
Genetic Differentiation and Demographic History of Three Cerris Oak Species in China Based on Nuclear Microsatellite Makers

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Population Sampling
2.2. DNA Extraction and Microsatellite Genotyping
2.3. Genetic Diversity, Genetic Structure and Gene Flow Analysis
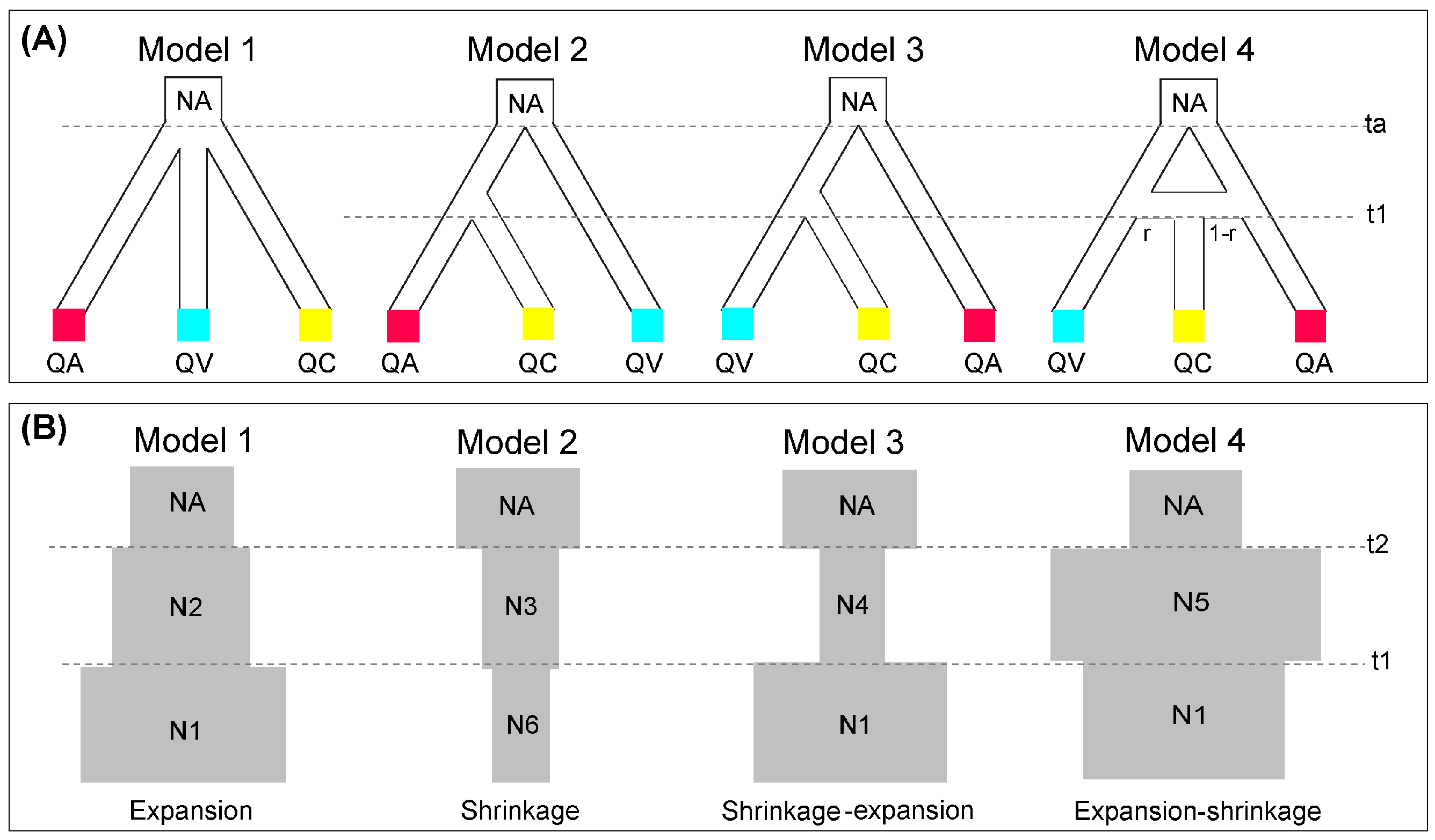
2.4. Species Divergence and Demographic History
2.5. Niche Comparison Analyses on Geographic Space (G-Space) Environment Space (E-Space)
3. Results
3.1. Genetic Diversity
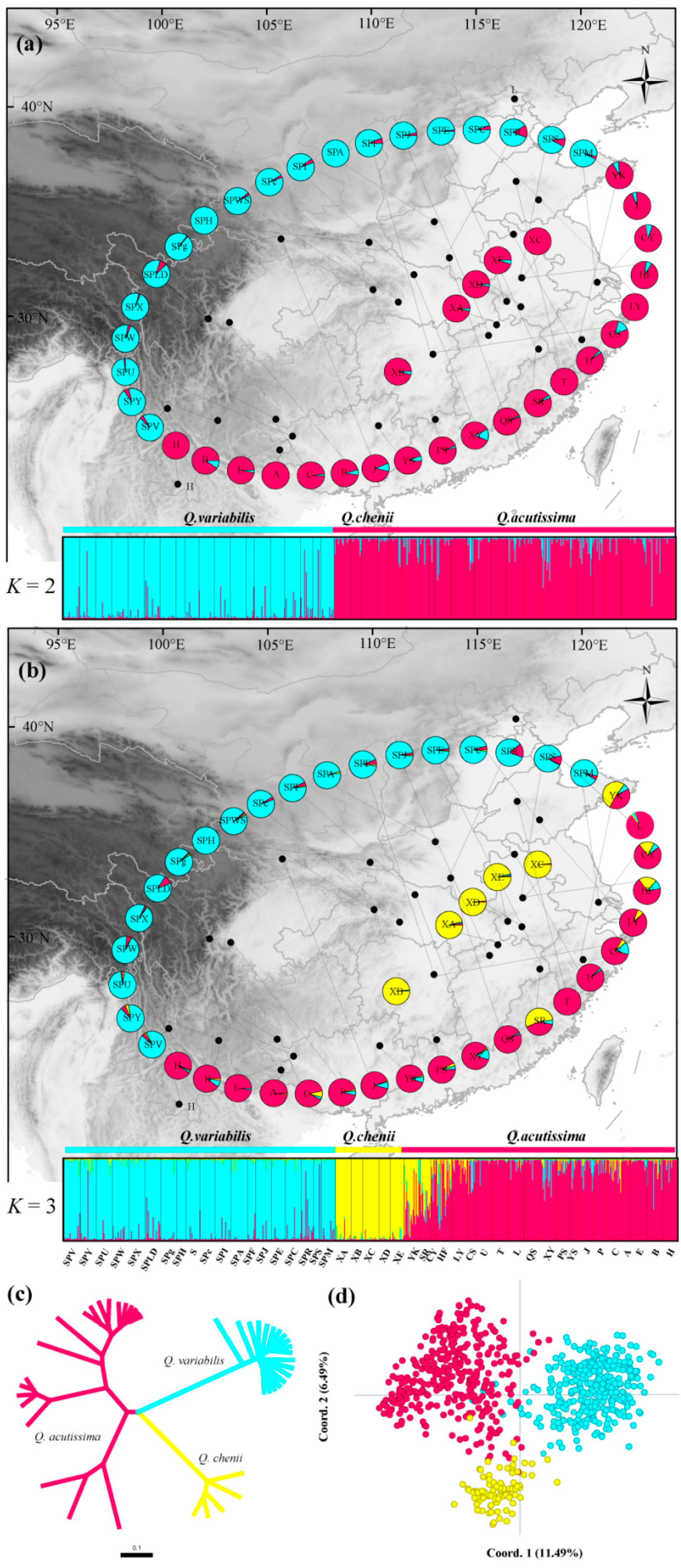
3.2. Genetic Differentiation and Structure Analysis
3.3. Genetic Migration among Species
3.4. Demographic History
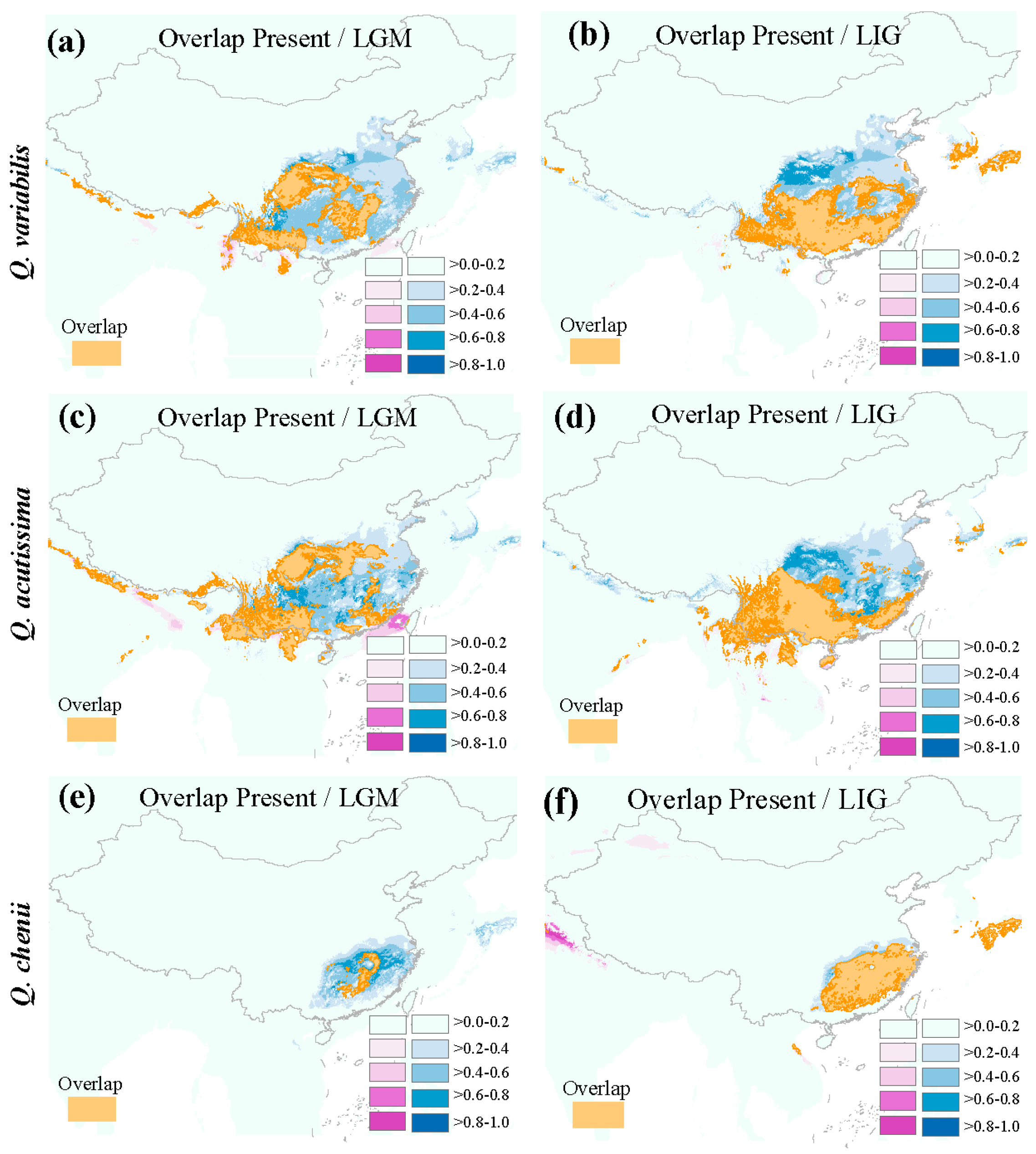
3.5. Ecological Niche Modeling
4. Discussion
4.1. Species Divergence
4.2. Demographic History
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Hewitt, G. The genetic legacy of the quaternary ice ages. Nature 2000, 405, 907–913. [Google Scholar] [CrossRef]
- Slatkin, M.; Excoffier, L. Serial Founder effects during range expansion: A Spatial Analog of Genetic Drift. Genetics 2012, 191, 171–181. [Google Scholar] [CrossRef] [Green Version]
- Lafontaine, G.D.; Napier, J.D.; Petit, R.J.; Feng, S.H. Invoking adaptation to decipher the genetic legacy of past climate change. Ecology 2018, 99, 1530–1546. [Google Scholar] [CrossRef] [PubMed]
- IPCC. Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change; Cambridge University Press: Cambridge, UK, 2013. [Google Scholar]
- Calsbeek, R.; Thompson, J.N.; Richardson, J.E. Patterns of molecular evolution and diversification in a biodiversity hotspot: The California Floristic Province. Mol. Ecol. 2003, 11, 1021–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, E.B.; Koo, M.S.; Conroy, C.; Patton, J.L.; Moritz, C. The California Hotspots Project: Identifying regions of rapid diversification of mammals. Mol. Ecol. 2008, 17, 120–138. [Google Scholar] [CrossRef]
- Lancaster, L.T.; Kay, K.M. Origin and diversification of the California flora: Re-examining classic hypotheses with molecular phylogenies. Evolution 2013, 67, 1041–1054. [Google Scholar] [CrossRef]
- Ortego, J.; Noguerales, V.; Gugger, P.F.; Sork, V.L. Evolutionary and demographic history of the Californian scrub white oak species complex: An integrative approach. Mol. Ecol. 2015, 24, 6188–6208. [Google Scholar] [CrossRef] [Green Version]
- Hampe, A.; Jump, A.S. Climate relicts: Past, present, future. Annu. Rev. Ecol. Evol. S 2011, 42, 313–333. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, A.A.; Sgrò, C.M. Climate change and evolutionary adaptation. Nature 2011, 470, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Marcott, S.A.; Shakun, J.D.; Clark, P.U.; Mix, A.C. A reconstruction of regional and global temperature for the past 11,300 years. Science 2013, 339, 1198–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kremer, A.; Hipp, A.L. Oaks: An evolutionary success story. New Phytol. 2020, 226, 987–1011. [Google Scholar] [CrossRef]
- Petit, R.J.; Bodénès, C.; Ducousso, A.; Roussel, G.; Kremer, A. Hybridization as a mechanism of invasion in oaks. New Phytol. 2004, 161, 151–164. [Google Scholar] [CrossRef] [Green Version]
- McVay, J.D.; Hipp, A.L.; Manos, P.S. A genetic legacy of introgression confounds phylogeny and biogeography in oaks. Proc. Biol. Sci. 2017, 284, 20170300. [Google Scholar] [CrossRef] [Green Version]
- Hipp, A.L.; Manos, P.S.; Hahn, M.; Avishai, M.; Bodénès, C.; Cavender-Bares, J.; Crowl, A.A.; Deng, M.; Denk, T.; Fitz-Gibbon, S.; et al. Genomic landscape of the global oak phylogeny. New Phytol. 2020, 226, 1198–1212. [Google Scholar] [CrossRef]
- Simeone, M.C.; Cardoni, S.; Piredda, R.; Imperatori, F.; Avishai, M.; Grimm, G.W.; Denk, T. Comparative systematics and phylogeography of Quercus Section Cerris in western Eurasia: Inferences from plastid and nuclear DNA variation. PeerJ 2018, 6, e5793. [Google Scholar] [CrossRef] [Green Version]
- Denk, T.; Grimm, G.W. Significance of pollen characteristics for infrageneric classification and phylogeny in Quercus (Fagaceae). Int. J. Plant Sci. 2009, 170, 926–940. [Google Scholar] [CrossRef]
- Denk, T.; Grimm, G.W. The oaks of western Eurasia: Traditional classifications and evidence from two nuclear markers. Taxon 2010, 59, 351–366. [Google Scholar] [CrossRef]
- Hubert, F.; Grimm, G.W.; Jousselin, E.; Berry, V.; Franc, A.; Kremer, A. Multiple nuclear genes stabilize the phylogenetic backbone of the genus Quercus. Syst. Biodivers. 2014, 12, 405–423. [Google Scholar] [CrossRef]
- Yang, Y.; Hu, Y.; Ren, T.; Sun, J.; Zhao, G. Remarkably conserved plastid genomes of Quercus group Cerris in China: Comparative and phylogenetic analyses. Nord. J. Bot. 2018, 36, e01921. [Google Scholar] [CrossRef]
- Chen, D.; Zhang, X.; Kang, H.; Sun, X.; Yin, S.; Du, H.; Liu, C. Phylogeography of Quercus variabilis based on chloroplast DNA sequence in East Asia: Multiple glacial refugia and mainland-migrated island populations. PLoS ONE 2012, 7, e47268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Li, Y.; Liu, C.; Xia, T.; Zhang, Q.; Fang, Y. Phylogeography of the temperate tree species Quercus acutissima in China: Inferences from chloroplast DNA variations. Biochem. Syst. Ecol. 2015, 63, 190–197. [Google Scholar] [CrossRef]
- Zhang, X.W.; Li, Y.; Zhang, Q.; Fang, Y.M. Ancient east-west divergence, recent admixture, and multiple marginal refugia shape genetic structure of a widespread oak species (Quercus acutissima) in China. Tree Genet. Genomes 2018, 14, 88. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, X.; Fang, Y. Landscape features and climatic forces shape the genetic structure and evolutionary history of an oak species (Quercus chenii) in East China. Front. Plant Sci. 2019, 10, 1060. [Google Scholar] [CrossRef]
- Gao, J.; Liu, Z.L.; Zhao, W.; Tomlinson, K.W.; Xia, S.W.; Zeng, Q.Y.; Wang, X.R.; Chen, J. Combined genotype and phenotype analyses reveal patterns of genomic adaptation to local environments in the subtropical oak Quercus acutissima. J. Syst. Evol. 2021, 59, 541–556. [Google Scholar] [CrossRef]
- Menitsky, Y.L. Oaks of Asia, Translated from Russian Science Publishers; Science Publishers, Inc.: Enfield, NH, USA; Plymouth, UK, 2005. [Google Scholar]
- Zhang, R.S.; Yang, J.; Hu, H.L.; Xia, R.X.; Li, Y.P.; Su, J.F.; Li, Q.; Liu, Y.Q.; Qin, L. A high level of chloroplast genome sequence variability in the Sawtooth Oak Quercus acutissima. Int. J. Biol. Macromol. 2020, 152, 340–348. [Google Scholar] [CrossRef]
- Song, S.Y.; Krajewska, K.; Wang, Y.F. The first occurrence of the Quercus section Cerris Spach fruits in the Miocene of China. Acta Palaeobot. 2000, 40, 153–163. [Google Scholar]
- Chun, W.; Huang, C. (Eds.) Flora Reipublicae Popularis Sinice; Science Press: Beijing, China, 1998; Volume 22. [Google Scholar]
- Papadopulos, A.S.; Baker, W.J.; Crayn, D.; Butlin, R.K.; Kynast, R.G.; Hutton, I.; Savolainen, V. Speciation with gene flow on Lord Howe Island. Proc. Natl. Acad. Sci. USA 2011, 108, 13188–13193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butlin, R.; Debelle, A.; Kerth, C.; Snook, R.R.; Beukeboom, L.W.; Castillo Cajas, R.; Diao, W.; Maan, M.E.; Paolucci, S.; Weissing, F.J.; et al. What do we need to know about speciation? Trends Ecol. Evol. 2012, 27, 27–39. [Google Scholar]
- You, F.M.; Huo, N.; Gu, Y.Q.; Luo, M.C.; Ma, Y.; Hane, D.; Lazo, G.R.; Dvorak, J.; Anderson, O.D. BatchPrimer3: A high throughput web application for PCR and sequencing primer design. BMC Bioinform. 2008, 9, 253. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Li, J.; Li, Y. Isolation and characterization of microsatellite markers for an endemic tree in East Asia, Quercus variabilis (Fagaceae). Appl. Plant Sci. 2015, 3, 1500032. [Google Scholar] [CrossRef]
- Marshall, T.C.; Slate, J.; Kruuk, L.E.B.; Pemberton, J.M. Statistical confidence for likelihood-based paternity inference in natural populations. Mol. Ecol. 1998, 7, 639–655. [Google Scholar] [CrossRef] [Green Version]
- Goudet, J. FSTAT, a Program to Estimate and Test Gene Diversities and Fixation Indices (Version 2.9.3). Available online: http://www.unil.ch/izea/softwares/fstat.html (accessed on 24 July 2017).
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [Green Version]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, N.A. DISTRUCT: A program for the graphical display of population structure. Mol. Ecol. Resour. 2004, 4, 137–138. [Google Scholar] [CrossRef]
- Excoffier, L.; Laval, G.; Schneider, S. Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evol. Bioinform. 2005, 1, 47–50. [Google Scholar] [CrossRef] [Green Version]
- Excoffier, L.; Smouse, P.E.; Quattro, J.M. Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics 1992, 131, 479–491. [Google Scholar] [CrossRef]
- Nei, M. Genetic distance between populations. Am. Nat. 1972, 106, 283–292. [Google Scholar] [CrossRef]
- Langella, O. Populations v.1.2.32: Population Genetic Software. Available online: http://www.bioinformatics.org/project/?group_id=84 (accessed on 3 July 2020).
- Page, R.D.M. Tree View: An application to display phylogenetic trees on personal computers. Bioinformatics 1996, 12, 357–358. [Google Scholar] [CrossRef] [Green Version]
- Beerli, P. Comparison of bayesian and maximum-likelihood inference of population genetic parameters. Bioinformatics 2006, 22, 341–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, G.A.; Rannala, B. Bayesian inference of recent migration rates using multilocus genotypes. Genetics 2003, 163, 1177–1191. [Google Scholar] [CrossRef] [PubMed]
- Cornuet, J.M.; Pudlo, P.; Veyssier, J.; Dehne-Garcia, A.; Gautier, M.; Raphaël, L.; Jean-Michel, M.; Arnaud, E. DIYABC v2.0: A software to make approximate Bayesian computation inferences about population history using single nucleotide polymorphism, DNA sequence and microsatellite data. Bioinformatics 2014, 30, 1187–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piry, S.; Luikart, G.; Cornuet, J.M. BOTTLENECK: A computer program for detecting recent reductions in effective population size using allele frequency data. J. Hered. 1999, 90, 502–503. [Google Scholar] [CrossRef]
- Luikart, G.; Cornuet, J.M. Empirical evaluation of a test for identifying recently bottlenecked populations from allele frequency data. Conserv. Biol. 1998, 12, 228–237. [Google Scholar] [CrossRef]
- Petitpierre, B.; Kueffer, C.; Broennimann, O.; Randin, C.; Daehler, C.; Guisan, A. Climatic niche shifts are rare among terrestrial plant invaders. Science 2012, 335, 1344–1348. [Google Scholar] [CrossRef] [Green Version]
- Phillips, S.J.; Anderson, R.P.; Schapire, R.E. Maximum entropy modeling of species geographic distributions. Ecol. Model. 2006, 190, 231–259. [Google Scholar] [CrossRef] [Green Version]
- Warren, D.L.; Glor, R.E.; Turelli, M. Environmental niche equivalency versus conservatism: Quantitative approaches to niche evolution. Evolution 2008, 62, 2868–2883. [Google Scholar] [CrossRef]
- Schoener, T.W. The Anolis lizards of bimini: Resource partitioning in a complex fauna. Ecology 1968, 49, 704–726. [Google Scholar] [CrossRef]
- Broennimann, O.; Fitzpatrick, M.C.; Pearman, P.B.; Petitpierre, B.; Pellissier, L.; Yoccoz, N.G.; Thuiller, W.; Fortin, M.; Randin, C.; Zimmermann, N.E.; et al. Measuring ecological niche overlap from occurrence and spatial environmental data. Glob. Ecol. Biogeogr. 2012, 21, 481–497. [Google Scholar] [CrossRef] [Green Version]
- Herrando-Moraira, S.; Nualart, N.; Herrando-Moraira, A.; Chung, M.Y.; Chung, M.G.; López-Pujol, J. Climatic niche characteristics of native and invasive Lilium Lancifolium. Sci. Rep. 2019, 9, 14334. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Di, X.; Meng, X.; Feng, L.; Liu, Z.; Zhao, G. Phylogeography and evolution of two closely related oak species (Quercus) from north and northeast China. Tree Genet. Genomes 2016, 12, 89. [Google Scholar] [CrossRef]
- Kou, Y.; Cheng, S.; Tian, S.; Li, B.; Fan, D.; Chen, Y.; Soltis, D.E.; Soltis, P.S.; Zhang, Z. The antiquity of Cyclocarya paliurus (Juglandaceae) provides new insights into the evolution of relict plants in subtropical China since the late Early Miocene. J. Biogeogr. 2016, 43, 351–360. [Google Scholar] [CrossRef]
- Deng, M.; Jiang, X.L.; Hipp, A.L.; Manos, P.S.; Hahn, M. Phylogeny and biogeography of East Asian evergreen oaks (Quercus section Cyclobalanopsis; Fagaceae): Insights into the Cenozoic history of evergreen broad-leaved forests in subtropical Asia. Mol. Phylogenet. Evol. 2018, 119, 170–181. [Google Scholar] [CrossRef]
- Yang, J.; Guo, Y.F.; Chen, X.D.; Zhang, X.; Ju, M.M.; Bai, G.Q.; Liu, Z.L.; Zhao, G.F. Framework phylogeny, evolution and complex diversification of Chinese oaks. Plants 2020, 9, 1024. [Google Scholar] [CrossRef]
- Harrison, T.M.; Copeland, P.; Kidd, W.S.F.; Yin, A.N. Raising Tibet. Science 1992, 255, 1663–1670. [Google Scholar] [CrossRef] [Green Version]
- Chung, S.L.; Lo, C.H.; Lee, T.Y.; Zhang, Y.; Xie, Y.; Li, X.; Wang, K.L.; Wang, P.L. Diachronous uplift of the Tibetan plateau starting 40? Myr ago. Nature 1998, 394, 769–773. [Google Scholar] [CrossRef]
- Shi, Y.F.; Li, J.J.; Li, B.Y. Uplift and Environmental Changes of Qinghai-Tibetan Plateau in the Late Cenozoic; Guangdong Science and Technology Press: Guangzhou, China, 1998. [Google Scholar]
- Guo, Z.; Ruddiman, W.; Hao, Q.Z.; Wu, H.B.; Qiao, Y.S.; Zhu, R.X.; Peng, S.Z.; Wei, J.J.; Yuan, B.Y.; Liu, T.S. Onset of Asian desertification by 22 Myr ago inferred from loess deposits in China. Nature 2002, 416, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Spicer, R.; Harris, N.; Widdowson, M.; Herman, A.; Guo, S.; Valdes, P.; Wolfe, J.; Kelley, S. Constant elevation of southern Tibet over the past 15 million years. Nature 2003, 421, 622–624. [Google Scholar] [CrossRef] [PubMed]
- Sobel, J.M.; Chen, G.F.; Watt, L.R.; Schemske, D.W. The biology of speciation. Evolution 2010, 64, 295–315. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.L.; Meng, H.H.; Zhang, H.X.; Vyacheslav, B.V.; Sanderson, S.C. Himalayan origin and evolution of Myricaria (Tamaricaeae) in the Neogene. PLoS ONE 2014, 9, e97582. [Google Scholar]
- Liu, M.L.; He, Y.L.; López-Pujol, J.; Jia, Y.; Li, Z.H. Complex population evolutionary history of four cold-tolerant Notopterygium herb species in the Qinghai-Tibetan Plateau and adjacent areas. Heredity 2019, 123, 242–263. [Google Scholar] [CrossRef]
- Liu, J.; Möller, M.; Provan, J.; Gao, L.M.; Poudel, R.C.; Li, D.Z. Geological and ecological factors drive cryptic speciation of yews in a biodiversity hotspot. New Phytol. 2013, 199, 1093–1108. [Google Scholar] [CrossRef]
- Feng, L.; Zheng, Q.J.; Qian, Z.Q.; Yang, J.; Zhang, Y.P.; Li, Z.H.; Zhao, G.F. Genetic structure and evolutionary history of three Alpine Sclerophyllous oaks in East Himalaya-Hengduan Mountains and adjacent regions. Front. Plant Sci. 2016, 7, 1688. [Google Scholar] [CrossRef] [Green Version]
- Hipp, A.L.; Eaton, D.A.; Cavender-Bares, J.; Fitzek, E.; Nipper, R.; Manos, P.S. A framework phylogeny of the American oak clade based on sequenced RAD data. PLoS ONE 2014, 9, e93975. [Google Scholar] [CrossRef] [Green Version]
- Riordan, E.C.; Gugger, P.F.; Ortego, J.; Smith, C.; Gaddis, K.; Thompson, P.; Sork, V.L. Association of genetic and phenotypic variability with geography and climate in three southern California oaks. Am. J. Bot. 2016, 103, 73–85. [Google Scholar] [CrossRef] [Green Version]
- Muir, G.; Fleming, C.C.; Schltterer, C. Species status of hybridizing oaks. Nature 2000, 405, 1016. [Google Scholar] [CrossRef]
- Ortego, J.; Gugger, P.F.; Riordan, E.C.; Sork, V.L. Influence of climatic niche suitability and geographical overlap on hybridization patterns among southern Californian oaks. J. Biogeogr. 2014, 41, 1895–1908. [Google Scholar] [CrossRef]
- Quintero, I.; González-Caro, S.; Zalamea, P.C.; Cadena, C.D. Asynchrony of seasons: Genetic differentiation associated with geographic variation in climatic seasonality and reproductive phenology. Am. Nat. 2014, 184, 352–363. [Google Scholar] [CrossRef]
- Yang, J.; Vázquez, L.; Feng, L.; Liu, Z.; Zhao, G. Climatic and soil factors shape the demographical history and genetic diversity of a deciduous oak (Quercus liaotungensis) in Northern China. Front. Plant Sci. 2018, 9, 1534. [Google Scholar] [CrossRef]
- Chen, X.D.; Yang, J.; Guo, Y.F.; Zhao, Y.M.; Zhou, T.; Zhang, X.; Ju, M.M.; Li, Z.H.; Zhao, G.F. Spatial genetic structure and demographic history of the dominant forest oak Quercus fabri hance in subtropical China. Front. Plant Sci. 2021, 11, 583284. [Google Scholar] [CrossRef]
- Comes, H.P.; Kadereit, J.W. The effect of Quaternary climatic changes on plant distribution and evolution. Trends Plant Sci. 1998, 3, 432–438. [Google Scholar] [CrossRef]
- Hewitt, G.M. Genetic consequences of climatic oscillations in the Quaternary. Philos. Trans. R. Soc. London. Ser. B Biol. Sci. 2004, 359, 183–195. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y. Characteristics of late Quaternary monsoonal glaciation on the Tibetan Plateau and in East Asia. Quatern. Int. 2002, 97, 79–91. [Google Scholar] [CrossRef]
- Zheng, B.; Xu, Q.; Shen, Y. The relationship between climate change and Quaternary glacial cycles on the Qinghai–Tibetan Plateau: Review and speculation. Quatern. Int. 2002, 97, 93–101. [Google Scholar] [CrossRef]
- Hao, Q.; Wang, L.; Oldfield, F.; Guo, Z. Extra-long interglacial in Northern Hemisphere during MISs 15-13 arising from limited extent of Arctic ice sheets in glacial MIS 14. Sci. Rep. 2015, 5, 1–8. [Google Scholar] [CrossRef]
- Guo, Z.; Liu, T.; Fedoroff, N.; Wei, L.; Ding, Z.; Wu, N.; Lu, H.; Jiang, W.; An, Z. Climate extremes in loess of China coupled with the strength of deep-water formation in the North Atlantic. Glob. Planet. Chang. 1998, 18, 113–128. [Google Scholar] [CrossRef]
- Chen, X.D.; Yang, J.; Feng, L.; Zhou, T.; Zhang, H.; Li, H.M.; Bai, G.Q.; Meng, X.; Zhao, G.F. Phylogeography and population dynamics of an endemic oak (Quercus fabri Hance) in subtropical China revealed by molecular data and ecological niche modeling. Tree Genet. Genomes 2020, 16, 2. [Google Scholar] [CrossRef]
- Tian, S.; Kou, Y.; Zhang, Z.; Yuan, L.; Li, D.; López-Pujol, J.; Fan, D.M.; Zhang, Z. Phylogeography of Eomecon chionantha in subtropical China: The dual roles of the Nanling Mountains as a glacial refugium and a dispersal corridor. BMC Evol. Biol. 2018, 18, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
| Source of Variation | Df | SS | Variation (%) | Fixation Index |
|---|---|---|---|---|
| Q. variabilis | ||||
| Among populations | 18 | 322.571 | 8.97 | FST = 0.090 *** |
| Within populations | 653 | 2613.898 | 91.03 | |
| Total | 671 | 2936.469 | ||
| Q. acutissima | ||||
| Among populations | 19 | 739.158 | 21.37 | FST = 0.214 *** |
| Within populations | 656 | 2513.000 | 78.63 | |
| Total | 675 | 3252.158 | ||
| Q. chenii | ||||
| Among populations | 4 | 54.134 | 5.45 | FST = 0.055 *** |
| Within populations | 165 | 757.501 | 94.55 | |
| Total | 169 | 811.635 | ||
| All samples | ||||
| Among species | 2 | 1056.084 | 19.52 | FCT = 0.195 *** |
| Among populations within species | 41 | 1104.497 | 11.76 | FSC = 0.146 *** |
| Within populations | 1474 | 5764.860 | 68.72 | FST = 0.313 *** |
| Total | 1517 | 7925.441 |
| M (m/μ) | ||||
|---|---|---|---|---|
| Species | θ | QV→ | QA→ | QC→ |
| QV | 0.896 (0.881–0.911) | 4.571 (4.349–4.790) | 7.2180 (6.963–7.479) | |
| QA | 1.540 (1.514–1.567) | 9.098 (8.819–9.382) | 5.2464 (5.039–5.459) | |
| QC | 3.412 (3.310–3.517) | 2.3863 (2.291–2.484) | 2.6501 (2.550–2.753) | |
| Parameter | N1 | N2 | N3 | NA | T1 (Generations) | Ta (Generations) | µ | p |
|---|---|---|---|---|---|---|---|---|
| Species referred to | QV | QA | QC | QV+QA+QC | QC from QA | Differentiation of QV and QA | ||
| Median | 5.52 × 105 | 4.45 × 105 | 8.78 × 105 | 7.14 × 104 | 1.20 × 105 | 2.48 × 105 | 8.33 × 10−6 | 2.69 × 10−1 |
| Lower_bound (2.5%) | 2.69 × 105 | 2.20 × 105 | 6.60 × 105 | 4.84 × 103 | 6.34 × 104 | 2.03 × 105 | 5.67 × 10−6 | 1.74 × 10−1 |
| Upper_bound (97.5%) | 9.28 × 105 | 8.45 × 105 | 9.91 × 105 | 6.00 × 105 | 1.91 × 105 | 2.97 × 105 | 1.00 × 10−5 | 3.00 × 10−1 |
| RMAE | 0.145 | 0.138 | 0.143 | 0.326 | 0.158 | 0.087 | 0.129 | 0.132 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, X.-D.; Zhang, X.; Zhang, H.; Zhou, T.; Zhao, Y.-M.; Yang, J.; Zhao, G.-F. Genetic Differentiation and Demographic History of Three Cerris Oak Species in China Based on Nuclear Microsatellite Makers. Forests 2021, 12, 1164. https://doi.org/10.3390/f12091164
Chen X-D, Zhang X, Zhang H, Zhou T, Zhao Y-M, Yang J, Zhao G-F. Genetic Differentiation and Demographic History of Three Cerris Oak Species in China Based on Nuclear Microsatellite Makers. Forests. 2021; 12(9):1164. https://doi.org/10.3390/f12091164
Chicago/Turabian StyleChen, Xiao-Dan, Xiao Zhang, Hao Zhang, Tao Zhou, Yue-Mei Zhao, Jia Yang, and Gui-Fang Zhao. 2021. "Genetic Differentiation and Demographic History of Three Cerris Oak Species in China Based on Nuclear Microsatellite Makers" Forests 12, no. 9: 1164. https://doi.org/10.3390/f12091164
APA StyleChen, X.-D., Zhang, X., Zhang, H., Zhou, T., Zhao, Y.-M., Yang, J., & Zhao, G.-F. (2021). Genetic Differentiation and Demographic History of Three Cerris Oak Species in China Based on Nuclear Microsatellite Makers. Forests, 12(9), 1164. https://doi.org/10.3390/f12091164