency [46,47,48]. The information about transcriptional dynamics during shoot regeneration could be used to optimize the combination of TFs to further improve plant regeneration.
As an inter-species hybrid poplar, the expression levels of two alleles can be precisely distinguished from each other because of the genomic variations between two subgenomes. The analysis of two alleles in hybrid poplar 84K during shoot regeneration indicated that most of the alleles exhibited a positive correlation in expression level, suggesting the expression patterns of these REG alleles are mostly determined by trans-acting factors, such as transcription factors. This also agrees with the fact that overexpression of single or fused transcription factors can improve regeneration efficiency in diverse species. Our results suggest that most of the alleles could also be synchronized when regeneration-inducing transcription factors are overexpressed in hybrid poplars.
Whole genome duplication (WGD) events are key driving forces in plant evolution. During the evolution history, WGD provided novel genetic materials with new genomic variations and contributed to the species diversification process [49]. After duplication, some genes are lost while others are retained, followed by neofunctionalization and subfunctionalization [50]. Distinct expression and methylation patterns have also been reported between duplicated genes [51]. In our analysis, we identified 895 REG-homologous genes (REGHs) in Arabidopsis, 1217 REGH alleles from the A subgenome of hybrid poplar, and 1229 REGH alleles from the G subgenome. Further analysis of these genes at different regeneration stages indicated that all members of some gene families are highly expressed in all the stages, whereas in some other gene families, the portions of expressed gene members vary by stage (Figure 3). In STEM clustering analysis, the REGHs in Arabidopsis and poplar were clustered into major groups based on expression patterns during regeneration stages. In each cluster, the gene numbers vary by gene family and distribution patterns. Gene numbers are quite different between Arabidopsis and hybrid poplar. These data suggest that the transcriptional diversity of duplicated genes could also contribute to the variation in regeneration efficiency among plant species.
The presence and absence variation (PAV) between two subgenomes in the hybrid results in some REG alleles being present only in one subgenome and exhibiting distinct expression in comparison to other paralogs (Figure 6). The PAV of REGs may contribute to transcriptional diversity during shoot regeneration. Recently, a model has been proposed to explain the transcriptional dynamics during plant regeneration [52]. The result showed that the process of protoplast isolation could induce the expression of driver genes in regeneration at a very low frequency. Some cells with certain combinations of transcription levels of key genes were further selected by the culture medium. The transcriptome chaos is proposed to be the key foundation for the selection of cells with regeneration ability [52]. In poplar, most of the transgenic systems were developed for hybrid poplars rather than pure species, suggesting the advance of hybrids in plant regeneration, which may be contributed by the transcriptional diversity conferred by the PAVs between subgenomes in the hybrids. Considering the genetic complexity caused by recombination in hybridization, the screening of plant tissues for explants through genotyping would further improve regeneration efficiency.
Single-cell RNA sequencing (scRNA-seq) technologies provide a revolutionary level for studying the transcriptional dynamics during plant regeneration [14,15]. A recent single-cell RNA-seq study in Arabidopsis callus indicates that, among the identified three major cell layers, the middle layer is crucial for organ regeneration [53]. The spatial transcriptome study of tomato callus also revealed the heterogeneous cell populations in callus during shoot regeneration and found the roles of chlorenchyma cells in shoot primordia formation and shoot regeneration [54]. Considering the heterozygous genome in forest trees, the transcriptional regulation at the single-cell level would be more complicated. Further analysis of single-cell and/or spatial transcriptome during shoot regeneration of forest trees at allele level would provide more clues to address the challenges to improve regeneration rate for subsequent genome editing.
5. Conclusions
In conclusion, our analysis identified REGs and REGHs in hybrid poplar and revealed the conservativeness and diversity of transcriptional regulation during shoot regeneration in poplar and Arabidopsis. The expression patterns of most allele pairs of REGs in hybrid poplar were positively correlated, suggesting the trans-acting regulation of these alleles. The expression patterns of gene families also suggest the contribution of gene duplication to transcriptional complexity in hybrid poplar. Further analysis using single-cell and/or spatial transcriptome techniques would provide more details about regulatory mechanisms during shoot regeneration. The screening of explants using genotype information would facilitate the improvement of regeneration efficiency.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/f14112195/s1, Figure S1. Expression trends of REG-homologous genes in dataset 1 in poplar 84K and Arabidopsis. Four clusters (C7 to C10) were identified through STEM (Short Time-series Expression Miner) trend analysis. In each cluster, the trend line plot on the left side was used to show the gene expression patterns of poplar and Arabidopsis genes. The bar plot of each cluster on the right side shows the gene number of each gene family. The coral bars above the x-axis indicated gene numbers in Arabidopsis (AT), and the cyan bars below the x-axis indicated gene numbers in hybrid poplar (84K). The names of gene families are shown in colored bars at the bottom right. In the trend line plot, the stages of shoot regeneration are indicated at the x-axis. S1 and S2 indicate the two datasets of poplar and Arabidopsis. S1_T1, S1_T2, S1_T3, and S1_T4 were from dataset S1, corresponding to S1_Pag_CIM0, S1_Pag_CIM8, S1_Pag_SIM4, and S1_Pag_SIM8 in poplar, and S1_At_CIM0, S1_At_CIM4, S1_At_SIM4, and S1_At_SIM6 in Arabidopsis. Figure S2. Expression trends of REG-homologous genes in dataset 2 in poplar 84K and Arabidopsis. The processes of data analysis and visualization are the same as in Figure S1. S2_T1, S2_T2, S2_T3, S2_T4, S2_T5, and S2_T6 were from dataset S2, corresponding to S2_Pag_CIM0, S2_Pag_CIM7, S2_Pag_CIM14, S2_Pag_SIM7, S2_Pag_SIM14, and S2_Pag_SIM21 in poplar, and S2_At_CIM0, S2_At_CIM3, S2_At_CIM7, S2_At_SIM3, S2_At_SIM6, and S2_At_SIM8 in Arabidopsis. Table S1. Regeneration-related genes in Arabidopsis. Table S2. Gene list of REGHs in Arabidopsis. Table S3. Regeneration-related genes in hybrid poplar 84K. Table S4. Gene list of REGHs in hybrid poplar 84K. Table S5. Names of genes in Figure 2a. Table S6. Identification of gene pairs in Figure 5.
Author Contributions
L.X. conceived and designed the experiments. L.X., X.D. and C.W. wrote the paper. X.D., C.W., G.Y. and Y.G. performed the experiments and data analysis. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (32171826 to L.X.).
Data Availability Statement
The transcriptome data of Arabidopsis were downloaded from NCBI SRA (https://www.ncbi.nlm.nih.gov/sra, accessed on 21 December 2022, accession number PRJNA592549) and CNCB NGDC (https://ngdc.cncb.ac.cn, accessed on 21 December 2022, accession number PRJCA005872). The data for the hybrid poplar were downloaded from NCBI SRA (accession number PRJNA395928), accessed date is 3 September 2022.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Perez-Garcia, P.; Moreno-Risueno, M.A. Stem cells and plant regeneration. Dev. Biol. 2018, 442, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Ikeuchi, M.; Favero, D.S.; Sakamoto, Y.; Iwase, A.; Coleman, D.; Rymen, B.; Sugimoto, K. Molecular Mechanisms of Plant Regeneration. Annu. Rev. Plant Biol. 2019, 70, 377–406. [Google Scholar] [CrossRef] [PubMed]
- Ince, Y.Ç.; Sugimoto, K. Illuminating the path to shoot meristem regeneration: Molecular insights into reprogramming cells into stem cells. Curr. Opin. Plant Biol. 2023, 102452. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Poretska, O.; Sieberer, T. ALTERED MERISTEM PROGRAM1 Restricts Shoot Meristem Proliferation and Regeneration by Limiting HD-ZIP III-Mediated Expression of RAP2.6L. Plant Physiol. 2018, 177, 1580–1594. [Google Scholar] [CrossRef] [PubMed]
- Ikeuchi, M.; Iwase, A.; Rymen, B.; Lambolez, A.; Kojima, M.; Takebayashi, Y.; Heyman, J.; Watanabe, S.; Seo, M.; De Veylder, L.; et al. Wounding Triggers Callus Formation via Dynamic Hormonal and Transcriptional Changes. Plant Physiol. 2017, 175, 1158–1174. [Google Scholar] [CrossRef] [PubMed]
- Eshed Williams, L. Genetics of Shoot Meristem and Shoot Regeneration. Annu. Rev. Genet. 2021, 55, 661–681. [Google Scholar] [CrossRef] [PubMed]
- Feher, A. Somatic embryogenesis—Stress-induced remodeling of plant cell fate. Biochim. Biophys. Acta 2015, 1849, 385–402. [Google Scholar] [CrossRef]
- Efferth, T. Biotechnology Applications of Plant Callus Cultures. Engineering 2019, 5, 50–59. [Google Scholar] [CrossRef]
- Liu, X.; Bie, X.M.; Lin, X.; Li, M.; Wang, H.; Zhang, X.; Yang, Y.; Zhang, C.; Zhang, X.S.; Xiao, J. Uncovering the transcriptional regulatory network involved in boosting wheat regeneration and transformation. Nat. Plants 2023, 9, 908–925. [Google Scholar] [CrossRef]
- Wang, F.X.; Shang, G.D.; Wang, J.W. Towards a hierarchical gene regulatory network underlying somatic embryogenesis. Trends Plant Sci. 2022, 27, 1209–1217. [Google Scholar] [CrossRef]
- Zhang, G.; Zhao, F.; Chen, L.; Pan, Y.; Sun, L.; Bao, N.; Zhang, T.; Cui, C.X.; Qiu, Z.; Zhang, Y.; et al. Jasmonate-mediated wound signalling promotes plant regeneration. Nat. Plants 2019, 5, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.H.; Zhang, X.S. The hormonal control of regeneration in plants. Curr. Top. Dev. Biol. 2014, 108, 35–69. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Liu, J.; Gao, A.; Chen, X.; Gao, L.; Liao, L.; Luo, B.; Ogutu, C.O.; Han, Y. Epigenetic reprogramming of H3K27me3 and DNA methylation during leaf-to-callus transition in peach. Hortic. Res. 2022, 9, uhac132. [Google Scholar] [CrossRef] [PubMed]
- Liao, R.-Y.; Wang, J.-W. Analysis of meristems and plant regeneration at single-cell resolution. Curr. Opin. Plant Biol. 2023, 74, 102378. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zhang, Y.; Fang, X.; Tran, S.; Zhai, N.; Yang, Z.; Guo, F.; Chen, L.; Yu, J.; Ison, M.S.; et al. Transcriptional landscapes of de novo root regeneration from detached Arabidopsis leaves revealed by time-lapse and single-cell RNA sequencing analyses. Plant Commun. 2022, 3, 100306. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Chen, X.; Huang, H.; Xu, L. Reprogramming of H3K27me3 is critical for acquisition of pluripotency from cultured Arabidopsis tissues. PLoS Genet. 2012, 8, e1002911. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, K.; Temman, H.; Kadokura, S.; Matsunaga, S. To regenerate or not to regenerate: Factors that drive plant regeneration. Curr. Opin. Plant Biol. 2019, 47, 138–150. [Google Scholar] [CrossRef] [PubMed]
- Mathew, M.M.; Prasad, K. Model systems for regeneration: Arabidopsis. Development 2021, 148, dev195347. [Google Scholar] [CrossRef]
- Gaj, M.D.; Trojanowska, A.; Ujczak, A.; Mędrek, M.; Kozioł, A.; Garbaciak, B. Hormone-response mutants of Arabidopsis thaliana (L.) Heynh. impaired in somatic embryogenesis. Plant Growth Regul. 2006, 49, 183–197. [Google Scholar] [CrossRef]
- Liang, Y.; Heyman, J.; Xiang, Y.; Vandendriessche, W.; Canher, B.; Goeminne, G.; De Veylder, L. The wound-activated ERF15 transcription factor drives Marchantia polymorpha regeneration by activating an oxylipin biosynthesis feedback loop. Sci. Adv. 2022, 8, eabo7737. [Google Scholar] [CrossRef]
- Ishikawa, M.; Hasebe, M. Molecular mechanisms of reprogramming of differentiated cells into stem cells in the moss Physcomitrium patens. Curr. Opin. Plant Biol. 2022, 65, 102123. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wei, Z.; Xing, S. Stable plastid transformation of rice, a monocot cereal crop. Biochem. Biophys. Res. Commun. 2018, 503, 2376–2379. [Google Scholar] [CrossRef] [PubMed]
- Ahmadabadi, M.; Ruf, S.; Bock, R. A leaf-based regeneration and transformation system for maize (Zea mays L.). Transgenic Res. 2007, 16, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Han, S.; Zhang, Y.; Hao, D. Genetic architecture of embryogenic callus induction in maize from the perspective of population genomics. Plant Cell Tissue Organ Cult. (PCTOC) 2022, 150, 345–359. [Google Scholar] [CrossRef]
- Pan, W.; Liu, X.; Li, D.; Zhang, H. Establishment of an Efficient Genome Editing System in Lettuce Without Sacrificing Specificity. Front. Plant Sci. 2022, 13, 930592. [Google Scholar] [CrossRef] [PubMed]
- Castellanos-Hernández, O.A.; Rodriguez-Sahagun, A.; Acevedo-Hernández, G.J.; Herrera-Estrella, L.R. Genetic Transformation of Forest Trees; InTechOpen: London, UK, 2011; pp. 190–214. [Google Scholar]
- Shao, L.; Xing, F.; Xu, C.; Zhang, Q.; Che, J.; Wang, X.; Song, J.; Li, X.; Xiao, J.; Chen, L.-L.; et al. Patterns of genome-wide allele-specific expression in hybrid rice and the implications on the genetic basis of heterosis. Proc. Natl. Acad. Sci. USA 2019, 116, 5653–5658. [Google Scholar] [CrossRef] [PubMed]
- Wen, S.S.; Ge, X.L.; Wang, R.; Yang, H.F.; Bai, Y.E.; Guo, Y.H.; Zhang, J.; Lu, M.Z.; Zhao, S.T.; Wang, L.Q. An Efficient Agrobacterium-Mediated Transformation Method for Hybrid Poplar 84K (Populus alba × P. glandulosa) Using Calli as Explants. Int. J. Mol. Sci. 2022, 23, 2216. [Google Scholar] [CrossRef]
- Gallardo, F.; Fu, J.; Cantón, F.R.; García-Gutiérrez, A.; Cánovas, F.M.; Kirby, E.G. Expression of a conifer glutamine synthetase gene in transgenic poplar. Planta 1999, 210, 19–26. [Google Scholar] [CrossRef]
- Liu, B.; Zhang, J.; Yang, Z.; Matsui, A.; Seki, M.; Li, S.; Yan, X.; Kohnen, M.V.; Gu, L.; Prasad, K.; et al. PtWOX11 acts as master regulator conducting the expression of key transcription factors to induce de novo shoot organogenesis in poplar. Plant Mol. Biol. 2018, 98, 389–406. [Google Scholar] [CrossRef]
- Coleman, D.; Kawamura, A.; Ikeuchi, M.; Favero, D.S.; Lambolez, A.; Rymen, B.; Iwase, A.; Suzuki, T.; Sugimoto, K. The SUMO E3 Ligase SIZ1 Negatively Regulates Shoot Regeneration. Plant Physiol. 2020, 184, 330–344. [Google Scholar] [CrossRef]
- Wu, L.Y.; Shang, G.D.; Wang, F.X.; Gao, J.; Wan, M.C.; Xu, Z.G.; Wang, J.W. Dynamic chromatin state profiling reveals regulatory roles of auxin and cytokinin in shoot regeneration. Dev. Cell 2022, 57, 526–542. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Qiu, D.; Bai, S.; Ma, J.; Zhang, L.; Shao, F.; Zhang, K.; Yang, Y.; Sun, T.; Huang, J.; Zhou, Y.; et al. The genome of Populus alba x Populus tremula var. glandulosa clone 84K. DNA Res. 2019, 26, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Jiao, C.; Sun, H.; Rosli, H.G.; Pombo, M.A.; Zhang, P.; Banf, M.; Dai, X.; Martin, G.B.; Giovannoni, J.J.; et al. iTAK: A Program for Genome-wide Prediction and Classification of Plant Transcription Factors, Transcriptional Regulators, and Protein Kinases. Mol. Plant 2016, 9, 1667–1670. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Jiao, B.; Yang, Y.; Shan, L.; Li, T.; Li, X.; Xi, Z.; Wang, X.; Liu, J. WGDI: A user-friendly toolkit for evolutionary analyses of whole-genome duplications and ancestral karyotypes. Mol. Plant 2022, 15, 1841–1851. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, Y.; Kawamura, A.; Suzuki, T.; Segami, S.; Maeshima, M.; Polyn, S.; De Veylder, L.; Sugimoto, K. Transcriptional activation of auxin biosynthesis drives developmental reprogramming of differentiated cells. Plant Cell 2022, 34, 4348–4365. [Google Scholar] [CrossRef]
- Shin, J.; Bae, S.; Seo, P.J. De novo shoot organogenesis during plant regeneration. J. Exp. Bot. 2020, 71, 63–72. [Google Scholar] [CrossRef]
- Iwase, A.; Mita, K.; Nonaka, S.; Ikeuchi, M.; Koizuka, C.; Ohnuma, M.; Ezura, H.; Imamura, J.; Sugimoto, K. WIND1-based acquisition of regeneration competency in Arabidopsis and rapeseed. J. Plant Res. 2015, 128, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Tong, J.; Xiao, L.; Ruan, Y.; Liu, J.; Zeng, M.; Huang, H.; Wang, J.W.; Xu, L. YUCCA-mediated auxin biogenesis is required for cell fate transition occurring during de novo root organogenesis in Arabidopsis. J. Exp. Bot. 2016, 67, 4273–4284. [Google Scholar] [CrossRef] [PubMed]
- Shi, B.; Zhang, C.; Tian, C.; Wang, J.; Wang, Q.; Xu, T.; Xu, Y.; Ohno, C.; Sablowski, R.; Heisler, M.G.; et al. Two-Step Regulation of a Meristematic Cell Population Acting in Shoot Branching in Arabidopsis. PLoS Genet. 2016, 12, e1006168. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Debernardi, J.M.; Dubcovsky, J.; Gallavotti, A. Recent advances in crop transformation technologies. Nat. Plants 2022, 8, 1343–1351. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, C.; Liu, Z.; Heidmann, I.; Supena, E.D.J.; Fukuoka, H.; Joosen, R.; Lambalk, J.; Angenent, G.; Scorza, R.; Custers, J.B.M.; et al. Heterologous expression of the BABY BOOM AP2/ERF transcription factor enhances the regeneration capacity of tobacco (Nicotiana tabacum L.). Planta 2007, 225, 341–351. [Google Scholar] [CrossRef]
- Debernardi, J.M.; Tricoli, D.M.; Ercoli, M.F.; Hayta, S.; Ronald, P.; Palatnik, J.F.; Dubcovsky, J. A GRF–GIF chimeric protein improves the regeneration efficiency of transgenic plants. Nat. Biotechnol. 2020, 38, 1274–1279. [Google Scholar] [CrossRef] [PubMed]
- Kong, J.; Martin-Ortigosa, S.; Finer, J.; Orchard, N.; Gunadi, A.; Batts, L.A.; Thakare, D.; Rush, B.; Schmitz, O.; Stuiver, M.; et al. Overexpression of the Transcription Factor GROWTH-REGULATING FACTOR5 Improves Transformation of Dicot and Monocot Species. Front. Plant Sci. 2020, 11, 572319. [Google Scholar] [CrossRef]
- Gao, B.; Chen, M.; Li, X.; Liang, Y.; Zhu, F.; Liu, T.; Zhang, D.; Wood, A.J.; Oliver, M.J.; Zhang, J. Evolution by duplication: Paleopolyploidy events in plants reconstructed by deciphering the evolutionary history of VOZ transcription factors. BMC Plant Biol. 2018, 18, 256. [Google Scholar] [CrossRef]
- Kuzmin, E.; Taylor, J.S.; Boone, C. Retention of duplicated genes in evolution. Trends Genet. 2022, 38, 59–72. [Google Scholar] [CrossRef]
- Shi, T.; Rahmani, R.S.; Gugger, P.F.; Wang, M.; Li, H.; Zhang, Y.; Li, Z.; Wang, Q.; Van de Peer, Y.; Marchal, K.; et al. Distinct Expression and Methylation Patterns for Genes with Different Fates following a Single Whole-Genome Duplication in Flowering Plants. Mol. Biol. Evol. 2020, 37, 2394–2413. [Google Scholar] [CrossRef]
- Xu, M.; Du, Q.; Tian, C.; Wang, Y.; Jiao, Y. Stochastic gene expression drives mesophyll protoplast regeneration. Sci. Adv. 2021, 7, eabg8466. [Google Scholar] [CrossRef]
- Zhai, N.; Xu, L. Pluripotency acquisition in the middle cell layer of callus is required for organ regeneration. Nat. Plants 2021, 7, 1453–1460. [Google Scholar] [CrossRef]
- Song, X.; Guo, P.; Xia, K.; Wang, M.; Liu, Y.; Chen, L.; Zhang, J.; Xu, M.; Liu, N.; Yue, Z.; et al. Spatial transcriptomics reveals light-induced chlorenchyma cells involved in promoting shoot regeneration in tomato callus. Proc. Natl. Acad. Sci. USA 2023, 120, e2310163120. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).