
Genetic Diversity and Structure of Quercus hondae, a Rare Evergreen Oak Species in Southwestern Japan

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
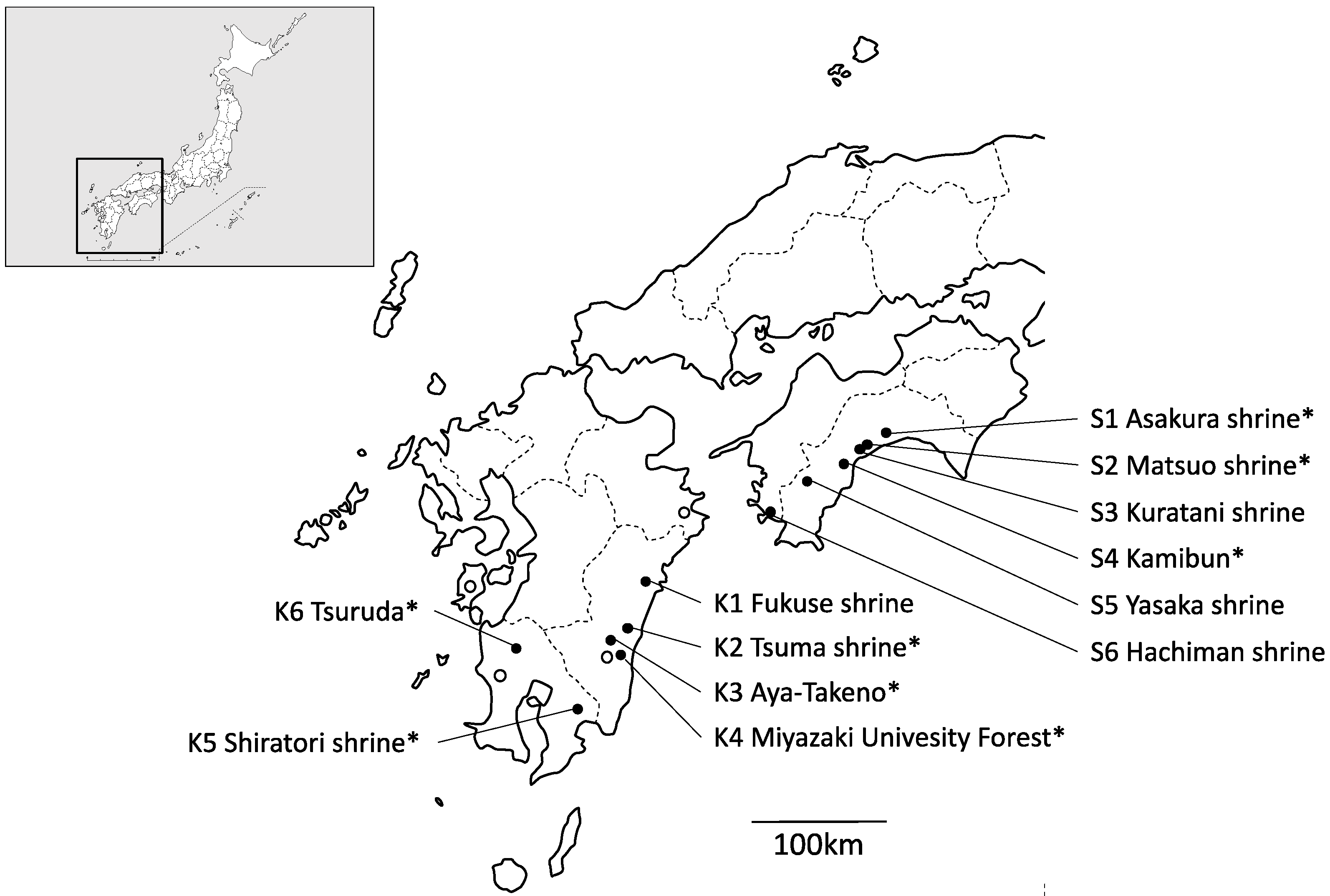
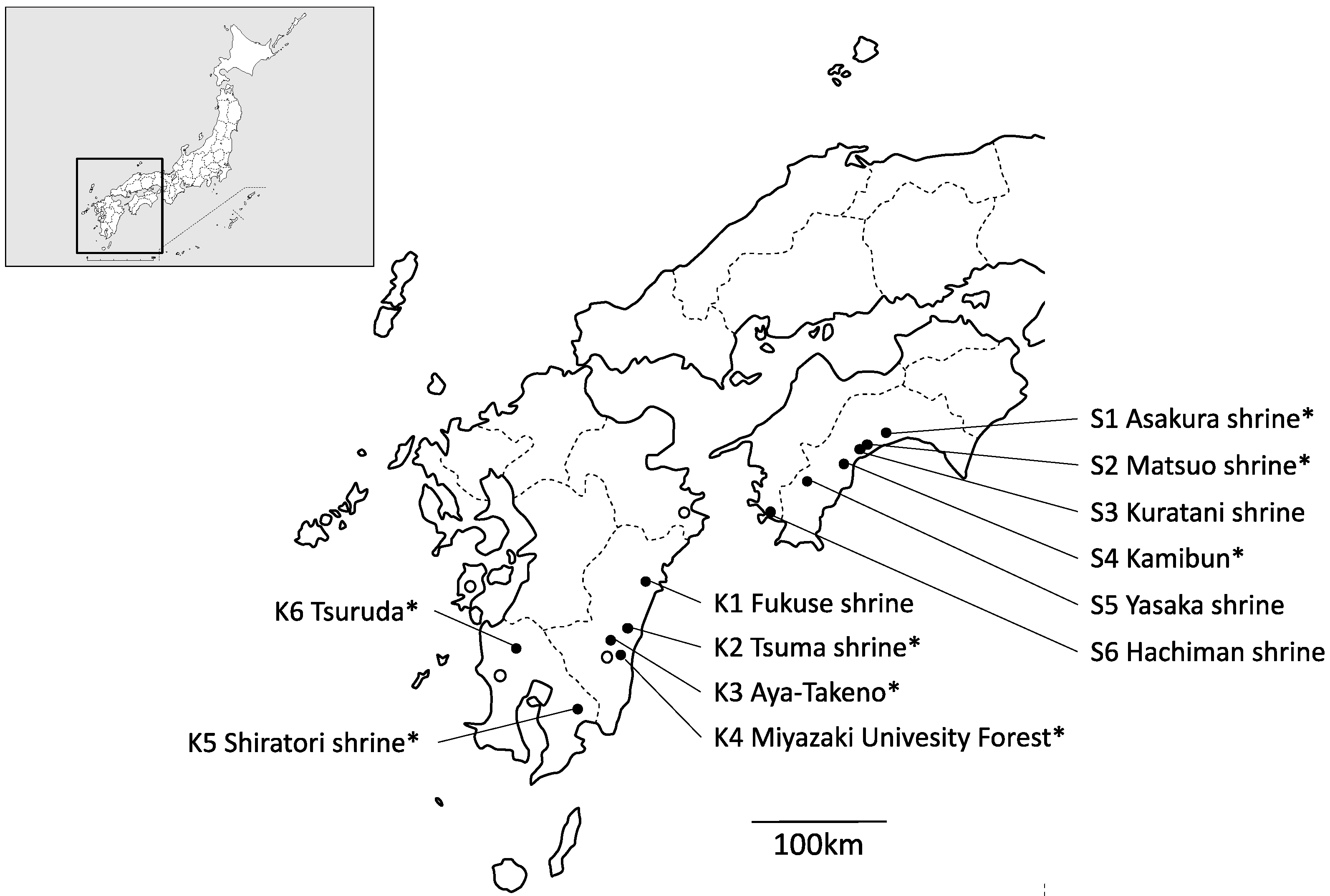
2.1. Collecting Population Samples and DNA Extraction
2.2. Chloroplast DNA Sequencing and Data Analysis
2.3. Microsatellite Genotyping and Data Analysis
3. Results
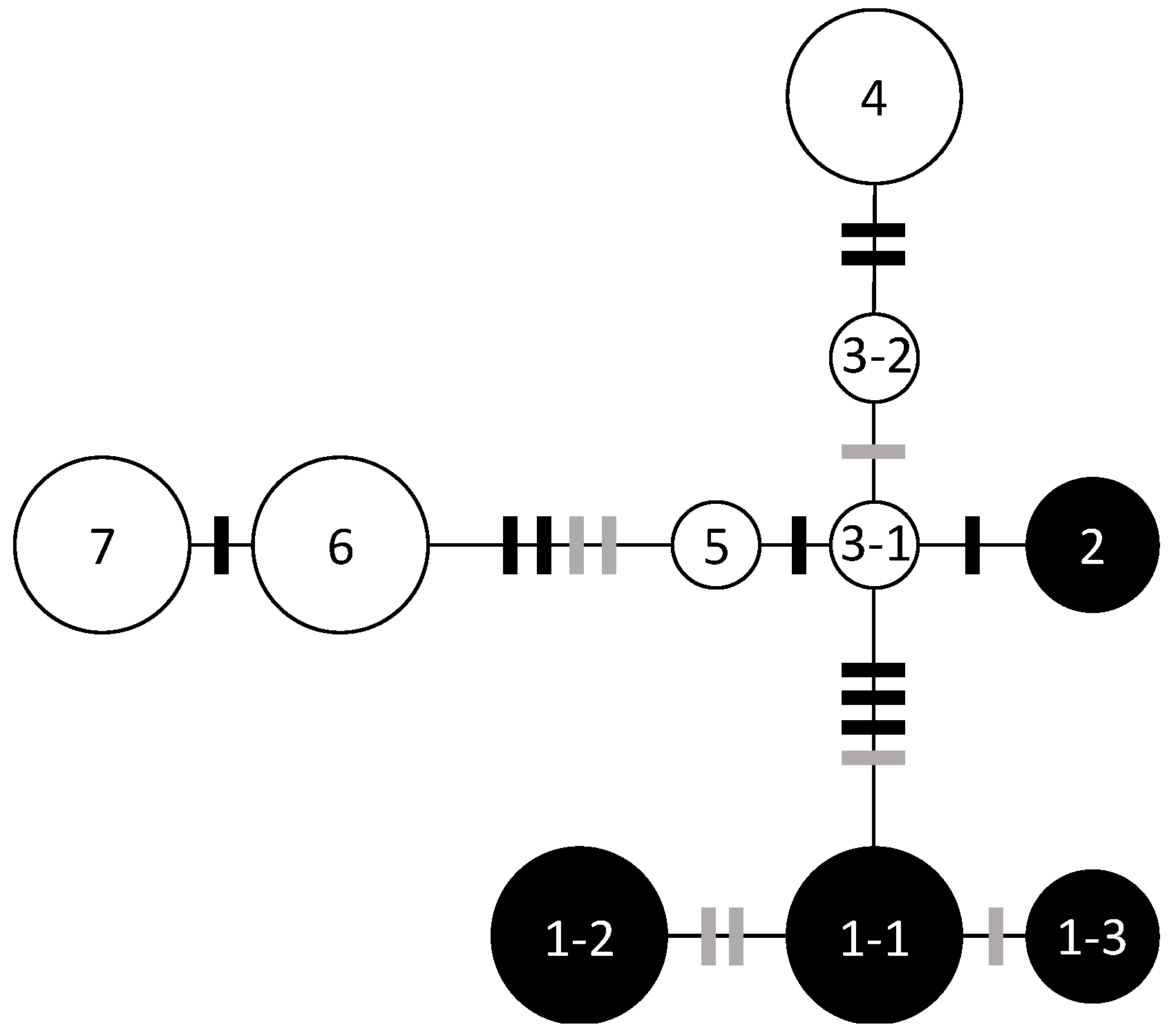
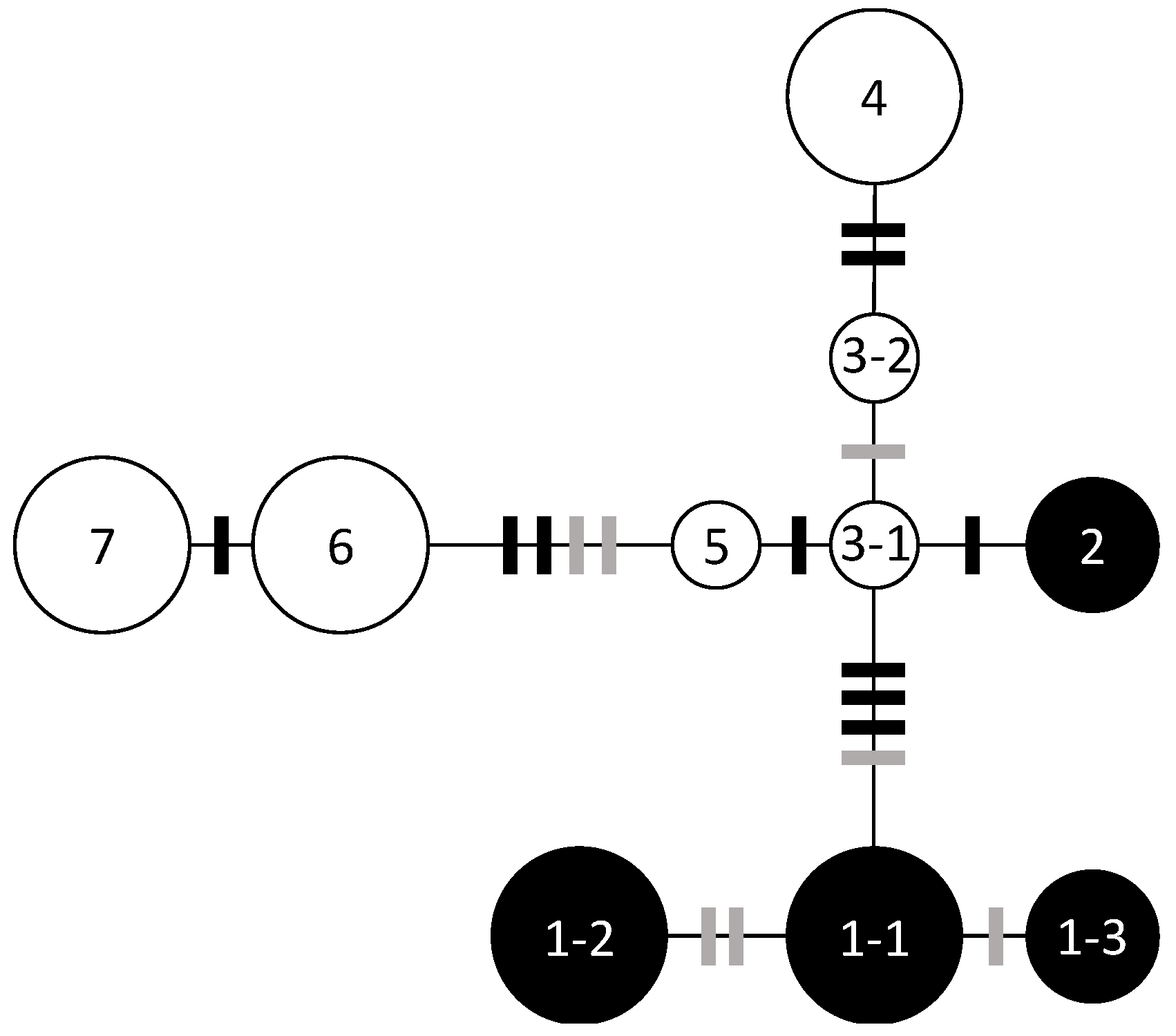
3.1. Chloroplast DNA Haplotype Variations
3.2. Genetic Diversity and Genetic Structure Based on Microsatellite Markers
4. Discussion
4.1. Low Genetic Diversity of Q. hondae
4.2. High Genetic Differentiation between Populations of Q. hondae
4.3. Implications for Conservation of Q. hondae
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hamrick, J.L.; Godt, M.J.W. Allozyme diversity in plant species. In Plant Population Genetics, Breeding, and Genetic Resources; Brown, A.H.D., Clegg, M.T., Kahler, A.L., Weir, B.S., Eds.; Sinouer Association: Sunderland, MA, USA, 1989; pp. 43–63. [Google Scholar]
- Karron, J.D. A comparison of levels of genetic polymorphism and self-compatibility in geographically restricted and widespread plant congeners. Evol. Ecol. 1987, 1, 47–58. [Google Scholar] [CrossRef]
- Gitzendanner, M.A.; Soltis, P.S. Patterns of genetic variation in rare and widespread plant congeners. Am. J. Bot. 2000, 87, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Yoichi, W.; Tomaru, N. Patterns of geographic distribution have a considerable influence on population genetic structure in one common and two rare species of Rhododendron (Ericaceae). Tree Genet. Genom. 2014, 10, 827–837. [Google Scholar] [CrossRef]
- Backs, J.R.; Ashley, M.V. Quercus conservation genetics and genomics: Past, present, and future. Forests 2021, 12, 882. [Google Scholar] [CrossRef]
- Cannon, C.H.; Petit, R.J. The oak syngameon: More than the sum of its parts. New Phytol. 2020, 226, 978–983. [Google Scholar] [CrossRef] [PubMed]
- Cannon, C.H.; Lerdau, M.T. Demography and destiny: The syngameon in hyperdiverse systems. Proc. Natl. Acad. Sci. USA 2019, 116, 8105. [Google Scholar] [CrossRef] [PubMed]
- Lazic, D.; Hipp, A.L.; Carlson, J.E.; Gailing, O. Use of genomic resources to assess adaptive divergence and introgression in oaks. Forests 2021, 12, 690. [Google Scholar] [CrossRef]
- Kamiya, K.; Harada, K.; Ogino, K.; Clyde, M.M.; Latiff, A.M. Phylogeny and genetic variation of Fagaceae in tropical montane forests. Tropics 2003, 13, 119–125. [Google Scholar] [CrossRef]
- Ito, S.; Ohtsuka, K.; Yamashita, T. Ecological distribution of seven evergreen Quercus species in southern and eastern Kyushu, Japan. Veg. Sci. 2007, 24, 53–63. [Google Scholar] [CrossRef]
- Han, E.K.; Cho, W.B.; Park, J.S.; Choi, I.S.; Kwak, M.; Kim, B.Y.; Lee, J.H. A Disjunctive Marginal Edge of Evergreen Broad-Leaved Oak (Quercus gilva) in East Asia: The High Genetic Distinctiveness and Unusual Diversity of Jeju Island Populations and Insight into a Massive, Independent Postglacial Colonization. Genes 2020, 11, 1114. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Lee, D.-H.; Choi, I.-S.; Choi, B.-H. Genetic diversity and historical migration patterns of an endemic evergreen oak, Quercus acuta, across Korea and Japan, inferred from nuclear microsatellites. Plant Syst. Evol. 2014, 300, 1913–1923. [Google Scholar] [CrossRef]
- Murray, M.G.; Thompson, W.F. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 1980, 8, 4321–4325. [Google Scholar] [CrossRef] [PubMed]
- Tate, J.A.; Simpson, B.B. Paraphyly of Tarasa (Malvaceae) and diverse origins of the polyploid species. Syst. Bot. 2003, 28, 723–737. [Google Scholar] [CrossRef]
- Sang, T.; Crawford, D.J.; Stuessy, T.F. Chloroplast DNA phylogeny, reticulate evolution, and biogeography of Paeonia (Paeoniaceae). Am. J. Bot. 1997, 84, 1120–1136. [Google Scholar] [CrossRef]
- Kanno, M.; Yokoyama, J.; Suyama, Y.; Ohyama, M.; Itoh, T.; Suzuki, M. Geographical distribution of two haplotypes of chloroplast DNA in four oak species (Quercus) in Japan. J. Plant Res. 2004, 117, 311–317. [Google Scholar] [CrossRef]
- Gouy, M.; Guindon, S.; Gascuel, O. SeaView version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 2010, 27, 221–224. [Google Scholar] [CrossRef]
- Clement, M.; Posada, D.; Crandall, K.A. TCS: A computer program to estimate gene genealogies. Mol. Ecol. 2000, 9, 1657–1659. [Google Scholar] [CrossRef]
- Tong, X.; Xu, N.N.; Li, L.; Chen, X.Y. Development and characterization of polymorphic microsatellite markers in Cyclobalanopsis glauca (Fagaceae). Am. J. Bot. 2012, 99, e120–e122. [Google Scholar] [CrossRef]
- Lee, J.-H.; Park, M.-H.; Min, G.-S.; Choi, B.-H. Isolation and Characterization of 13 Microsatellite Loci from Korean Quercus acuta (Fagaceae). J. Plant Biol. 2010, 53, 201–204. [Google Scholar] [CrossRef]
- Meirmans, P.G. GENODIVE version 3.0: Easy-to-use software for the analysis of genetic data of diploids and polyploids. Mol. Ecol. Resour. 2020, 20, 1126–1131. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef]
- Goudet, J. FSTAT (version 1.2): A computer program to calculate F-statistics. J. Hered. 1995, 86, 485–486. [Google Scholar] [CrossRef]
- Chybicki, I.J.; Burczyk, J. Simultaneous estimation of null alleles and inbreeding coefficients. J. Hered. 2009, 100, 106–113. [Google Scholar] [CrossRef]
- Nei, M.; Maruyama, T.; Chakraborty, R. The Bottleneck Effect and Genetic Variability in Populations. Evolution 1975, 29, 1–10. [Google Scholar] [CrossRef]
- Cornuet, J.M.; Luikart, G. Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 1996, 144, 2001–2014. [Google Scholar] [CrossRef]
- Piry, S.; Luikart, G.; Cornuet, J.M. Computer note. BOTTLENECK: A computer program for detecting recent reductions in the effective size using allele frequency data. J. Hered. 1999, 90, 502–503. [Google Scholar] [CrossRef]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef]
- Nei, M. Molecular Evolutionary Genetics; Columbia University Press: New York, NY, USA, 1987. [Google Scholar]
- Langella, O. Populations 1.2.30: Population Genetic Software (Individuals or Population Distances, Phylogenetic Trees). 2007. Available online: https://bioinformatics.org/populations/ (accessed on 22 February 2022).
- Weir, B.S.; Cockerham, C.C. Estimating F-Statistics for the analysis of population structure. Evolution 1984, 38, 1358–1370. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics 2003, 164, 1567–1587. [Google Scholar] [CrossRef] [PubMed]
- Hubisz, M.J.; Falush, D.; Stephens, M.; Pritchard, J.K. Inferring weak population structure with the assistance of sample group information. Mol. Ecol. Resour. 2009, 9, 1322–1332. [Google Scholar] [CrossRef]
- Earl, D.A.; von Holdt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2011, 4, 359–361. [Google Scholar] [CrossRef]
- Kopelman, N.M.; Mayzel, J.; Jakobsson, M.; Rosenberg, N.A.; Mayrose, I. Clumpak: A program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Resour. 2015, 15, 1179–1191. [Google Scholar] [CrossRef] [PubMed]
- Cole, C.T. Genetic variation in rare and common plants. Annu. Rev. Ecol. Evol. Syst. 2003, 34, 213–237. [Google Scholar] [CrossRef]
- Ito, S.; Nakagawa, M.; Buckley, G.P.; Nogami, K. Species richness in sugi (Cryptomeria japonica D. DON) plantations in southeastern Kyushu, Japan: The effects of stand type and age on understory trees and shrubs. J. For. Res. 2003, 8, 49–57. [Google Scholar] [CrossRef]
- Whittemore, A.T.; Schaal, B.A. Interspecific gene flow in sympatric oaks. Proc. Natl. Acad. Sci. USA 1991, 88, 2540–2544. [Google Scholar] [CrossRef] [PubMed]
- Dumolin-Lapegue, S.; Demesure, B.; Fineschi, S.; Le Corre, V.; Petit, R.J. Phylogeographic structure of white oaks throughout the European continent. Genetics 1997, 146, 1475–1487. [Google Scholar] [CrossRef] [PubMed]
- Okaura, T.; Quang, N.D.; Ubukata, M.; Harada, K. Phylogeographic structure and late Quaternary population history of the Japanese oak Quercus mongolica var. crispula and related species revealed by chloroplast DNA variation. Genes Genet. Syst. 2007, 82, 465–477. [Google Scholar] [CrossRef]
- Tsukada, M. Map of vegetation during the last glacial maximum in Japan. Quatern. Res. 1985, 23, 369–381. [Google Scholar] [CrossRef]
- Aoki, K.; Suzuki, T.; Hsu, T.W.; Murakami, N. Phylogeography of the component species of broad-leaved evergreen forests in Japan, based on chloroplast DNA variation. J. Plant Res. 2004, 117, 77–94. [Google Scholar] [CrossRef]
- Yoshida, T.; Nagai, H.; Yahara, T.; Tachida, H. Genetic structure and putative selective sweep in the pioneer tree, Zanthoxylum ailanthoides. J. Plant Res. 2010, 123, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Aoki, K.; Ueno, S.; Kamijo, T.; Setoguchi, H.; Murakami, N.; Kato, M.; Tsumura, Y. Genetic differentiation and genetic diversity of Castanopsis (Fagaceae), the dominant tree species in Japanese broadleaved evergreen forests, revealed by analysis of EST-associated microsatellites. PLoS ONE 2014, 9, e87429. [Google Scholar] [CrossRef]
- Hamrick, J.L.; Godt, M.J.W.; Sherman-Broyles, S.L. Factors influencing levels of genetic diversity in woody plant species. New For. 1992, 6, 95–124. [Google Scholar] [CrossRef]
- Rosenberg, N.A.; Burke, T.; Elo, K.; Feldman, M.W.; Freidlin, P.J.; Groenen, M.A.M.; Hillel, J.; Maki-Tanila, A.; Tixier-Boichard, M.l.; Vignal, A.; et al. Empirical Evaluation of Genetic Clustering Methods Using Multilocus Genotypes from 20 Chicken Breeds. Genetics 2001, 159, 699–713. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, N.A.; Pritchard, J.K.; Weber, J.L.; Cann, H.M.; Kidd, K.K.; Zhivotovsky, L.A.; Feldman, M.W. Genetic structure of human populations. Science 2002, 298, 2381–2385. [Google Scholar] [CrossRef] [PubMed]
- Chalupa, V. Vegetative propagation of oak (Quercus robur and Q. petraea) by cutting and tissue culture. Ann. Des Sci. For. INRA/EDP Sci. 1993, 50, 295s–307s. [Google Scholar] [CrossRef]
- Drew, J.J.; Dirr, M.A. Propagation of Quercus L. Species by Cuttings. J. Environ. Hort. 1989, 7, 115–117. [Google Scholar] [CrossRef]
- Kenzo, T.; Ichie, T.; Kamiya, K.; Ngo, K.M.; Lum, S.K.Y. Rooting Ability of Leafy-stem cuttings of hybrid Shorea (Dipterocarpaceae). J. Trop. For. Sci. 2019, 31, 324–331. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
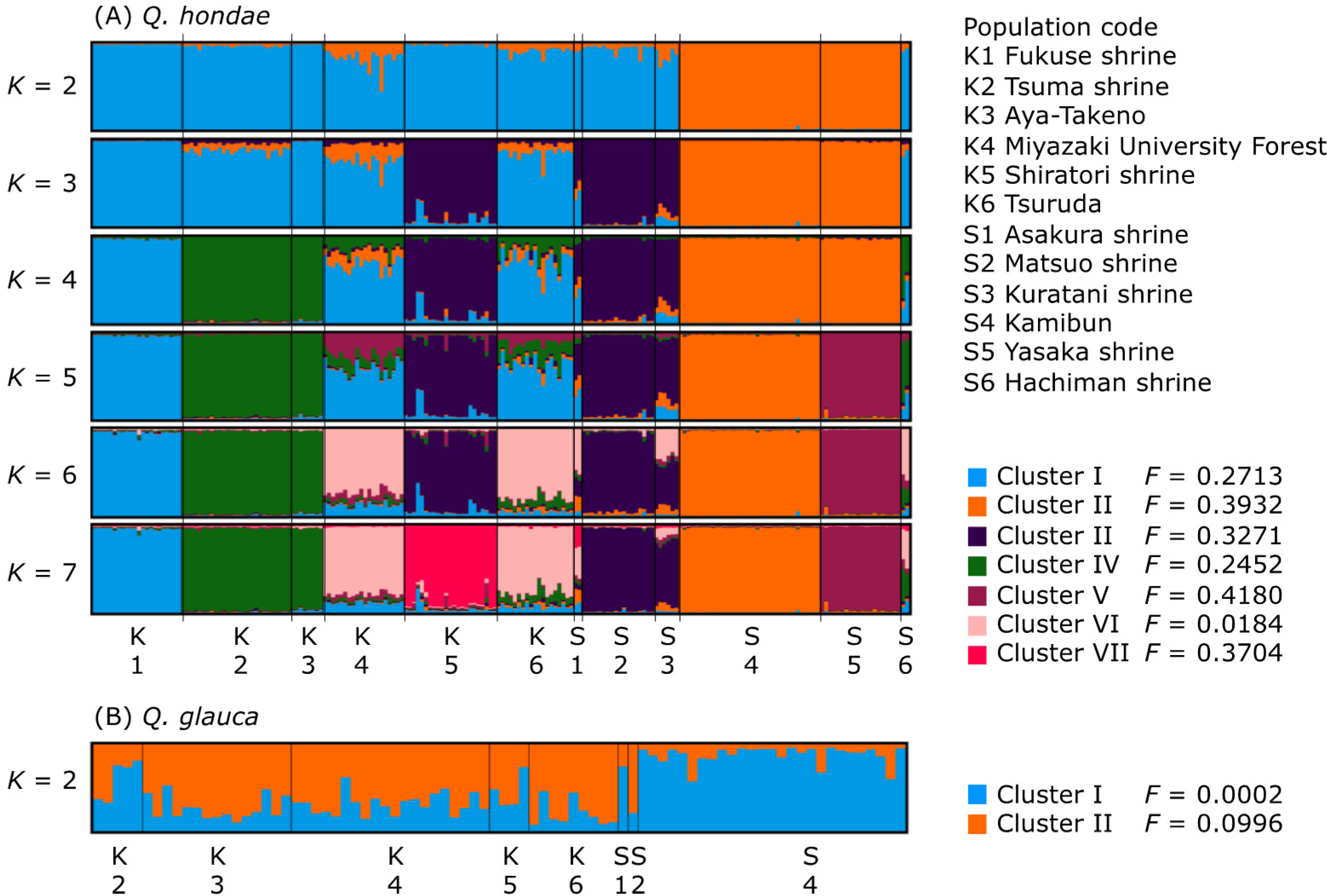{kind=link}
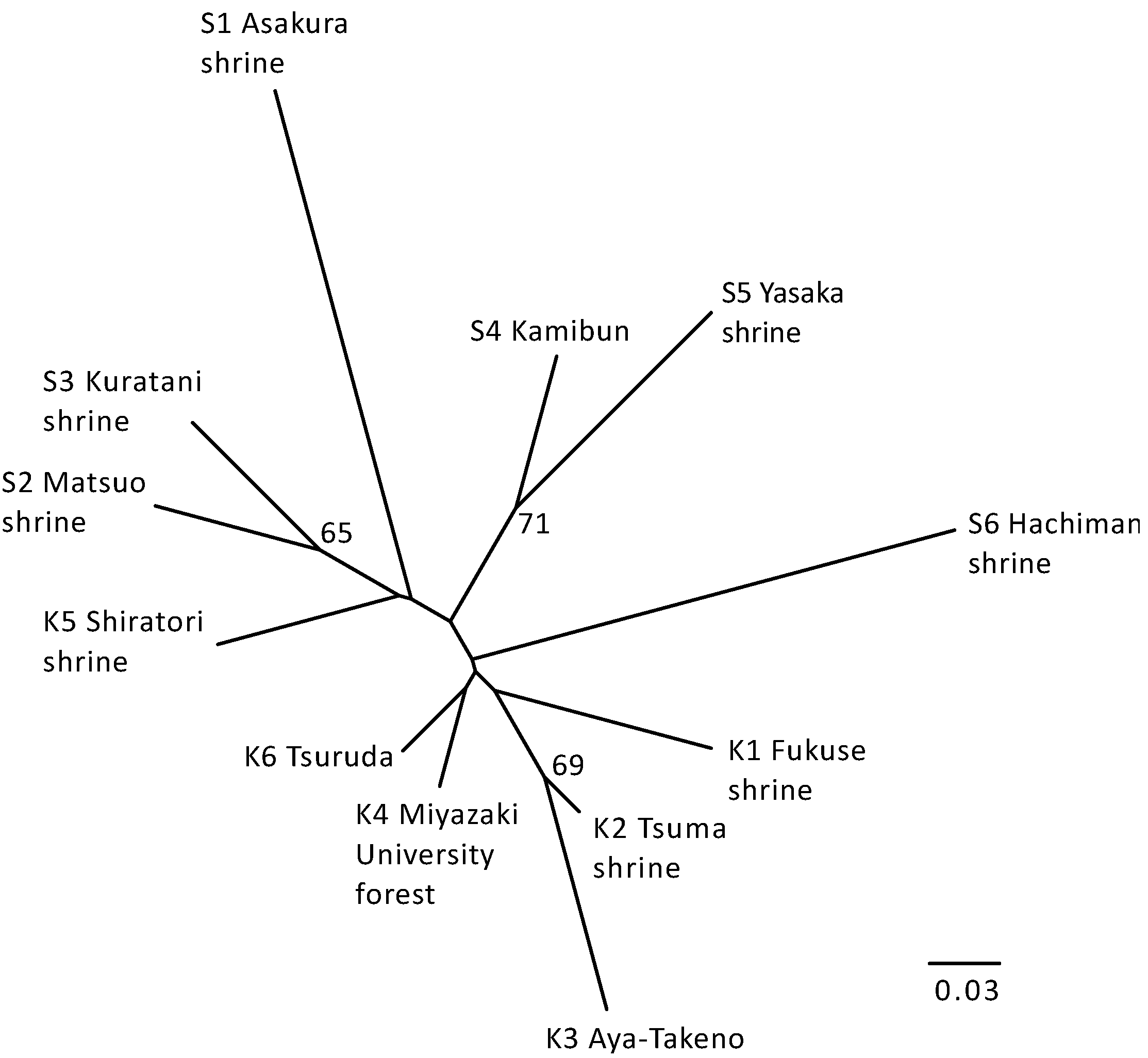{kind=link}
| Population Code | Location | N | Na | Ne | Ho | uHe | Fis (FSTAT) | Fis (INEST) | BOTTLENECK Wilcoxon Test Probability 1 | BOTTLENECK Mode-Shift 2 | cpDNA Haplotypes (n) 3 | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IAM | TPM | SMM | |||||||||||
| Q. hondae | |||||||||||||
| K1 | Miyazaki, Fukuse shrine | 22 | 3.22 | 2.11 | 0.465 | 0.421 | −0.107 | 0.019 | 0.006 | 0.020 | 0.422 | Normal L-shaped | 2 (8) |
| K2 | Miyazaki, Tsuma shrine | 27 | 3.11 | 2.40 | 0.481 | 0.481 | −0.030 | 0.017 | 0.010 | 0.027 | 0.098 | Shifted mode | 1-1 (8) |
| K3 | Miyazaki, Aya-Takeno | 8 | 2.44 | 1.81 | 0.431 | 0.431 | −0.042 | 0.034 | – | – | – | – | 1-2 (8) |
| K4 | Miyazaki, Miyazaki University Forest | 20 | 5.11 | 2.85 | 0.594 | 0.575 | −0.036 | 0.024 | 0.064 | 0.455 | 0.997 | Normal L-shaped | 1-1 (8) |
| K5 | Kagoshima, Shiratori shrine | 23 | 3.44 | 2.02 | 0.527 | 0.482 | −0.095 | 0.014 | 0.150 | 0.410 | 0.674 | Normal L-shaped | 1-3 (8) |
| K6 | Kagoshima, Tsuruda | 19 | 4.67 | 2.71 | 0.503 | 0.530 | 0.052 | 0.040 | 0.180 | 0.674 | 1.000 | Normal L-shaped | 1-1 (4), 1-3 (4) |
| S1 | Kochi, Asakura shrine | 2 | – | – | – | – | – | – | – | – | – | – | 1-2 (2) |
| S2 | Kochi, Matsuo shrine | 18 | 2.78 | 1.95 | 0.370 | 0.369 | −0.003 | 0.047 | 0.219 | 0.219 | 0.219 | Normal L-shaped | 1-2 (8) |
| S3 | Kochi, Kuratani shrine | 6 | 2.11 | 1.66 | 0.333 | 0.318 | −0.053 | 0.052 | – | – | – | – | 1-2 (6) |
| S4 | Kochi, Kamibun | 35 | 2.67 | 2.31 | 0.505 | 0.451 | −0.121 | 0.008 | 0.008 | 0.012 | 0.012 | Shifted mode | 1-1 (8) |
| S5 | Kochi, Yasaka shrine | 20 | 2.56 | 2.00 | 0.411 | 0.412 | 0.001 | 0.031 | 0.004 | 0.020 | 0.027 | Shifted mode | 1-2 (8) |
| S6 | Ehime, Hachiman shrine | 2 | – | – | – | – | – | – | – | – | – | – | 1-1 (2) |
| Q. glauca | |||||||||||||
| K2 | Miyazaki, Tsuma shrine | 5 | 4.40 | 3.41 | 0.485 | 0.755 | 0.387 * | 0.104 | – | – | – | – | 4 (4), 7 (1) |
| K3 | Miyazaki, Aya-Takeno | 15 | 6.10 | 4.19 | 0.595 | 0.700 | 0.155 * | 0.025 | 0.002 | 0.005 | 0.248 | Normal L-shaped | 6 (2), 7 (10) |
| K4 | Miyazaki, Miyazaki University Forest | 20 | 8.30 | 4.34 | 0.548 | 0.697 | 0.218 * | 0.049 | 0.500 | 0.958 | 0.999 | Normal L-shaped | 3-1 (1), 4 (7) |
| K5 | Kagoshima, Shiratori shrine | 4 | 3.80 | 3.16 | 0.650 | 0.739 | 0.138 | 0.036 | – | – | – | – | 6 (4) |
| K6 | Kagoshima, Tsuruda | 9 | 5.70 | 4.04 | 0.557 | 0.708 | 0.224 * | 0.033 | – | – | – | – | 3-1 (1), 5 (2), 6 (5) |
| S1 | Kochi, Asakura shrine | 1 | – | – | – | – | – | – | – | – | – | – | 4 (1) |
| S2 | Kochi, Matsuo shrine | 1 | – | – | – | – | – | – | – | – | – | – | 6 (1) |
| S4 | Kochi, Kamibun | 27 | 9.50 | 4.83 | 0.606 | 0.715 | 0.155 * | 0.038 | 0.116 | 0.313 | 1.000 | Normal L-shaped | 3-2 (1), 4 (6) |
| Locus | Na | Ne | Ho | Hs | Ht | Gis | Na | Ne | Ho | Hs | Ht | Gis |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Q. hondae | Q. glauca | |||||||||||
| CG75 | 4 | 1.47 | 0.385 | 0.339 | 0.433 | −0.135 | 9 | 3.02 | 0.744 | 0.821 | 0.816 | 0.093 |
| CG105 | 3 | 2.01 | 0.989 | 0.503 | 0.502 | −0.967 | 5 | 2.20 | 0.817 | 0.615 | 0.655 | −0.329 |
| CG139 | 3 | 1.36 | 0.299 | 0.281 | 0.337 | −0.064 | 5 | 1.19 | 0.150 | 0.201 | 0.182 | 0.252 |
| CG258 | 1 | 1.00 | 0.000 | 0.000 | 0.000 | – | 2 | 1.02 | 0.011 | 0.026 | 0.020 | 0.582 |
| CG371 | 10 | 2.29 | 0.573 | 0.608 | 0.722 | 0.059 | 21 | 3.91 | 0.562 | 0.895 | 0.911 | 0.373 |
| MSQa1 | 5 | 2.18 | 0.674 | 0.573 | 0.707 | −0.175 | 5 | 2.10 | 0.430 | 0.681 | 0.706 | 0.369 |
| MSQa2 | 8 | 2.11 | 0.582 | 0.564 | 0.769 | −0.033 | 16 | 3.74 | 0.875 | 0.885 | 0.869 | 0.011 |
| MSQa4 | 16 | 2.69 | 0.581 | 0.684 | 0.877 | 0.150 | 20 | 2.21 | 0.146 | 0.804 | 0.843 | 0.819 |
| MSQa10 | 5 | 1.35 | 0.287 | 0.278 | 0.329 | −0.032 | 10 | 2.30 | 0.542 | 0.718 | 0.742 | 0.245 |
| MSQa11 | 5 | 1.87 | 0.563 | 0.495 | 0.665 | −0.137 | 15 | 3.71 | 0.822 | 0.894 | 0.890 | 0.081 |
| MSQa13 | 3 | 1.11 | 0.148 | 0.102 | 0.156 | −0.442 | 17 | 3.94 | 0.908 | 0.896 | 0.880 | −0.013 |
| Mean | 5.73 | 1.77 | 0.462 | 0.402 | 0.500 | −0.178 | 11.36 | 2.67 | 0.546 | 0.676 | 0.683 | 0.226 |
| Population | K2 | K3 | K4 | K5 | K6 | S2 | S3 | S4 | S5 | |
|---|---|---|---|---|---|---|---|---|---|---|
| K1 | Miyazaki, Fukuse shrine | 0.1435 *** | 0.2280 *** | 0.0987 *** | 0.1909 *** | 0.0877 *** | 0.2250 *** | 0.2502 *** | 0.1917 *** | 0.3451 *** |
| K2 | Miyazaki, Tsuma shrine | – | 0.0766 *** | 0.0969 *** | 0.1487 *** | 0.0757 *** | 0.1789 *** | 0.1663 ** | 0.1736 *** | 0.2752 *** |
| K3 | Miyazaki, Aya-Takeno | – | 0.1598 *** | 0.2733 *** | 0.1184 *** | 0.3028 *** | 0.2884 * | 0.2475 *** | 0.3716 *** | |
| K4 | Miyazaki, Miyazaki University Forest | – | 0.1278 *** | 0.0310 * | 0.1400 *** | 0.1413 *** | 0.1347 *** | 0.1471 *** | ||
| K5 | Kagoshima, Shiratori shrine | – | 0.1268 *** | 0.1321 *** | 0.1798 *** | 0.2603 *** | 0.2805 *** | |||
| K6 | Kagoshima, Tsuruda | – | 0.1477 *** | 0.0929 * | 0.1422 *** | 0.2244 *** | ||||
| S2 | Kochi, Matsuo shrine | – | 0.1808 *** | 0.2217 *** | 0.2714 *** | |||||
| S3 | Kochi, Kuratani shrine | – | 0.2278 *** | 0.3340 *** | ||||||
| S4 | Kochi, Kamibun | – | 0.2250 *** | |||||||
| S5 | Kochi, Yasaka shrine | – |
| Population | K3 | K4 | K6 | S4 | |
|---|---|---|---|---|---|
| K2 | Miyazaki, Tsuma shrine | 0.0236 NS | 0.0184 NS | 0.0205 NS | 0.0413 NS |
| K3 | Miyazaki, Aya-Takeno | – | 0.0015 NS | 0.0003 NS | 0.0220 * |
| K4 | Miyazaki, Miyazaki University Forest | – | −0.0087 NS | 0.0236 ** | |
| K6 | Kagoshima, Tsuruda | – | 0.0324 ** | ||
| S4 | Kochi, Kamibun | – |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kamiya, K.; Ogasahara, M.; Kenzo, T.; Muramoto, Y.; Araki, T.; Ichie, T. Genetic Diversity and Structure of Quercus hondae, a Rare Evergreen Oak Species in Southwestern Japan. Forests 2022, 13, 579. https://doi.org/10.3390/f13040579
Kamiya K, Ogasahara M, Kenzo T, Muramoto Y, Araki T, Ichie T. Genetic Diversity and Structure of Quercus hondae, a Rare Evergreen Oak Species in Southwestern Japan. Forests. 2022; 13(4):579. https://doi.org/10.3390/f13040579
Chicago/Turabian StyleKamiya, Koichi, Misato Ogasahara, Tanaka Kenzo, Yasuharu Muramoto, Takuya Araki, and Tomoaki Ichie. 2022. "Genetic Diversity and Structure of Quercus hondae, a Rare Evergreen Oak Species in Southwestern Japan" Forests 13, no. 4: 579. https://doi.org/10.3390/f13040579
APA StyleKamiya, K., Ogasahara, M., Kenzo, T., Muramoto, Y., Araki, T., & Ichie, T. (2022). Genetic Diversity and Structure of Quercus hondae, a Rare Evergreen Oak Species in Southwestern Japan. Forests, 13(4), 579. https://doi.org/10.3390/f13040579