
Evaluation of Soda Lignin from Wheat Straw/Sarkanda Grass as a Potential Future Consolidant for Archaeological Wood

 ,
,  ,
, 
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Wood Specimens
2.2. Density and Maximum Moisture Content Measurements
2.3. Materials Used for Impregnation
2.4. Impregnation Set-Up
2.5. Change in Dimensions and Weight
2.6. Chemical Characterization and Distribution
2.6.1. SEM
2.6.2. ATR-FTIR
2.6.3. Analytical Pyrolysis Py-GC/MS
3. Results and Discussion
3.1. Characterization of Wood Specimens before Treatment
3.1.1. Macroscopic Observations
3.1.2. Density and Maximum Moisture Content
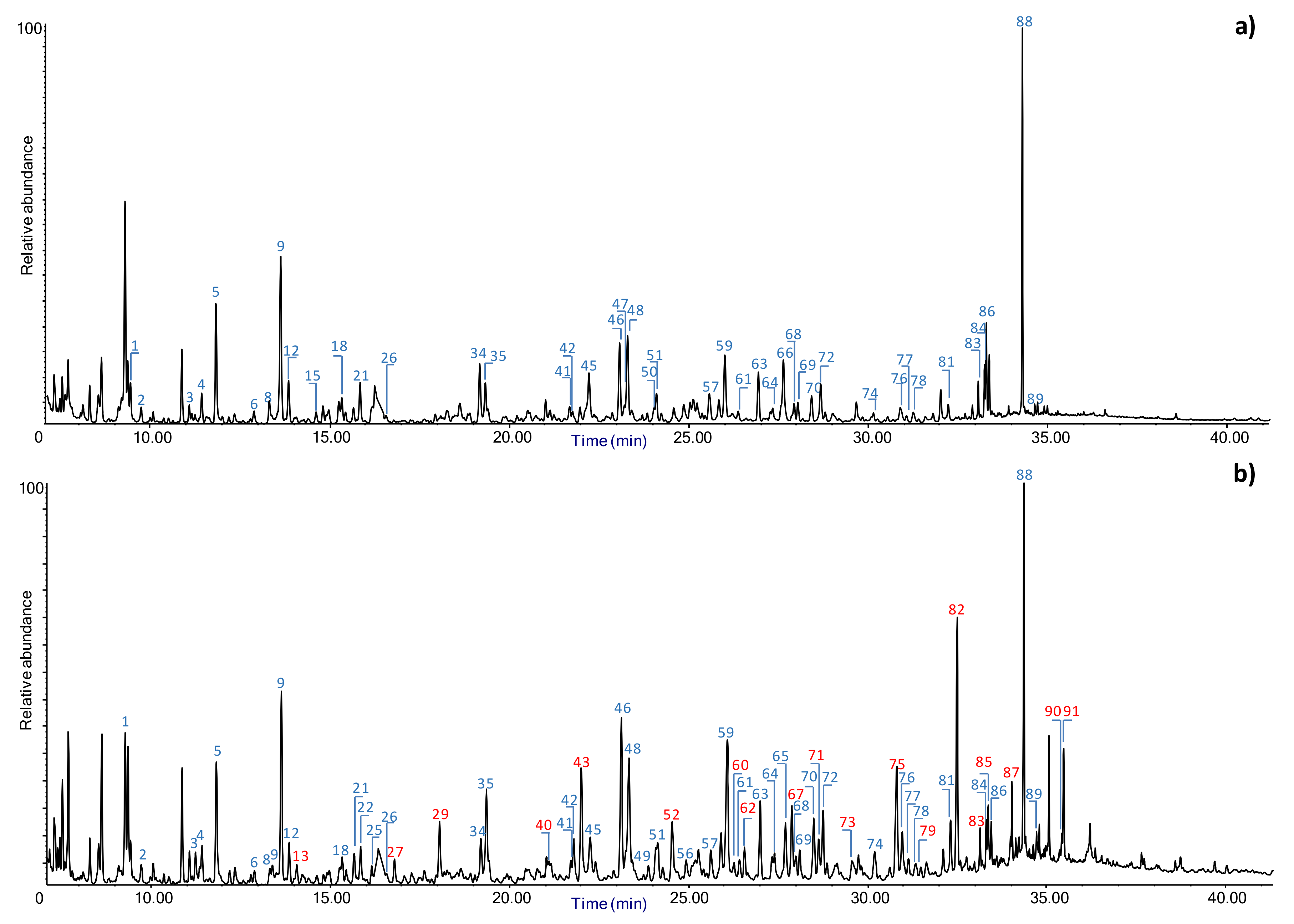
3.1.3. Analytical Pyrolysis (Py-GC/MS)
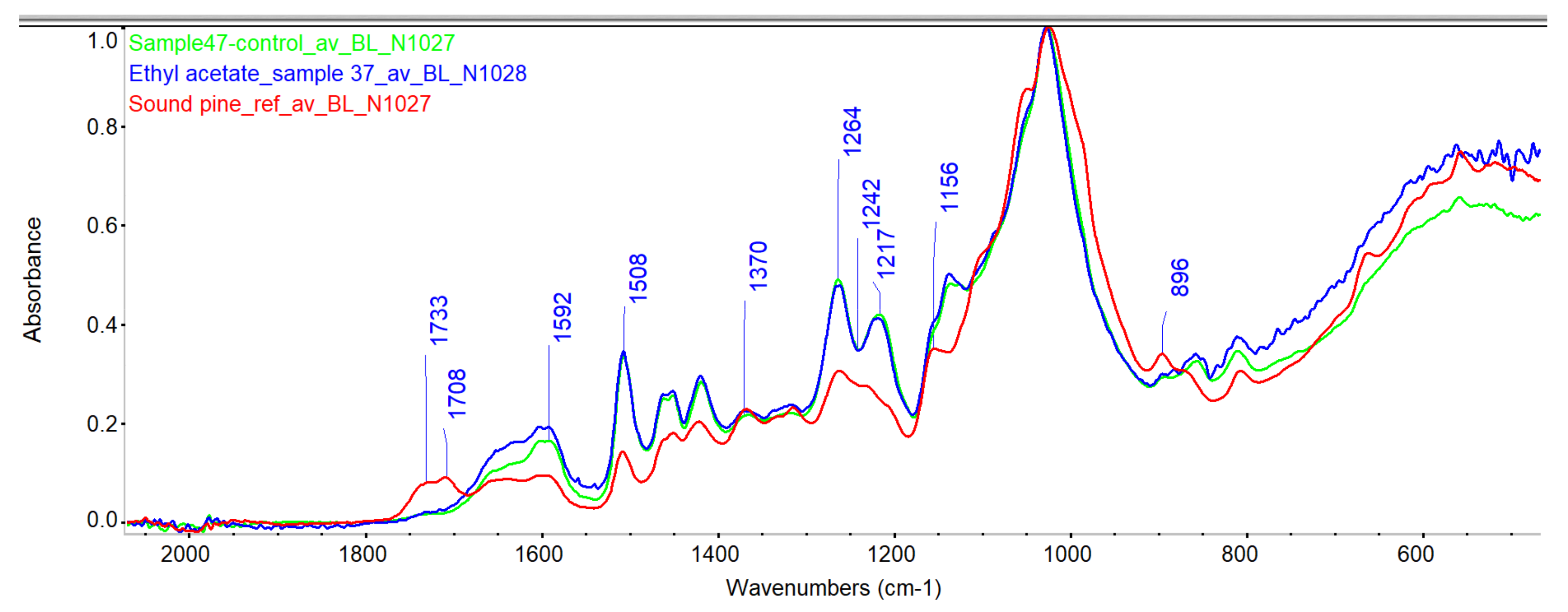
3.1.4. Infrared Spectroscopy (ATR-FTIR)
3.2. After Treatment
3.2.1. Weight Gain
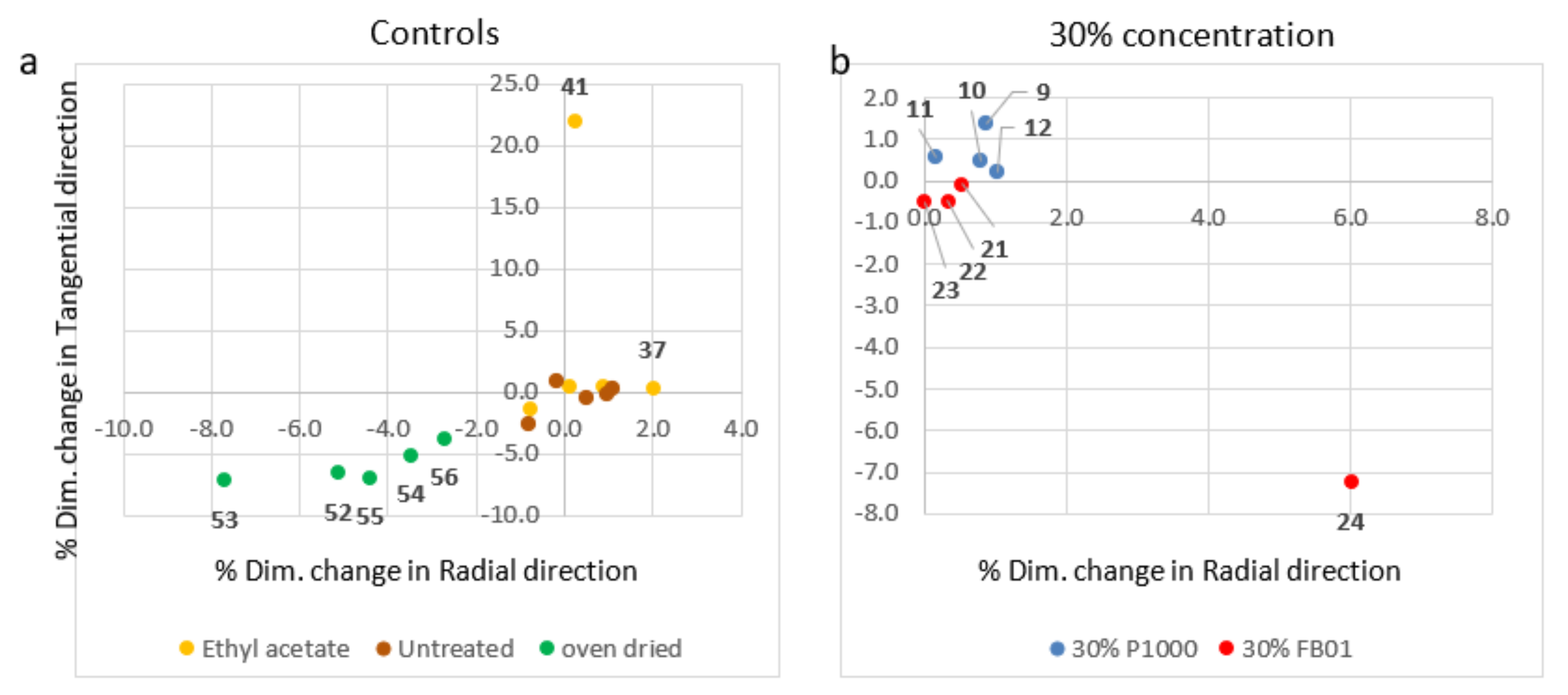
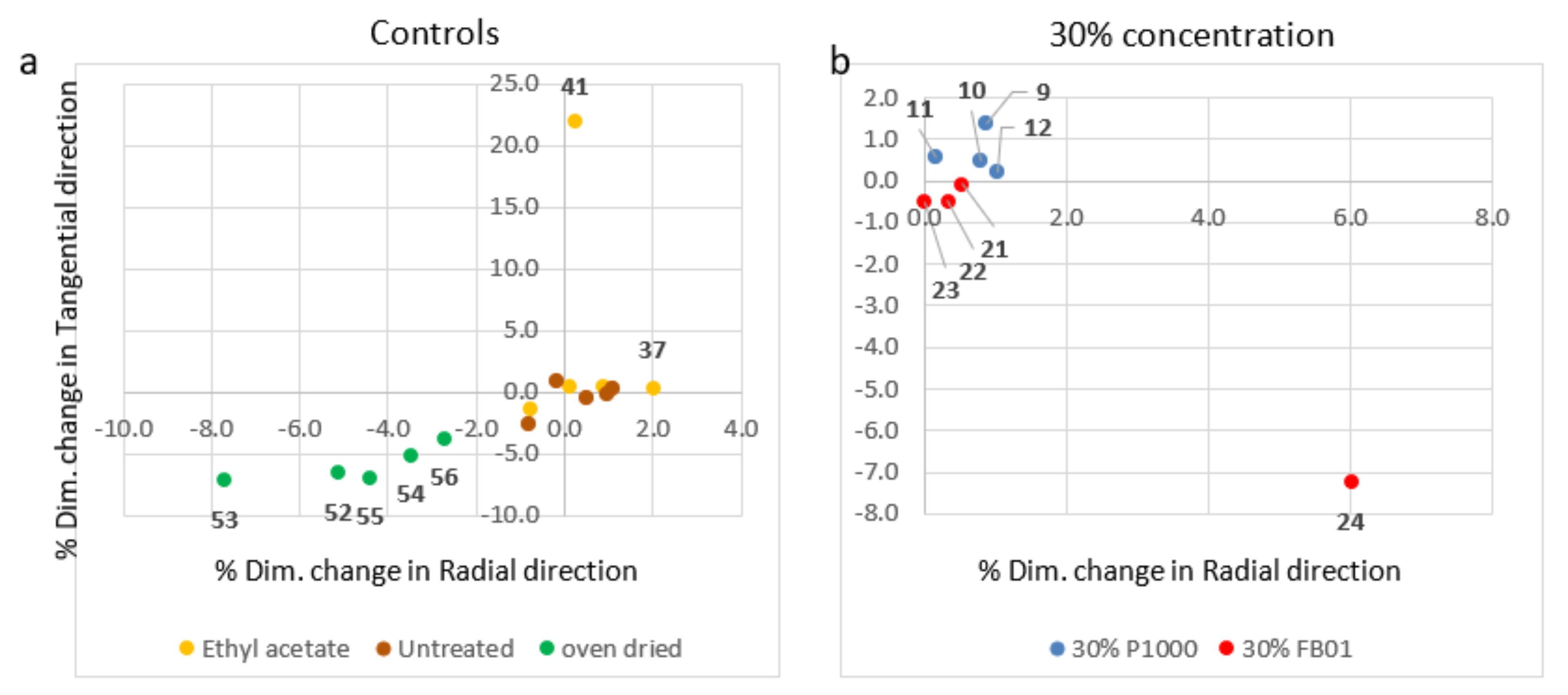
3.2.2. Dimensional Changes
3.2.3. Morphology
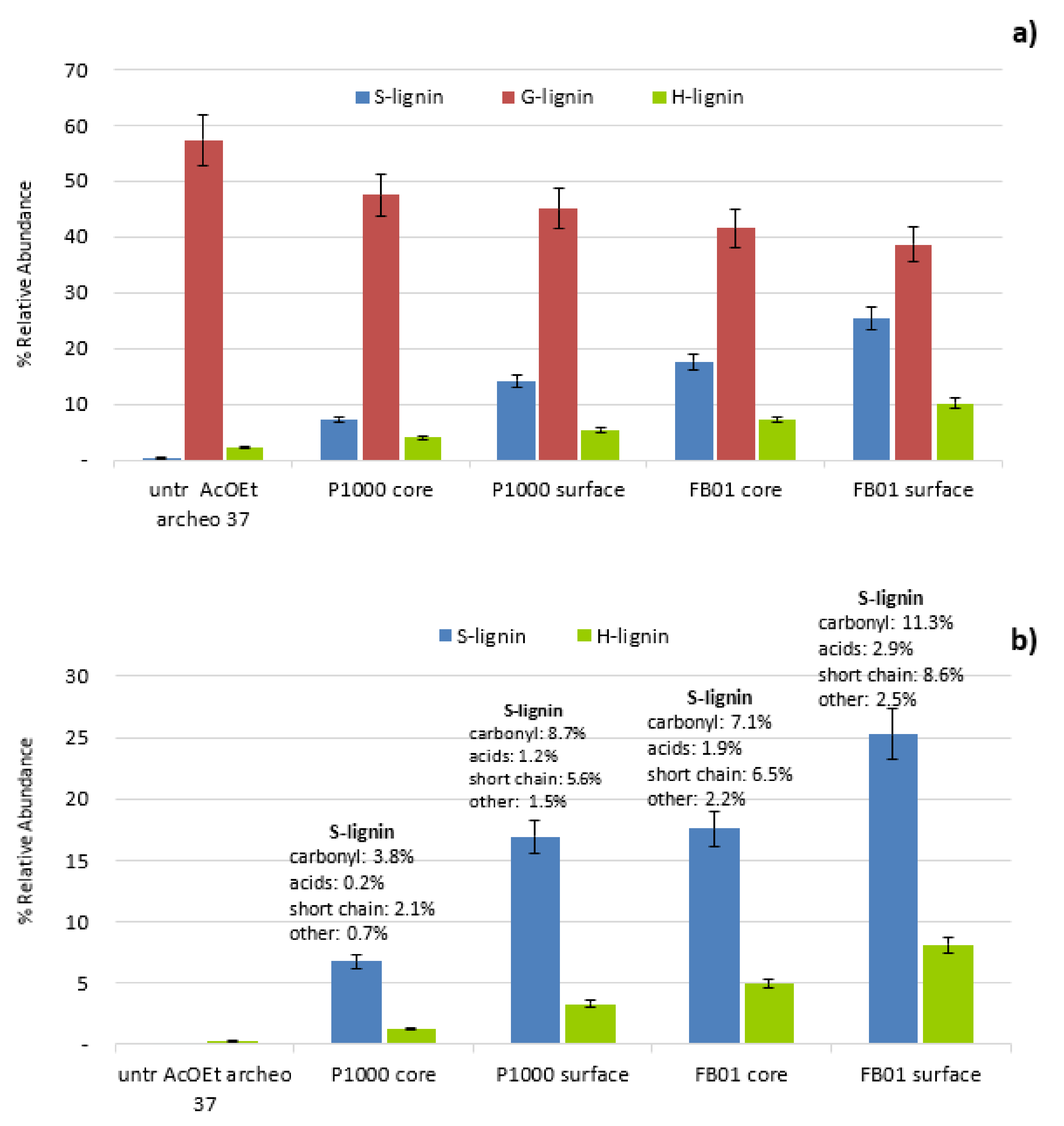
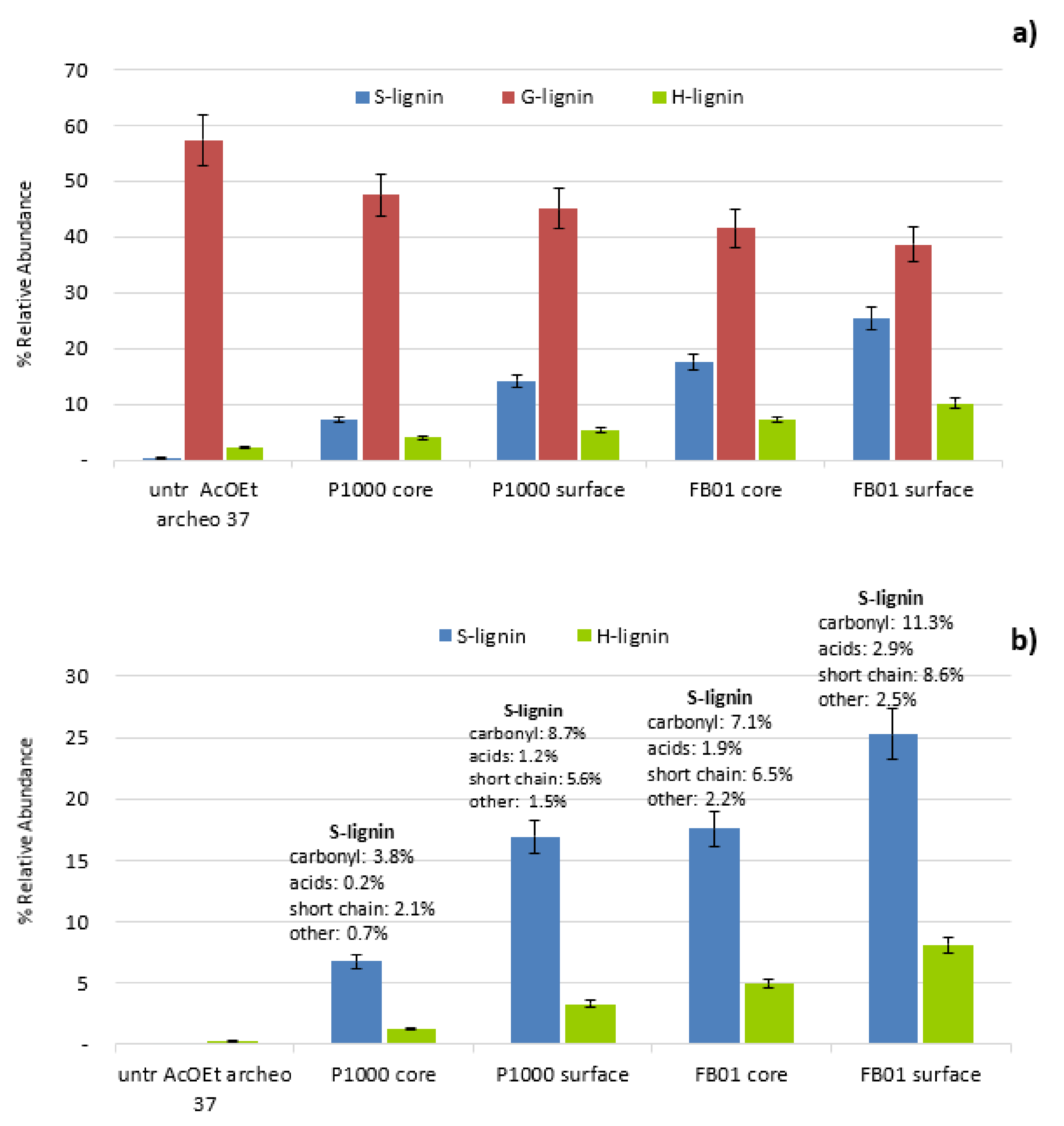
3.2.4. Extent of Penetration of Soda Lignins in Treated Specimens
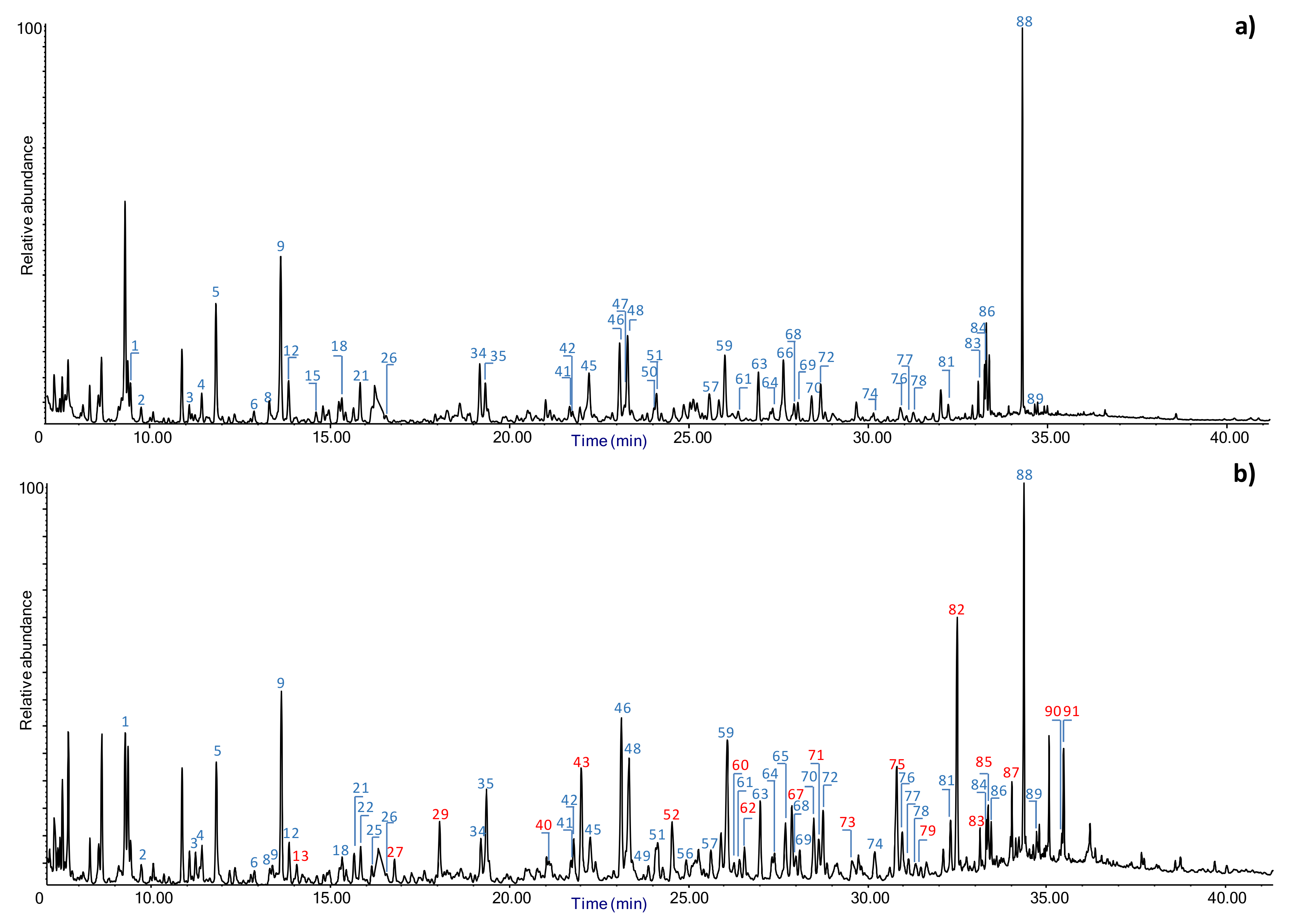
Evaluation of Penetration by Py-GC/MS
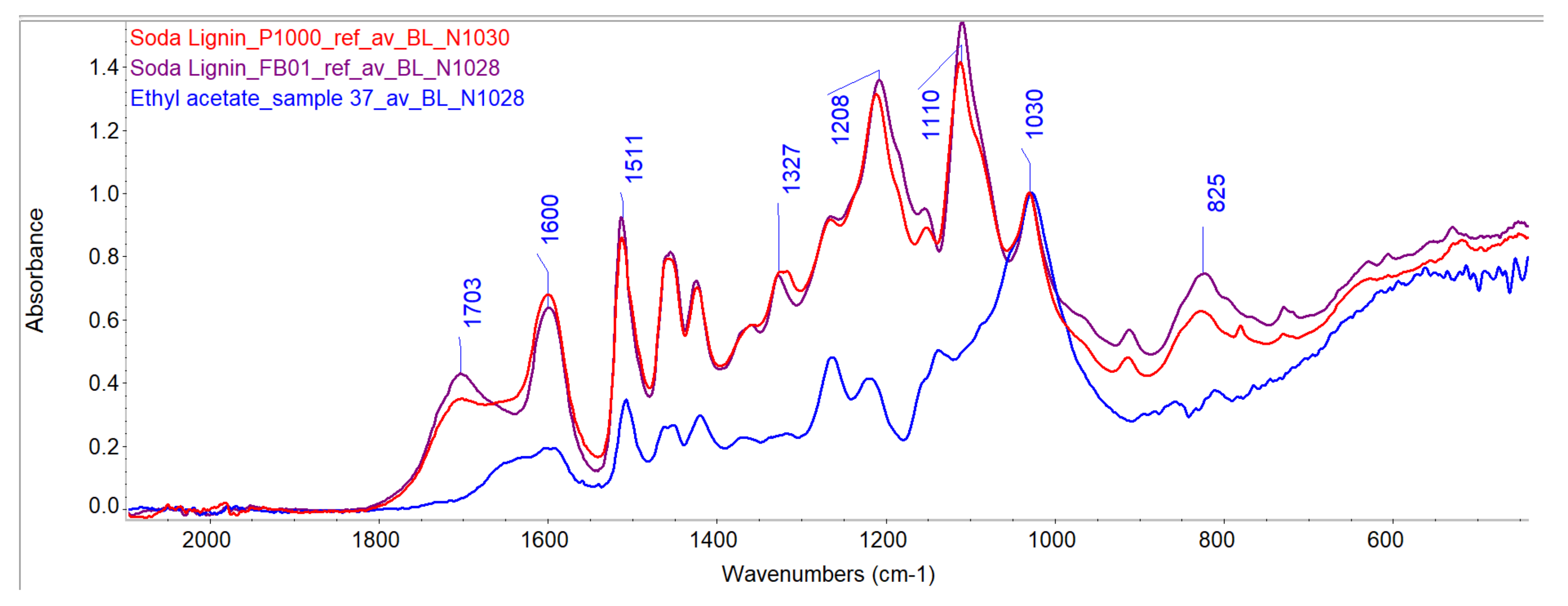
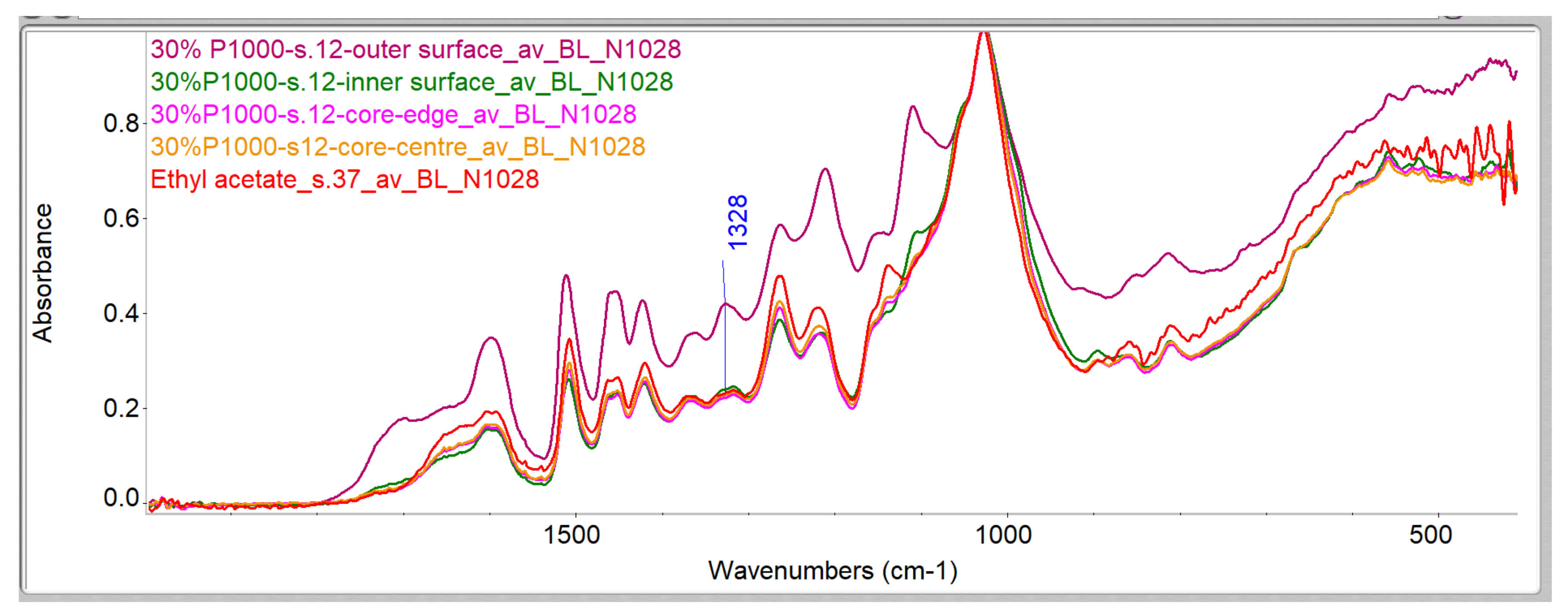
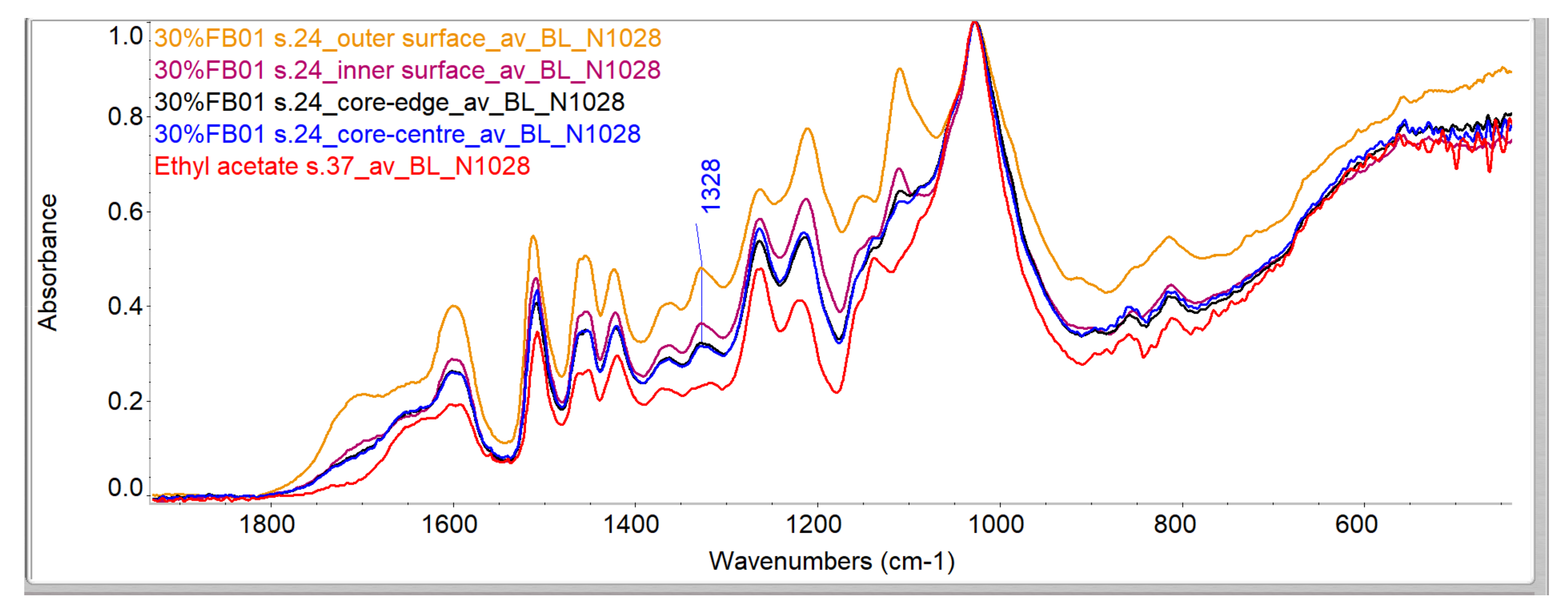
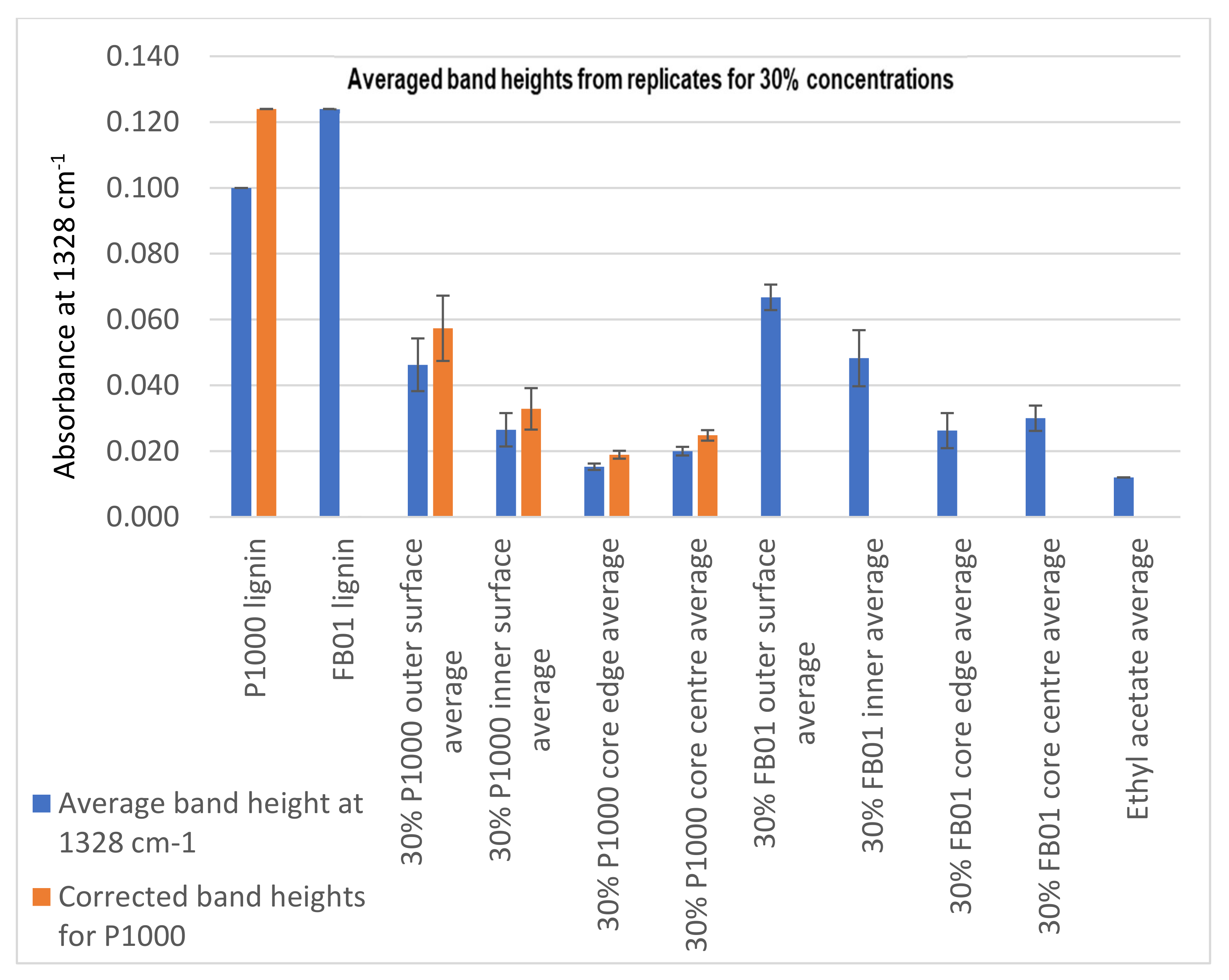
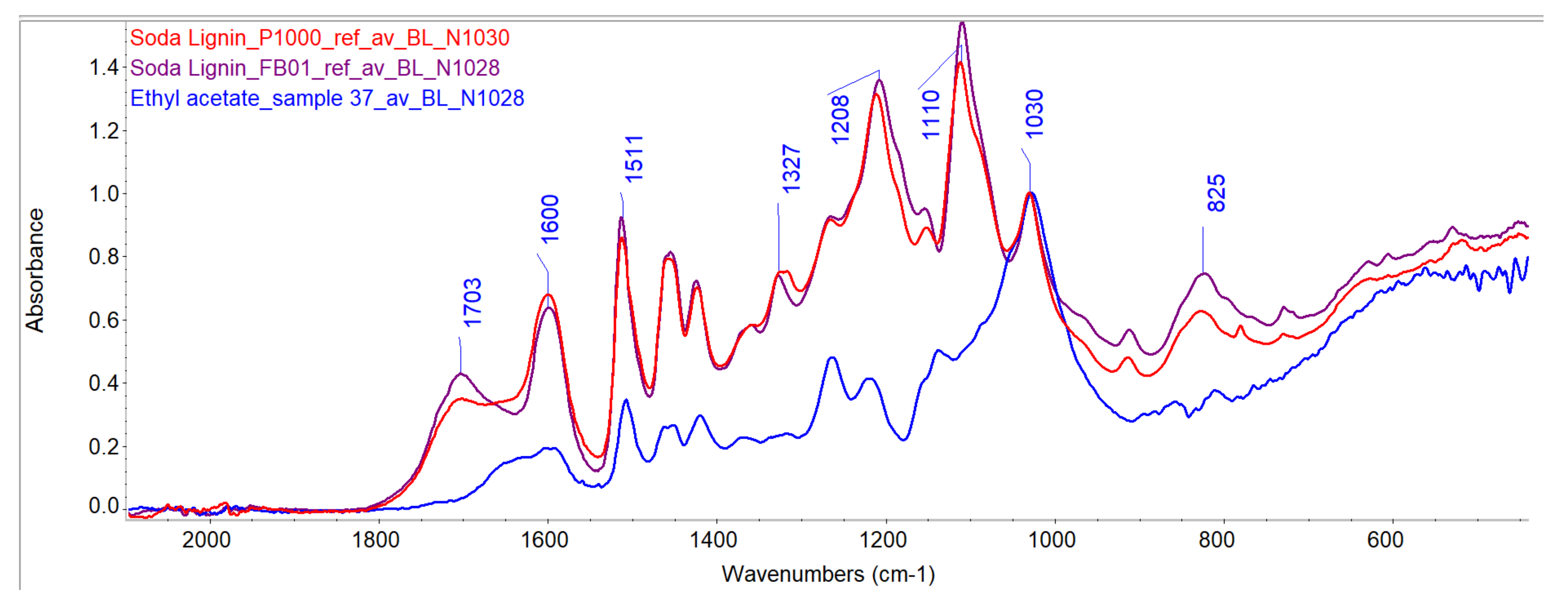
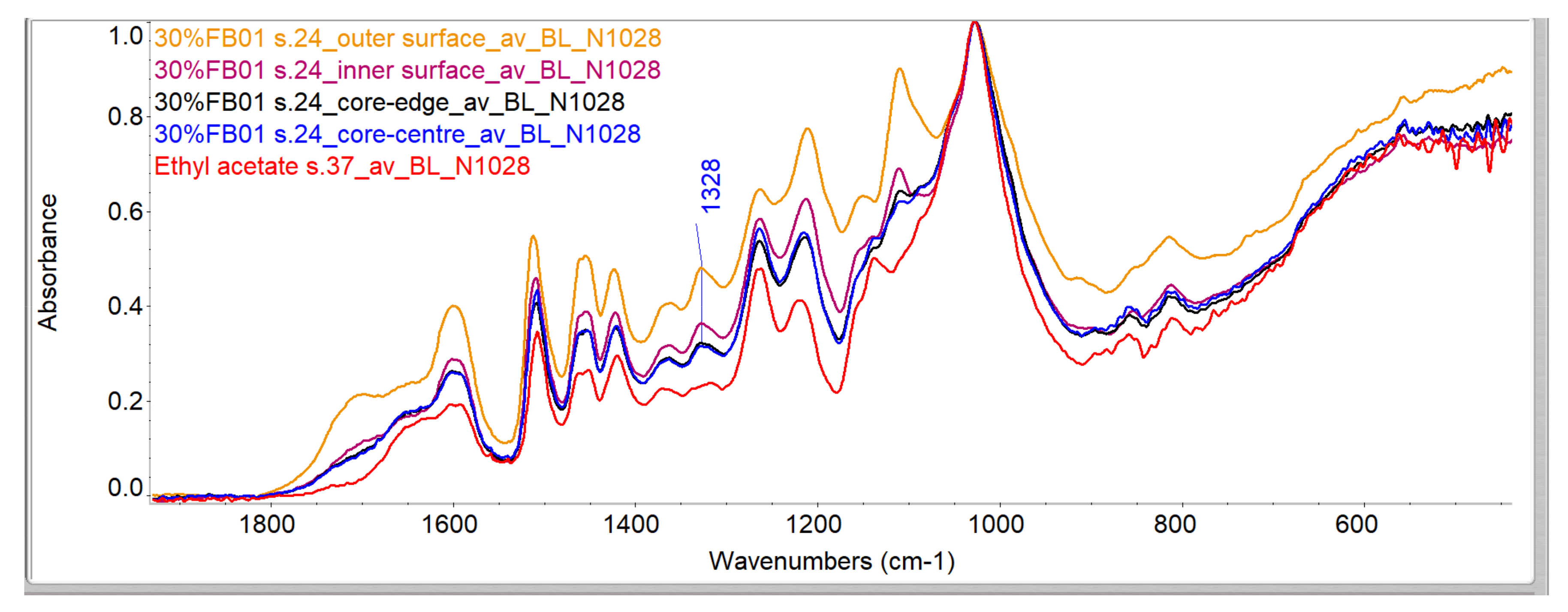
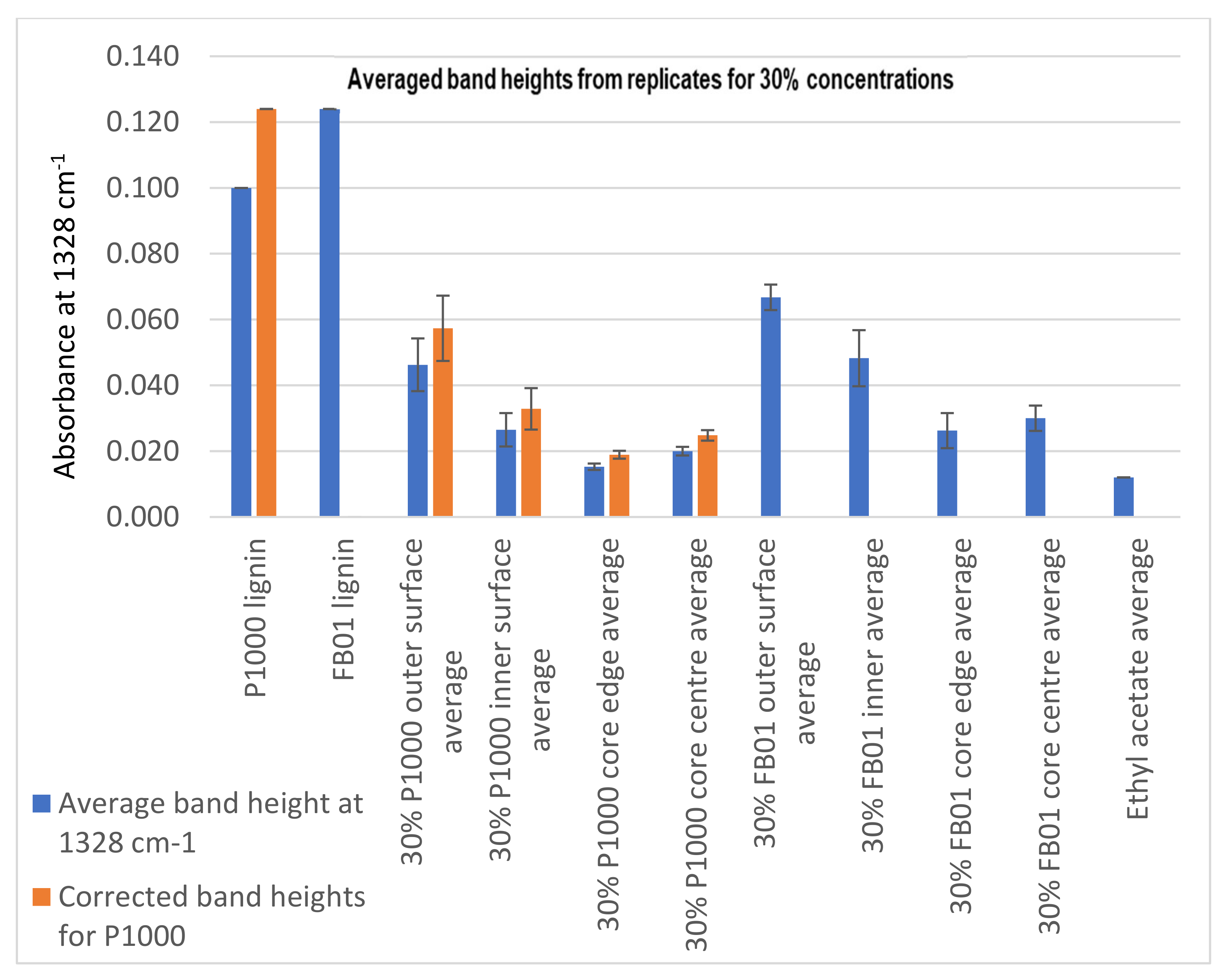
Evaluation of Penetration by ATR-FTIR
4. Conclusions and Perspectives
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Herbst, C.F. Om bevaring af oldsager af træ fundne i törvemoser. In Antiquarisk Tidsskrift; Det Kongelige Nordiske Oldskrift-selskab: Kjøbenhavn, Danmark, 1861; pp. 174–176. [Google Scholar]
- Speerschneider, C.A. Behandling af oldsager af træ, som ere fundne i moser for at bevare dem i deres oprindelige form og farve. In Antiquarisk Tidsskrift; Det Kongelige Nordiske Oldskrift-Selskab: Kjøbenhavn, Danmark, 1861; pp. 176–178. [Google Scholar]
- Christensen, B.B. The Conservation of Waterlogged Wood in the National Museum of Denmark: With a Report on the Methods Chosen for the Stabilization of the Timbers of the Viking Ships from Roskilde Fjord, and a Report on Experiments Carried out in Order to Improve upen These Methods; The National Museum of Denmark: Copenhagen, Denmark, 1970; Volume 1. [Google Scholar]
- Rosenqvist, A.M. The Stabilizing of Wood Found in the Viking Ship of Oseberg: Part I. Stud. Conserv. 1959, 4, 13–22. [Google Scholar]
- Eaton, J.W. The preservation of wood by the alum process. Fla. Anthropol. 1962, XV, 115–117. [Google Scholar]
- McQueen, C.M.A.; Tamburini, D.; Braovac, S. Identification of inorganic compounds in composite alum-treated wooden artefacts from the Oseberg collection. Sci. Rep. 2018, 8, 2901. [Google Scholar] [CrossRef] [Green Version]
- McQueen, C.M.A.; Tamburini, D.; Łucejko, J.J.; Braovac, S.; Gambineri, F.; Modugno, F.; Colombini, M.P.; Kutzke, H. New insights into the degradation processes and influence of the conservation treatment in alum-treated wood from the Oseberg collection. Microchem. J. 2017, 132, 119–129. [Google Scholar] [CrossRef] [Green Version]
- Braovac, S.; Tamburini, D.; Łucejko, J.J.; McQueen, C.M.A.; Kutzke, H.; Colombini, M.P. Chemical analyses of extremely degraded wood using analytical pyrolysis and inductively coupled plasma atomic emission spectroscopy. Microchem. J. 2016, 124, 368–379. [Google Scholar] [CrossRef]
- Braovac, S.; Kutzke, H. The presence of sulfuric acid in alum-conserved wood—Origin and consequences. J. Cult. Herit. 2012, 13, S203–S208. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, L.; Zhou, T.; Chen, J.; Xu, F. Synergy of Lewis and Brønsted acids on catalytic hydrothermal decomposition of carbohydrates and corncob acid hydrolysis residues to 5-hydroxymethylfurfural. Sci. Rep. 2017, 7, 40908. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Zou, H.; Nan, J.; Wu, L.; Yang, X.; Su, Y.; Lu, T.; Xu, J. Conversion of carbohydrate biomass to methyl levulinate with Al2(SO4)3 as a simple, cheap and efficient catalyst. Catal. Commun. 2014, 50, 13–16. [Google Scholar] [CrossRef]
- Brazdausks, P.; Paze, A.; Rizhikovs, J.; Puke, M.; Meile, K.; Vedernikovs, N.; Tupciauskas, R.; Andzs, M. Effect of aluminium sulphate-catalysed hydrolysis process on furfural yield and cellulose degradation of Cannabis sativa L. shives. Biomass Bioenergy 2016, 89, 98–104. [Google Scholar] [CrossRef]
- Chamberlain, D. Anion mediation of aluminium-catalysed degradation of paper. Polym. Degrad. Stab. 2007, 92, 1417–1420. [Google Scholar] [CrossRef]
- Baty, J.; Minter, W.; Lee, S. The role of electrophilic metal ions aluminum (III) and magnesium (II) in paper degradation and deacidification. In American Institute for the Conservation of Historic and Artistic Works. In Proceedings of the 38th Annual Meeting, Milwaukee, WI, USA, 11–14 May 2010. [Google Scholar]
- Baty, J.; Sinnott, M.L. Efficient electrophilic catalysis of 1,5-anhydrocellobiitol hydrolysis by AlIII; implications for the conservation of ?rosin-alum? sized paper. Chem. Commun. 2004, 2004, 866–867. [Google Scholar] [CrossRef] [PubMed]
- Łucejko, J.J.; McQueen, C.M.A.; Sahlstedt, M.; Modugno, F.; Colombini, M.P.; Braovac, S. Comparative chemical investigations of alum treated archaeological wood from various museum collections. Herit. Sci. 2021, 9, 69. [Google Scholar] [CrossRef]
- Häggström, C.; Sandström, T.; Lindahl, K.; Sahlstedt, M. Alum-Treated Archaeological Wood: Characterization and Re-Conservation; Riksantikvarieämbetet: Gotland, Sweden, 2013; p. 131. [Google Scholar]
- Bojesen-Koefoed, I.M. Re-conservation of wood treated with alum in the 1920s—Challenges and strategies. In Proceedings of the 11th ICOM Group on Wet Organic Archaeological Materials Conference, Greenville 2010, Greenville, NC, USA, 20 April 2012; Strætkvern, K., Williams, E., Eds.; Springer: Berlin/Heidelberg, Germany, 2012; pp. 497–502. [Google Scholar]
- Braovac, S.; Wittköpper, M.; Sahlstedt, M. Retreatment testing of alum-treated woods from the Oseberg collection—Results from first trials. In Proceedings of the 14th ICOM-CC Wet Organic Archaeological Materials Conference, Portsmouth, UK, 20–23 May 2019. [Google Scholar]
- Brøgger, A.W.; Shetelig, H.; Falk, H. Osebergfundet; Den norske Stat: Kristiania, Norge, 1917. [Google Scholar]
- Braovac, S.; McQueen, C.M.A.; Sahlstedt, M.; Kutzke, H.; Łucejko, J.J.; Klokkernes, T. Navigating conservation strategies: Linking material research on alum-treated wood from the Oseberg collection to conservation decisions. Herit. Sci. 2018, 6, 77. [Google Scholar] [CrossRef]
- Lora, J. Chapter 10-Industrial Commercial Lignins: Sources, Properties and Applications. In Monomers, Polymers and Composites from Renewable Resources; Elsevier: Amsterdam, The Netherlands, 2008; pp. 225–241. [Google Scholar]
- Ang, A.F.; Ashaari, Z.; Lee, S.H.; Tahir, P.M.; Halis, R. Lignin-Based Copolymer Adhesives for Composite Wood Panels—A Review. Int. J. Adhes. Adhes. 2019, 95, 102408. [Google Scholar] [CrossRef]
- Ghaffar, S.H.; Fan, M. Lignin in straw and its applications as an adhesive. Int. J. Adhes. Adhes. 2014, 48, 92–101. [Google Scholar] [CrossRef]
- Vachon, J.; Assad-Alkhateb, D.; Baumberger, S.; van Haveren, J.; Gosselink, R.J.A.; Monedero, M.; Bermudez, J.M. Use of lignin as additive in polyethylene for food protection: Insect repelling effect of an ethyl acetate phenolic extract. Compos. Part. C Open Access 2020, 2, 100044. [Google Scholar] [CrossRef]
- Lora, J.H. Characteristics, Industrial Sources, and Utilization of Lignins from Non-Wood Plants. In Chemical Modification, Properties, and Usage of Lignin; Springer Science and Business Media LLC: Boston, MA, USA, 2002; pp. 267–282. [Google Scholar]
- Gosselink, R.J.A.; Putten, J.C.; van Es, D.S. Fractionation of Technical Lignin. Patent WO2015 178771, 26 November 2015. [Google Scholar]
- Browning, B.L. Methods of Wood Chemistry; Interscience Publishers: New York, NY, USA, 1967; Volume I–II. [Google Scholar]
- Gosselink, R.J.A.; Van Dam, J.E.G.; de Jong, E.; Scott, E.L.; Sanders, J.P.M.; Li, J.; Gellerstedt, G. Fractionation, analysis, and PCA modeling of properties of four technical lignins for prediction of their application potential in binders. Holzforschung 2010, 64, 193–200. [Google Scholar] [CrossRef]
- Lu, Y.; Joosten, L.; Donkers, J.; Andriulo, F.; Slaghek, T.M.; Phillips-Jones, M.K.; Gosselink, R.J.A.; Harding, S.E. Characterisation of mass distributions of solvent-fractionated lignins using analytical ultracentrifugation and size exclusion chromatography methods. Sci. Rep. 2021, 11, 1–12. [Google Scholar] [CrossRef]
- Tamburini, D.; Łucejko, J.J.; Zborowska, M.; Modugno, F.; Cantisani, E.; Mamoňová, M.; Colombini, M.P. The short-term degradation of cellulosic pulp in lake water and peat soil: A multi-analytical study from the micro to the molecular level. Int. Biodeterior. Biodegrad. 2017, 116, 243–259. [Google Scholar] [CrossRef]
- Tamburini, D.; Lucejko, J.J.; Ribechini, E.; Colombini, M.P. New markers of natural and anthropogenic chemical alteration of archaeological lignin revealed by in situ pyrolysis/silylation-gas chromatography-mass spectrometry. J. Anal. Appl. Pyrolysis 2016, 118, 249–258. [Google Scholar] [CrossRef]
- Tamburini, D.; Łucejko, J.J.; Zborowska, M.; Modugno, F.; Prądzyński, W.; Colombini, M.P. Archaeological wood degradation at the site of Biskupin (Poland): Wet chemical analysis and evaluation of specific Py-GC/MS profiles. J. Anal. Appl. Pyrolysis 2015, 115, 7–15. [Google Scholar] [CrossRef]
- Grattan, D.W.; Clarke, R.W. Conservation of Waterlogged wood. In Conservation of Marine Archaeological Objects; Pearson, C., Ed.; Butterworth-Heinemann: Oxford, UK, 1987; pp. 164–206. [Google Scholar]
- Schwanninger, M.; Rodrigues, J.; Pereira, H.; Hinterstoisser, B. Effects of short-time vibratory ball milling on the shape of FT-IR spectra of wood and cellulose. Vib. Spectrosc. 2004, 36, 23–40. [Google Scholar] [CrossRef]
- Faix, O.; Bremer, J.; Schmidt, O.; Stevanovic, T.J. Monitoring of chemical changes in white-rot degraded beech wood by pyrolysis—Gas chromatography and Fourier-transform infrared spectroscopy. J. Anal. Appl. Pyrolysis 1991, 21, 147–162. [Google Scholar] [CrossRef]
- Boeriu, C.; Bravo, D.; Gosselink, R.J.A.; van Dam, J.E.G. Characterisation of structure-dependent functional properties of lignin with infrared spectroscopy. Ind. Crop. Prod. 2004, 20, 205–218. [Google Scholar] [CrossRef]
- Mohebby, B. Attenuated total reflection infrared spectroscopy of white-rot decayed beech wood. Int. Biodeterior. Biodegrad. 2005, 55, 247–251. [Google Scholar] [CrossRef]
- Faix, O. Classification of Lignins from Different Botanical Origins by FT-IR Spectroscopy. Holzforschung 1991, 45, 21–28. [Google Scholar] [CrossRef]
- Blomberg, J.; Persson, B.; Bexell, U. Effects of semi-isostatic densification on anatomy and cell-shape recovery on soaking. Holzforschung 2006, 60, 322–331. [Google Scholar] [CrossRef]
- Horie, V. Materials for Conservation: Organic Consolidants, Adhesives and Coatings, 2nd ed.; Butterworth-Heinemann: Oxford, UK, 2010. [Google Scholar]
- Kavvouras, P.K.; Kostarelou, C.; Zisi, A.; Petrou, M.; Moraitou, G. Use of Silanol-Terminated Polydimethylsiloxane in the Conservation of Waterlogged Archaeological Wood. Stud. Conserv. 2009, 54, 65–76. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer/Solvent | Sample | Analysis | ||
|---|---|---|---|---|
| FTIR | Py-GC/MS | SEM | ||
| 30% P1000 (w/v) | 9 | x | x | x |
| 10 | x | x | ||
| 11 | x | x | ||
| 12 | x | x | ||
| 30% FB01 (w/v) | 21 | x | x | x |
| 22 | x | x | ||
| 23 | x | x | ||
| 24 | x | x | ||
| ethyl acetate control | 37 | x | x | x |
| 38 | x | |||
| 39 | ||||
| 40 | ||||
| 41 | x | |||
| no treatment, control | 47 | x | x | |
| 48 | x | |||
| 49 | x | |||
| 50 | x | |||
| 51 | x | |||
| oven-dry for characterization | 52 | x | ||
| 53 | x | |||
| 54 | x | |||
| 55 | x | |||
| 56 | x | |||
| Compound | Fragment Ions (m/z) | Category | Origin | |
|---|---|---|---|---|
| 1 | 1,2-dihydroxyethane (2TMS) | 73, 103, 147, 191 | small molecules | H/L |
| 2 | 2-hydroxymethylfuran (TMS) | 53, 73, 81, 111, 125, 142, 155, 170 | furan | H |
| 3 | phenol (TMS) | 75, 151, 166 | short chain | H-lignin |
| 4 | 2-hydroxypropanoic acid (2TMS) | 73, 117, 147, 190 | small molecules | H/L |
| 5 | 2-hydroxyacetic acid (2TMS) | 73, 147, 177, 205 | small molecules | H/L |
| 6 | 1-hydroxy-1-cyclopenten-3-one (TMS) | 53, 73, 81, 101, 111, 127, 155, 169 | cyclopentenone | H |
| 7 | 3-hydroxymethylfuran (TMS) | 53, 75, 81, 111, 125, 142, 155, 170 | furan | H |
| 8 | o-cresol (TMS) | 73, 91, 135, 149, 165, 180 | short chain | H-lignin |
| 9 | 2-furancarboxylic acid (TMS) | 73, 95, 125, 169, 184 | furan | H |
| 10 | unknown I | 73, 152, 167 | small molecules | H |
| 11 | m-cresol (TMS) | 73, 91, 165, 180 | short chain | H-lignin |
| 12 | 2-hydroxy-1-cyclopenten-3-one (TMS) | 53, 73, 81, 101, 111, 127, 155, 170 | cyclopentenone | H |
| 13 | p-cresol (TMS) | 73, 91, 165, 180 | short chain | H-lignin |
| 14 | 3-hydroxy-(2H)-pyran-2-one (TMS) | 75, 95, 125, 151, 169, 184 | pyran | H |
| 15 | unknown II | 59, 73, 85, 101, 115, 131, 159 | small molecules | H |
| 16 | unknown III | 59, 73, 85, 103, 115, 129, 145, 173, 188 | small molecules | H |
| 17 | Z-2,3-dihydroxy-cyclopent-2-enone (TMS) | 59, 73, 115, 143, 171, 186 | cyclopentenone | H |
| 18 | E-2,3-dihydroxy-cyclopent-2-enone (TMS) | 75, 101, 143, 171, 186 | cyclopentenone | H |
| 19 | 1,2-dihydroxybenzene (TMS) | 75, 91, 136, 151, 167, 182 | hydroxybenzene | H/L |
| 20 | 3-hydroxy-(4H)-pyran-4-one (TMS) | 75, 95, 139, 151, 169, 184 | pyran | H |
| 21 | 5-hydroxy-2H-pyran-4(3H)-one (TMS) | 59, 75, 101, 129, 143, 171, 186 | pyran | H |
| 22 | 2-hydroxymethyl-3-methy-2-cyclopentenone (TMS) | 73, 103, 129, 173, 183, 198 | cyclopentenone | H |
| 23 | 1-hydroxy-2-methyl-1-cyclopenten-3-one (TMS) | 73, 97, 125, 139, 169, 184 | cyclopentenone | H |
| 24 | 1-methy-2-hydroxy-1-cyclopenten-3-one (TMS) | 73, 97, 125, 139, 169, 184 | cyclopentenone | H |
| 25 | 1,3-dihydroxyacetone (2TMS) | 73, 103, 147, 189, 219 | small molecules | H/L |
| 26 | guaiacol (TMS) | 73, 151, 166, 181, 196 | short chain | G-lignin |
| 27 | ethyl phenol TMS | 73, 135, 179, 194 | short chain | H-lignin |
| 28 | 3-hydroxy-6-methyl-(2H)-pyran-2-one (TMS) | 73, 109, 139, 168, 183, 198 | pyran | H |
| 29 | vinyl phenol (TMS) | 73, 151, 177, 192 | short chain | H-lignin |
| 30 | 2-methyl-3-hydroxy-(4H)-pyran-4-one (TMS) | 73, 101, 153, 183, 198 | pyran | H |
| 31 | 2-methyl-3-hydroxymethyl-2-cyclopentenone (TMS) | 73, 103, 129, 173, 183, 198 | cyclopentenone | H |
| 32 | 2,3-dihydrofuran-2,3-diol (2TMS) | 73, 147, 231, 246 | furan | H |
| 33 | 2-furyl-hydroxymethylketone (TMS) | 73, 81, 103, 125, 183, 198 | furan | H |
| 34 | 5-hydroxymethyl-2-furaldehyde (TMS) | 73, 81, 109, 111, 139, 169, 183, 198 | furan | H |
| 35 | 4-methylguaiacol (TMS) | 73, 149, 180, 195, 210 | short chain | G-lignin |
| 36 | 1,2-dihydroxybenzene (2TMS) | 73, 151, 239, 254 | hydroxybenzene | H/L |
| 37 | 2-hydroxymethyl-2,3-dihydropyran-4-one (TMS) | 73, 142, 170, 185, 200 | pyran | H |
| 38 | 1,4:3,6-dianhydro-α-D-glucopyranose (TMS) | 73, 103, 129, 155, 170, 171, 186 | anhydrosugars | H |
| 39 | Z-2,3-dihydroxy-cyclopent-2-enone (2TMS) | 73, 147, 230, 243, 258 | cyclopentenone | H |
| 40 | p-hydroxy benzaldehyde TMS | 73, 151, 179, 194 | carbonyl | H-lignin |
| 41 | 4-methylcatechol (2TMS) | 73,180, 253, 268 | demethylated | G-lignin |
| 42 | 4-ethylguaiacol (TMS) | 73, 149, 179, 194, 209, 224 | short chain | G-lignin |
| 43 | syringol (TMS) | 73, 153, 181, 196, 211, 226 | short chain | S-lignin |
| 44 | 1,4-dihydroxybenzene (2TMS) | 73, 112, 239, 354 | hydroxybenzene | H/L |
| 45 | arabinofuranose (4TMS) | 73, 147, 217, 230 | H | |
| 46 | 4-vinylguaiacol (TMS) | 73, 162, 177, 192, 207, 222 | short chain | G-lignin |
| 47 | 3-hydroxy-2-hydroxymethyl-2-cyclopentenone (2TMS) | 73, 147, 257, 272 | cyclopentenone | H |
| 48 | E-2,3-dihydroxy-cyclopent-2-enone (2TMS) | 73, 147, 243, 258 | cyclopentenone | H |
| 49 | 4-ethylcatechol (2TMS) | 73, 147, 179, 231, 267, 282 | demethylated | G-lignin |
| 50 | 3-hydroxy-2-(hydroxymethyl) cyclopenta-2,4-dienone (2TMS) | 73, 147, 255, 270 | cyclopentenone | H |
| 51 | eugenol (TMS) | 73, 147, 179, 206, 221, 236 | long chain | G-lignin |
| 52 | 4-methylsyringol (TMS) | 73, 167, 210, 225, 240 | short chain | S-lignin |
| 53 | 3-methoxy-1,2-benzenediol (2TMS) | 73, 153, 254, 269, 284 | demethylated | G-lignin |
| 54 | 3,5-dihydroxy-2-methyl-(4H)-pyran-4-one (2TMS) | 73, 128, 147, 183, 271, 286 | pyran | H |
| 55 | 1,6-anydro-beta-D-glucopyranose (TMS at position 4) | 73, 103, 117, 129, 145, 155, 171 | anhydrosugars | H |
| 56 | 1,6-anydro-beta-D-glucopyranose (TMS at position 2) | 73, 101, 116, 129, 132, 145, 155, 171 | anhydrosugars | H |
| 57 | Z-isoeugenol (TMS) | 73, 179, 206, 221, 236 | long chain | G-lignin |
| 58 | vanillyl alcohol (2TMS) | 73, 151, 210, 253, 268, 283, 298 | long chain | G-lignin |
| 59 | vanillin (TMS) | 73, 194, 209, 224 | carbonyl | G-lignin |
| 60 | methyl 4-hydroxy-3,5-dimethoxybenzoate TMS | 73, 223, 254, 269, 284 | ester | S-lignin |
| 61 | 1,2,3-trihydroxybenzene (3TMS) | 73, 133, 147, 239, 327, 342 | hydroxybenzene | H |
| 62 | 4-ethylsyringol (TMS) | 73, 191, 209, 224, 239, 254 | short chain | S-lignin |
| 63 | E-isoeugenol (TMS) | 73, 179, 206, 221, 236 | long chain | G-lignin |
| 64 | 1,4-anydro-D-galactopyranose (2TMS) | 73, 101, 116, 129, 145, 155, 171, 217 | anhydrosugars | H |
| 65 | 1,6-anydro-D-galactopyranose (2TMS) | 73, 101, 116, 129, 145, 161, 189, 204, 217 | anhydrosugars | H |
| 66 | 2-hydroxymethyl-5-hydroxy-2,3-dihydro-(4H)-pyran-4-one (2TMS) | 73, 129, 147, 155, 183, 273, 288 | pyran | H |
| 67 | 4-vinylsyringol (TMS) | 73, 179, 222, 237, 252 | short chain | S-lignin |
| 68 | 1,4-anydro-D-glucopyranose (2TMS at position 2 and 4) | 73, 101, 116, 129, 155, 191, 204, 217 | anhydrosugars | H |
| 69 | 1,2,4-trihydroxybenzene (3TMS) | 73, 133, 147, 239, 327, 342 | hydroxybenzene | H |
| 70 | acetovanillone (TMS) | 73, 193, 208, 223, 238 | carbonyl | G-lignin |
| 71 | 4-hydroxy benzoic acid (2TMS) | 73, 147, 193, 223, 267, 282 | acid | H-lignin |
| 72 | 1,6-anydro-beta-D-glucopyranose (2TMS at position 2 and 4) | 73, 101, 116, 129, 155, 191, 204, 217 | anhydrosugars | H |
| 73 | propenyl-syringol (TMS) | 73, 205, 236, 251, 266 | long chain | S-lignin |
| 74 | 1,4-anydro-D-galactopyranose (3TMS) | 73, 129, 147, 157, 191, 204, 217, 243, 332 | anhydrosugars | H |
| 75 | syringaldehyde (TMS) | 73, 224, 239, 254 | carbonyl | S-lignin |
| 76 | 2,3,5-trihydroxy-4H-pyran-4-one (3TMS) | 73, 133, 147, 239, 255, 270, 330, 345, 360 | pyran | H |
| 77 | 1,6-anydro-beta-D-glucopyranose (3TMS) | 73, 103, 129, 147, 191, 204, 217, 243, 333 | anhydrosugars | H |
| 78 | 1,4-anhydro-D-glucopyranose (3TMS) | 73, 103, 129, 147, 191, 204, 217, 243, 332 | anhydrosugars | H |
| 79 | E-propenylsyringol (2TMS) | 73, 205, 236, 251, 266 | long chain | S-lignin |
| 80 | 1,6-anydro-beta-D-glucofuranose (3TMS) | 73, 103, 129, 147, 191, 204, 217, 243, 319 | anhydrosugars | H |
| 81 | vanillic acid (2TMS) | 73, 253, 282, 297, 312 | acid | G-lignin |
| 82 | acetosyringone (TMS) | 73, 223, 238, 253, 268 | carbonyl | S-lignin |
| 83 | vanillylpropanol (2TMS) | 73, 179, 206, 221, 236, 311, 326 | long chain | G-lignin |
| 84 | Z-coniferyl alcohol (2TMS) | 73, 204, 252, 293, 309, 324 | monomer | G-lignin |
| 85 | coumaryl alcohol (2 TMS) | 73, 189, 205, 267, 279, 294 | monomer | H-lignin |
| 86 | coniferylaldehyde (TMS) | 73, 192, 220, 235, 250 | carbonyl | G-lignin |
| 87 | syringic acid (2TMS) | 73, 253, 297, 312, 327, 342 | acid | S-lignin |
| 88 | E-coniferyl alcohol (2 TMS) | 73, 204, 252, 293, 309, 324 | monomer | G-lignin |
| 89 | 3,4-dihydroxy cinnamyl alcohol (3TMS) | 73, 205, 293, 355, 382 | demethylated | G-lignin |
| 90 | E-synapyl alcohol (2TMS) | 73, 234, 323, 339, 354 | monomer | S-lignin |
| 91 | Ferulic acid (2TMS) | 73, 249, 308, 323, 338 | acid | G-lignin |
| Specimen Nr. | Density (g/cm3) | Max MC, % |
|---|---|---|
| Archaeological pine, oven-dried | ||
| 52 | 0.443 | 140 |
| 53 | 0.431 | 153 |
| 54 | 0.217 | 393 |
| 55 | 0.225 | 381 |
| 56 | 0.210 | 410 |
| Archaeological pine, freeze-dried, acclimatized to 50% RH, 20 °C | ||
| 47 | 0.258 | |
| 48 | 0.237 | |
| 49 | 0.361 | |
| 50 | 0.244 | |
| 51 | 0.381 | |
| Sound Pine | Archaeological Pine Untreated | Archaeological Pine Ethyl Acetate Treated | |
|---|---|---|---|
| Specimen | / | 47 | 37 |
| H% | 60.7 | 49.9 | 40.1 |
| L% | 39.3 | 50.1 | 59.9 |
| H/L | 1.54 | 1 | 0.67 |
| Band Position (cm−1) | Assignment | Reference |
|---|---|---|
| 1738–1709 | C=O stretch in unconjugated ketones, carbonyls and in ester groups, frequently of carbohydrate origin; conjugated aldehydes and carboxylic acids absorb around and below 1700 cm−1 | [35] |
| 1646 | conjugated C=O groups, mainly originating from lignin | [36] |
| 1635 | adsorbed water | [35] |
| 1605–1593 | aromatic skeletal vibrations plus C=O stretch | [35] |
| 1515–1505 | aromatic skeletal vibrations | |
| 1375–1374 | CH deformation vibration (cellulose) | |
| 1326 | syringyl plus guaiacyl ring condensed | [37] |
| 1270–1266 | G ring plus C=O stretch | [35] |
| 1235–1225 | OH plane deformation, also COOH | |
| 1230–1221 | C-C plus C-O plus C=O stretch; G-condensed > G-etherified | |
| 1162–1125 | C-O-C asymmetric valence vibration (cellulose) | |
| 1151 | C-O-C asymmetric valence vibration in cellulose and hemicelluloses | [38] |
| 1110–1107 | ring asymmetric valence vibration | [35] |
| 1024 | C-O stretch in cellulose and hemicelluloses; C-O of primary alcohol | [38] |
| 892 | C-H deformation in cellulose; C1 group frequency in cellulose and hemicelluloses | |
| 835–834 | C-H out-of-plane in position 2 and 6 of S and in all positions of H units | [39] |
| 832–817 | C-H out-of-plane in positions 2, 5 and 6 of G units |
| Polymer/Solvent | Sample | Before Treatment (g) * | After Treatment (g) * | % Change Relative to Start | Comment |
|---|---|---|---|---|---|
| 30% P1000 (w/v) | 9 | 2.1580 | 2.4369 | 12.9 | |
| 10 | 2.2111 | 2.6099 | 18.0 | hard to cut | |
| 11 | 3.5335 | 3.8734 | 9.6 | hard to cut | |
| 12 | 2.1890 | 2.6101 | 19.2 | ||
| 30% FB01 (w/v) | 21 | 2.1147 | 3.5541 | 68.1 | |
| 22 | 2.3584 | 3.8057 | 61.4 | very hard to cut | |
| 23 | 3.7985 | 4.6819 | 23.3 | very hard to cut | |
| 24 | 2.1844 | 3.6044 | 65.0 | ||
| ethyl acetate control | 37 | 2.2583 | 2.2555 | −0.1 | easy to cut |
| 38 | 2.5720 | 2.5306 | −1.6 | easy to cut | |
| 39 | 4.2203 | 4.1607 | −1.4 | orange end grain (corrosion) | |
| 40 | 2.1703 | 2.1559 | −0.7 | easy to cut | |
| 41 | 5.1835 | 4.5726 | −11.8 | knot |
| Polymer/Solvent | Sample | Before Treatment (Dry State) | After Impregnation and Drying | % Change Relative to Start | |||
|---|---|---|---|---|---|---|---|
| (mm) | (mm) | ||||||
| Rad | Tan | Rad | Tan | Rad | Tan | ||
| 30% P1000 (w/v) | 9 | 7.97 | 8.74 | 8.04 | 8.86 | 0.9 | 1.4 |
| 10 | 8.99 | 10.39 | 9.06 | 10.44 | 0.8 | 0.5 | |
| 11 | 6.19 | 10.25 | 6.20 | 10.31 | 0.2 | 0.6 | |
| 12 | 8.77 | 10.66 | 8.86 | 10.68 | 1.0 | 0.2 | |
| 30% FB01 (w/v) | 21 | 7.64 | 12.49 | 7.68 | 12.48 | 0.5 | −0.1 |
| 22 | 9.12 | 10.76 | 9.15 | 10.71 | 0.3 | −0.5 | |
| 23 | 9.40 | 10.19 | 9.40 | 10.14 | 0.0 | −0.5 | |
| 24 | 9.00 | 9.67 | 9.54 | 8.97 | 6.0 | −7.2 | |
| ethyl acetate control | 37 | 7.06 | 11.65 | 7.20 | 11.68 | 2.0 | 0.3 |
| 38 | 6.50 | 8.53 | 6.45 | 8.41 | −0.8 | −1.4 | |
| 39 | 8.26 | 7.36 | 8.33 | 7.39 | 0.8 | 0.4 | |
| 40 | 9.30 | 11.40 | 9.31 | 11.45 | 0.1 | 0.4 | |
| 41 | 8.72 | 4.58 | 8.74 | 5.59 | 0.2 | 22.1 | |
| no treatment, control | 47 | 9.28 | 9.53 | 9.38 | 9.55 | 1.1 | 0.2 |
| 48 | 10.40 | 11.17 | 10.38 | 11.26 | −0.2 | 0.8 | |
| 49 | 5.10 | 7.91 | 5.15 | 7.89 | 1.0 | −0.3 | |
| 50 | 6.12 | 10.40 | 6.15 | 10.34 | 0.5 | −0.6 | |
| 51 | 11.17 | 13.72 | 11.08 | 13.36 | −0.8 | −2.6 | |
| oven-dry for characterization | 52 | 12.28 | 13.01 | 11.65 | 12.16 | −5.1 | −6.5 |
| 53 | 9.09 | 15.03 | 8.39 | 13.96 | −7.7 | −7.1 | |
| 54 | 10.32 | 13.04 | 9.96 | 12.38 | −3.5 | −5.1 | |
| 55 | 10.44 | 15.36 | 9.98 | 14.30 | −4.4 | −6.9 | |
| 56 | 13.92 | 14.83 | 13.54 | 14.28 | −2.7 | −3.7 | |
| Specimens Treated with P1000 | Specimens Treated with FB01 | |||
|---|---|---|---|---|
| Core Centre | Inner Surface | Core Centre | Inner Surface | |
| H% | 41.3 | 35.5 | 33.6 | 25.9 |
| L% | 58.7 | 64.5 | 66.4 | 74.1 |
| H/L | 0.7 ± 0.13 | 0.6±0.09 | 0.5 ± 0.03 | 0.4 ± 0.04 |
| H-lignin | 6.8 | 8.3 | 10.8 | 13.7 |
| S-lignin | 12.3 | 21.9 | 26.6 | 34.1 |
| G-lignin | 81.0 | 69.8 | 62.6 | 52.1 |
| S/G | 0.2 ± 0.01 | 0.3 ± 0.04 | 0.4 ± 0.07 | 0.7 ± 0.10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Łucejko, J.J.; de Lamotte, A.; Andriulo, F.; Kutzke, H.; Harding, S.; Phillips-Jones, M.; Modugno, F.; Slaghek, T.M.; Gosselink, R.J.A.; Braovac, S. Evaluation of Soda Lignin from Wheat Straw/Sarkanda Grass as a Potential Future Consolidant for Archaeological Wood. Forests 2021, 12, 911. https://doi.org/10.3390/f12070911
Łucejko JJ, de Lamotte A, Andriulo F, Kutzke H, Harding S, Phillips-Jones M, Modugno F, Slaghek TM, Gosselink RJA, Braovac S. Evaluation of Soda Lignin from Wheat Straw/Sarkanda Grass as a Potential Future Consolidant for Archaeological Wood. Forests. 2021; 12(7):911. https://doi.org/10.3390/f12070911
Chicago/Turabian StyleŁucejko, Jeannette J., Anne de Lamotte, Fabrizio Andriulo, Hartmut Kutzke, Stephen Harding, Mary Phillips-Jones, Francesca Modugno, Ted M. Slaghek, Richard J. A. Gosselink, and Susan Braovac. 2021. "Evaluation of Soda Lignin from Wheat Straw/Sarkanda Grass as a Potential Future Consolidant for Archaeological Wood" Forests 12, no. 7: 911. https://doi.org/10.3390/f12070911
APA StyleŁucejko, J. J., de Lamotte, A., Andriulo, F., Kutzke, H., Harding, S., Phillips-Jones, M., Modugno, F., Slaghek, T. M., Gosselink, R. J. A., & Braovac, S. (2021). Evaluation of Soda Lignin from Wheat Straw/Sarkanda Grass as a Potential Future Consolidant for Archaeological Wood. Forests, 12(7), 911. https://doi.org/10.3390/f12070911