
Complete Chloroplast Genome of Clethra fargesii Franch., an Original Sympetalous Plant from Central China: Comparative Analysis, Adaptive Evolution, and Phylogenetic Relationships

 ,
,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling, Extraction, and Genome Sequencing
2.2. Assembly and Annotation of Chloroplast Genome
2.3. Comparative Analysis of the Chloroplast Genome
2.4. SSRs and Nucleotide Diversity Analysis
2.5. Adaptive Evolution Analysis
2.6. Phylogenetic Analysis
3. Results and Discussion
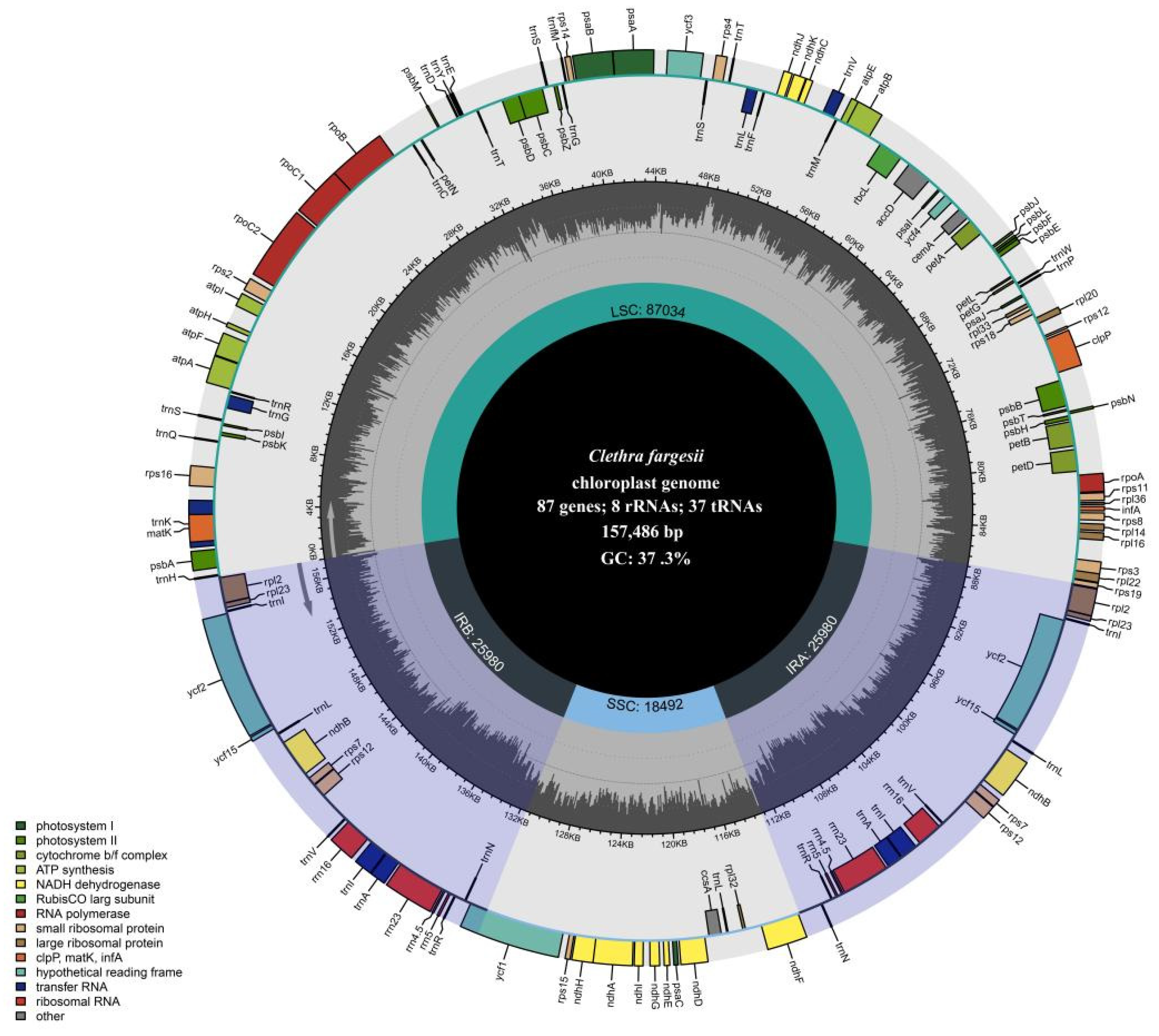
3.1. Chloroplast Genome Features
3.2. Comparative Chloroplast Genome Structure
3.3. Junction Characteristics
3.4. Codon Usage Analysis
3.5. Analysis of Simple Sequence Repeats (SSRs) and Nucleotide Diversity
3.6. Adaptive Evolution Analysis
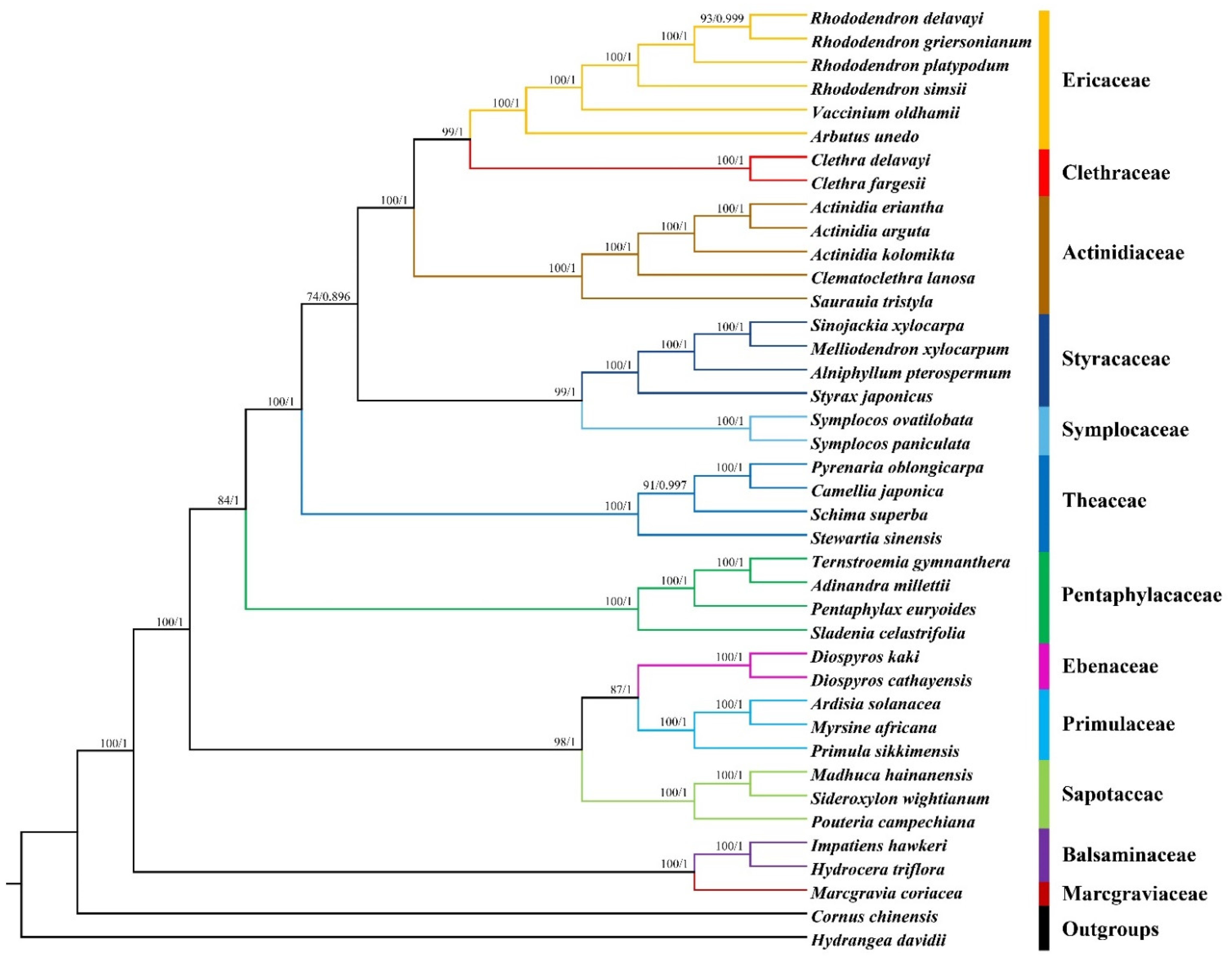
3.7. Phylogenetic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hu, L.C. Clethraceae. In Flora Reipublicae Popularis Sinicae; Fang, W.P., Hu, W.K., Eds.; Science Press: Beijing, China, 1990; Volume 56, pp. 120–156. [Google Scholar]
- Wu, Z.-Y. The areal-types of Chinese genera of seed plants. Acta Bot. Yunnanica 1991, 4, 1–139. [Google Scholar]
- Zhu, D.M. Sowing and seedling cultivation of Clethra barbinervis. Pract. For. Technol. 2011, 9, 33–34. [Google Scholar]
- Ikeda, H.; Natori, T.; Totsuka, T.; Iwaki, H. High SO2 resistance of Clethra barbinervis established in a smoke-polluted area of Ashio, Tochigi Prefecture, Japan. Ecol. Res. 1992, 7, 363–370. [Google Scholar] [CrossRef]
- Yamaji, K.; Watanabe, Y.; Masuya, H.; Shigeto, A.; Yui, H.; Haruma, T. Root fungal endophytes enhance heavy-metal stress tolerance of Clethra barbinervis growing naturally at mining sites via growth enhancement, promotion of nutrient uptake and decrease of heavy-metal concentration. PLoS ONE 2016, 11, e0169089. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Takenaka, C.; Tomioka, R. Accumulation of cobalt and nickel in tissues of Clethra barbinervis in a metal dosing trial. Plant Soil 2017, 421, 273–283. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Tsukada, C.; Takahama, K.; Hirotomo, T.; Tomioka, R.; Takenaka, C. Localization and speciation of cobalt and nickel in the leaves of the cobalt-hyperaccumulating tree Clethra barbinervis. Trees 2019, 33, 521–532. [Google Scholar] [CrossRef]
- Qin, H.N.; Fritsch, P. Clethraceae. In Flora of China; Wu, Z.Y., Raven, P.H., Hong, D.Y., Eds.; Science Press: Beijing, China; Missouri Botanical Garden Press: St. Louis, MO, USA, 2005; Volume 14, pp. 238–241. [Google Scholar]
- Anderberg, A.A.; Zhang, X. Phylogenetic relationships of Cyrillaceae and Clethraceae (Ericales) with special emphasis on the genus Purdiaea Planch. Org. Divers. Evol. 2002, 2, 127–137. [Google Scholar] [CrossRef]
- Fior, S.; Karis, P.O.; Anderberg, A.A. Phylogeny, taxonomy, and systematic position of Clethra (Clethraceae, Ericales) with notes on biogeography: Evidence from plastid and nuclear DNA sequences. Int. J. Plant Sci. 2003, 164, 997–1006. [Google Scholar] [CrossRef]
- Wicke, S.; Schneeweiss, G.M.; dePamphilis, C.W.; Muller, K.F.; Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef]
- Daniell, H.; Lin, C.S.; Yu, M.; Chang, W.J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 134. [Google Scholar] [CrossRef]
- Ravi, V.; Khurana, J.P.; Tyagi, A.K.; Khurana, P. An update on chloroplast genomes. Plant Syst. Evol. 2007, 271, 101–122. [Google Scholar] [CrossRef]
- Parks, M.; Cronn, R.; Liston, A. Increasing phylogenetic resolution at low taxonomic levels using massively parallel sequencing of chloroplast genomes. BMC Biol. 2009, 7, 84. [Google Scholar] [CrossRef]
- Dobrogojski, J.; Adamiec, M.; Luciński, R. The chloroplast genome: A review. Acta Physiol. Plant. 2020, 42, 98. [Google Scholar] [CrossRef]
- Shaw, J.; Lickey, E.B.; Schilling, E.E.; Small, R.L. Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: The tortoise and the hare III. Am. J. Bot. 2007, 94, 275–288. [Google Scholar] [CrossRef]
- Moore, M.J.; Bell, C.D.; Soltis, P.S.; Soltis, D.E. Using plastid genome-scale data to resolve enigmatic relationships among basal angiosperms. Proc. Natl. Acad. Sci. USA 2007, 104, 19363–19368. [Google Scholar] [CrossRef]
- Liu, Y.C.; Lin, B.Y.; Lin, J.Y.; Wu, W.L.; Chang, C.C. Evaluation of chloroplast DNA markers for intraspecific identification of Phalaenopsis equestris cultivars. Sci. Hortic. 2016, 203, 86–94. [Google Scholar] [CrossRef]
- CBOL Plant Working Group. A DNA barcode for land plants. Proc. Natl. Acad. Sci. USA 2009, 10, 12794–12797. [Google Scholar]
- Moore, M.J.; Soltis, P.S.; Bell, C.D.; Burleigh, J.G.; Soltis, D.E. Phylogenetic analysis of 83 plastid genes further resolves the early diversification of eudicots. Proc. Natl. Acad. Sci. USA 2010, 107, 4623–4628. [Google Scholar] [CrossRef] [PubMed]
- Mardis, E.R. Next-generation sequencing platforms. Annu. Rev. Anal. Chem. 2013, 6, 287–303. [Google Scholar] [CrossRef] [PubMed]
- Chase, M.W.; Hills, H.H. Silica gel: An ideal material for field preservation of leaf samples for DNA studies. Taxon 1991, 40, 215–220. [Google Scholar] [CrossRef]
- Doyle, J.J. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Jin, J.J.; Yu, W.B.; Yang, J.B.; Song, Y.; dePamphilis, C.W.; Yi, T.S.; Li, D.Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef] [PubMed]
- Wick, R.R.; Schultz, M.B.; Zobel, J.; Holt, K.E. Bandage: Interactive visualization of de novo genome assemblies. Bioinformatics 2015, 31, 3350–3352. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.J.; Moore, M.J.; Li, D.Z.; Yi, T.S. PGA: A software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods 2019, 15, 50. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Poczai, P.; Hyvonen, J.; Tang, J.; Amiryousefi, A. Chloroplot an online program for the versatile plotting of organelle genomes. Front. Genet. 2020, 11, 576124. [Google Scholar] [CrossRef]
- Darling, A.C.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Thiel, T.; Michalek, W.; Varshney, R.K.; Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Munch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Gao, F.; Chen, C.; Arab, D.A.; Du, Z.; He, Y.; Ho, S.Y.W. EasyCodeML: A visual tool for analysis of selection using CodeML. Ecol. Evol. 2019, 9, 3891–3898. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wong, W.S.; Nielsen, R. Bayes empirical bayes inference of amino acid sites under positive selection. Mol. Biol. Evol. 2005, 22, 1107–1118. [Google Scholar] [CrossRef] [PubMed]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Hohna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Minh, B.Q.; Nguyen, M.A.; von Haeseler, A. Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 2013, 30, 1188–1195. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2019, 20, 348–355. [Google Scholar] [CrossRef]
- He, Y.; Xiao, H.; Deng, C.; Xiong, L.; Yang, J.; Peng, C. The complete chloroplast genome sequences of the medicinal plant Pogostemon cablin. Int. J. Mol. Sci. 2016, 17, 820. [Google Scholar] [CrossRef]
- Shen, X.; Wu, M.; Liao, B.; Liu, Z.; Bai, R.; Xiao, S.; Li, X.; Zhang, B.; Xu, J.; Chen, S. Complete chloroplast genome sequence and phylogenetic analysis of the medicinal plant Artemisia annua. Molecules 2017, 22, 1330. [Google Scholar] [CrossRef]
- Necsulea, A.; Lobry, J.R. A new method for assessing the effect of replication on DNA base composition asymmetry. Mol. Biol. Evol. 2007, 24, 2169–2179. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-C.; Baek, S.-H.; Lee, J.-W.; Hyun, H.J. Complete chloroplast genome of Vaccinium oldhamii and phylogenetic analysis. Mitochondrial DNA B 2019, 4, 902–903. [Google Scholar] [CrossRef]
- Liu, D.; Fu, C.; Yin, L.; Ma, Y. Complete plastid genome of Rhododendron griersonianum, a critically endangered plant with extremely small populations (PSESP) from southwest China. Mitochondrial DNA B 2020, 5, 3086–3087. [Google Scholar] [CrossRef] [PubMed]
- Wakasugi, T.; Tsudzuki, T.; Sugiura, M. The genomics of land plant chloroplasts: Gene content and alteration of genomic information by RNA editing. Photosynth. Res. 2001, 70, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Kahlau, S.; Aspinall, S.; Gray, J.C.; Bock, R. Sequence of the tomato chloroplast DNA and evolutionary comparison of solanaceous plastid genomes. J. Mol. Evol. 2006, 63, 194–207. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Feng, D.; Song, G.; Wei, X.; Chen, L.; Wu, X.; Li, X.; Zhu, Z. The first intron of rice EPSP synthase enhances expression of foreign gene. Sci. China Life Sci. 2003, 46, 561–569. [Google Scholar] [CrossRef]
- Li, X.; Zuo, Y.; Zhu, X.; Liao, S.; Ma, J. Complete chloroplast genomes and comparative analysis of sequences evolution among seven Aristolochia (Aristolochiaceae) medicinal species. Int. J. Mol. Sci. 2019, 20, 1045. [Google Scholar] [CrossRef] [PubMed]
- Souza, U.J.B.D.; Vitorino, L.C.; Bessa, L.A.; Silva, F.G. The complete plastid genome of Artocarpus camansi: A high degree of conservation of the plastome structure in the family Moraceae. Forests 2020, 11, 1179. [Google Scholar] [CrossRef]
- The Angiosperm Phylogeny Group. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 2016, 181, 1–20. [Google Scholar] [CrossRef]
- Martin, W.; Herrmann, R.G. Gene transfer from organelles to the nucleus: How much, what happens, and why? Plant Physiol. 1998, 118, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Martin, W. Gene transfer from organelles to the nucleus: Frequent and in big chunks. Proc. Natl. Acad. Sci. USA 2003, 100, 8612–8614. [Google Scholar] [CrossRef]
- Chung, H.J.; Jung, J.D.; Park, H.W.; Kim, J.H.; Cha, H.W.; Min, S.R.; Jeong, W.J.; Liu, J.R. The complete chloroplast genome sequences of Solanum tuberosum and comparative analysis with Solanaceae species identified the presence of a 241-bp deletion in cultivated potato chloroplast DNA sequence. Plant Cell Rep. 2006, 25, 1369–1379. [Google Scholar] [CrossRef]
- Timme, R.E.; Kuehl, J.V.; Boore, J.L.; Jansen, R.K. A comparative analysis of the Lactuca and Helianthus (Asteraceae) plastid genomes identification of divergent regions and categorization of shared repeats. Am. J. Bot. 2007, 94, 302–312. [Google Scholar] [CrossRef]
- Dugas, D.V.; Hernandez, D.; Koenen, E.J.; Schwarz, E.; Straub, S.; Hughes, C.E.; Jansen, R.K.; Nageswara-Rao, M.; Staats, M.; Trujillo, J.T.; et al. Mimosoid legume plastome evolution: IR expansion, tandem repeat expansions, and accelerated rate of evolution in clpP. Sci. Rep. 2015, 5, 16958. [Google Scholar] [CrossRef]
- Li, Y.; Sylvester, S.P.; Li, M.; Zhang, C.; Li, X.; Duan, Y.; Wang, X. The complete plastid genome of Magnolia zenii and genetic comparison to Magnoliaceae species. Molecules 2019, 24, 261. [Google Scholar] [CrossRef]
- Munyao, J.N.; Dong, X.; Yang, J.X.; Mbandi, E.M.; Wanga, V.O.; Oulo, M.A.; Saina, J.K.; Musili, P.M.; Hu, G.W. Complete chloroplast genomes of Chlorophytum comosum and Chlorophytum gallabatense: Genome structures, comparative and phylogenetic analysis. Plants 2020, 9, 296. [Google Scholar] [CrossRef]
- Li, D.M.; Zhu, G.F.; Xu, Y.C.; Ye, Y.J.; Liu, J.M. Complete chloroplast genomes of three medicinal Alpinia Species: Genome organization, comparative analyses and phylogenetic relationships in family Zingiberaceae. Plants 2020, 9, 286. [Google Scholar] [CrossRef] [PubMed]
- Provan, J.; Powell, W.; Hollingsworth, P.M. Chloroplast microsatellites: New tools for studies in plant ecology and evolution. Trends Ecol. Evol. 2001, 16, 142–147. [Google Scholar] [CrossRef]
- Oulo, M.A.; Yang, J.X.; Dong, X.; Wanga, V.O.; Mkala, E.M.; Munyao, J.N.; Onjolo, V.O.; Rono, P.C.; Hu, G.W.; Wang, Q.F. Complete chloroplast genome of Rhipsalis baccifera, the only cactus with natural distribution in the old world: Genome rearrangement, intron gain and loss, and implications for phylogenetic studies. Plants 2020, 9, 979. [Google Scholar] [CrossRef]
- Jiang, K.; Miao, L.Y.; Wang, Z.W.; Ni, Z.Y.; Hu, C.; Zeng, X.H.; Huang, W.C. Chloroplast genome analysis of two medicinal coelogyne spp. (Orchidaceae) shed light on the genetic information, comparative genomics, and species identification. Plants 2020, 9, 1332. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.L.; Zheng, H.Y.; Price, M.; Zhou, S.D.; He, X.J. Phylogeny and comparative analysis of Chinese Chamaesium species revealed by the complete plastid genome. Plants 2020, 9, 965. [Google Scholar] [CrossRef]
- Li, X.; Li, Y.; Zang, M.; Li, M.; Fang, Y. Complete chloroplast genome sequence and phylogenetic analysis of Quercus acutissima. Int. J. Mol. Sci. 2018, 19, 2443. [Google Scholar] [CrossRef]
- Liu, H.; Hu, H.; Zhang, S.; Jin, J.; Liang, X.; Huang, B.; Wang, L. The complete chloroplast genome of the rare species Epimedium tianmenshanensis and comparative analysis with related species. Physiol. Mol. Biol. Plants 2020, 26, 2075–2083. [Google Scholar] [CrossRef]
- Park, J.; Xi, H.; Kim, Y. The complete chloroplast genome of Arabidopsis thaliana isolated in Korea (Brassicaceae): An investigation of intraspecific variations of the chloroplast genome of Korean A. thaliana. Int. J. Genom. 2020, 2020, 3236461. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Ruhsam, M.; Wang, J.; Zhu, H.; Li, W.; Zhang, X.; Xu, Y.; Xu, F.; Wang, X. The complete chloroplast genome of Euphrasia regelii, pseudogenization of ndh genes and the phylogenetic relationships within Orobanchaceae. Front. Genet. 2019, 10, 444. [Google Scholar] [CrossRef]
- Wu, L.; Nie, L.; Xu, Z.; Li, P.; Wang, Y.; He, C.; Song, J.; Yao, H. Comparative and phylogenetic analysis of the complete chloroplast genomes of three Paeonia Section moutan species (Paeoniaceae). Front. Genet. 2020, 11, 980. [Google Scholar] [CrossRef]
- Yu, X.Q.; Drew, B.T.; Yang, J.B.; Gao, L.M.; Li, D.Z. Comparative chloroplast genomes of eleven Schima (Theaceae) species: Insights into DNA barcoding and phylogeny. PLoS ONE 2017, 12, e0178026. [Google Scholar] [CrossRef]
- Li, D.M.; Ye, Y.J.; Xu, Y.C.; Liu, J.M.; Zhu, G.F. Complete chloroplast genomes of Zingiber montanum and Zingiber zerumbet: Genome structure, comparative and phylogenetic analyses. PLoS ONE 2020, 15, e0236590. [Google Scholar] [CrossRef]
- Khakhlova, O.; Bock, R. Elimination of deleterious mutations in plastid genomes by gene conversion. Plant J. 2006, 46, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Gao, H.; Wang, Y.; Song, J.; Henry, R.; Wu, H.; Hu, Z.; Yao, H.; Luo, H.; Luo, K.; et al. Complete chloroplast genome sequence of Magnolia grandiflora and comparative analysis with related species. Sci. China Life Sci. 2013, 56, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Li, J.; Zhang, H.; Cai, B.; Gao, Z.; Qiao, Y.; Mi, L. The complete chloroplast genome sequence of strawberry (Fragaria x ananassa Duch.) and comparison with related species of Rosaceae. PeerJ 2017, 5, e3919. [Google Scholar] [CrossRef]
- Li, D.M.; Zhao, C.Y.; Liu, X.F. Complete chloroplast genomesequences of Kaempferia galanga and Kaempferia elegans: Molecular structures and comparative analysis. Molecules 2019, 24, 474. [Google Scholar] [CrossRef] [PubMed]
- Souza, U.J.B.; Nunes, R.; Targueta, C.P.; Diniz-Filho, J.A.F.; Telles, M.P.C. The complete chloroplast genome of Stryphnodendron adstringens (Leguminosae-Caesalpinioideae): Comparative analysis with related Mimosoid species. Sci. Rep. 2019, 9, 14206. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Nielsen, R. Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol. Biol. Evol. 2000, 17, 32–43. [Google Scholar] [CrossRef]
- Tonti-Filippini, J.; Nevill, P.G.; Dixon, K.; Small, I. What can we do with 1000 plastid genomes? Plant J. 2017, 90, 808–818. [Google Scholar] [CrossRef]
- Anderberg, A.A.; Rydin, C.; Kallersjo, M. Phylogenetic relationships in the order Ericales s.l.: Analyses of molecular data from five genes from the plastid and mitochondrial genomes. Am. J. Bot. 2002, 89, 677–687. [Google Scholar] [CrossRef]
- Yan, M.; Fritsch, P.W.; Moore, M.J.; Feng, T.; Meng, A.; Yang, J.; Deng, T.; Zhao, C.; Yao, X.; Sun, H.; et al. Plastid phylogenomics resolves infrafamilial relationships of the Styracaceae and sheds light on the backbone relationships of the Ericales. Mol. Phylogenet. Evol. 2018, 121, 198–211. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genome Features | Clethra fargesii Franch. | Clethra delavayi Franch. | Rhododendron griersonianum Balf. F. et Forrest | Vaccinium oldhamii Miquel | Saurauia tristyla DC. | Actinidia eriantha Benth. | Styrax japonicus Sieb. et Zucc. | Schima superba Gardn. et Champ. |
|---|---|---|---|---|---|---|---|---|
| Total size (bp) | 157,486 | 157,253 | 206,467 | 173,245 | 156,676 | 156,964 | 157,929 | 157,254 |
| LSC size (bp) | 87,034 | 86,870 | 108,922 | 105,498 | 88,274 | 88,639 | 87,540 | 87,202 |
| SSC size (bp) | 18,492 | 18,469 | 2611 | 3067 | 20,482 | 20,541 | 18,281 | 18,100 |
| IR size (bp) | 25,980 | 25,957 | 47,467 | 32,340 | 23960 | 23,892 | 26,054 | 25,976 |
| Total GC (%) | 37.3 | 37.4 | 35.8 | 36.8 | 36.9 | 37.2 | 37.0 | 37.4 |
| LSC of GC (%) | 35.4 | 35.4 | 35.3 | 35.8 | 35.2 | 35.4 | 34.8 | 35.5 |
| SSC of GC (%) | 30.7 | 30.8 | 30.0 | 29.2 | 30.6 | 31.1 | 30.3 | 30.8 |
| IR of GC (%) | 43.0 | 43.0 | 36.5 | 38.7 | 42.9 | 43.6 | 42.9 | 42.8 |
| Total genes | 132 | 132 | 150 | 130 | 132 | 132 | 132 | 132 |
| Unique genes | 114 | 114 | 118 | 108 | 113 | 113 | 114 | 114 |
| PCGs | 87 | 87 | 95 | 85 | 84 | 84 | 87 | 87 |
| tRNA genes | 37 | 37 | 47 | 37 | 39 | 39 | 37 | 37 |
| rRNA genes | 8 | 8 | 8 | 8 | 8 | 8 | 8 | 8 |
| Duplicated PCGs | 7 | 7 | 17 | 11 | 5 | 5 | 7 | 7 |
| Unique PCGs | 80 | 80 | 78 | 74 | 79 | 79 | 80 | 80 |
| Category | Groups of Genes | Name of Genes |
|---|---|---|
| Self-replication | Ribosomal RNA | rrn4.5c, rrn5c, rrn16c, rrn23c |
| Transfer RNA | trnA-UGCa,c, trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnG-GCC, trnG-UCCa, trnH-GUG, trnI-CAUc, trnI-GAU a,c, trnK-UUUa, trnL-CAAc, trnL-UAAa, trnL-UAG, trnM-CAU, trnfM-CAU, trnN-GUUc, trnP-UGG, trnQ-UUG, trnR-UCU, trnR-ACGc, trnS-UGA, trnS-GCU, trnS-GGA, trnT-GGU, trnT-UGU, trnV-UACa, trnV-GACc, trnW-CCA, trnY-GUA | |
| Small subunit of ribosome | rps2, rps3, rps4, rps7c, rps8, rps11, rps12a,c, rps14, rps15, rps16a, rps18, rps19 | |
| Large subunit of ribosome | rpl2a,c, rpl14, rpl16a, rpl20, rpl22, rpl23c, rpl32, rpl33, rpl36 | |
| RNA polymerase subunits | rpoA, rpoB, rpoC1a, rpoC2 | |
| Photosynthesis | Photosystem I | pasA, pasB, pasC, pasI, pasJ |
| Photosystem II | psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbT, psbZ | |
| Subunits of cytochrome | petA, petBa, petDa, petG, petL, petN | |
| ATP synthase | atpA, atpB, atpE, atpFa, atpH, atpI | |
| NADH-dehydrogenase | ndhAa, ndhBa,c, ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK | |
| Other genes | Rubisco large subunit | rbcL |
| Translational initiation factor | infA | |
| Maturase K | matK | |
| Envelope membrane protein | cemA | |
| Acetyl-CoA carboxylase | accD | |
| Proteolysis | clpPb | |
| Cytochrome c biogenesis | ccsA | |
| Conserved open reading frames | ycf1, ycf2c, ycf3b, ycf4, ycf15c |
| M8 | Gene Name | Region | Selected Sites | Pr (w > 1) | Number of Selected Sites |
|---|---|---|---|---|---|
| Bayes Empirical Bayes (BEB) | ycf1 | SSC | 3027 I/3113 Y | 0.953 */1.000 ** | 37 |
| rps4 | LSC | 7916 P | 0.993 ** | 1 | |
| Naive Empirical Bayes (NEB) | ycf1 | SSC | 2544 P/3119 E | 1.000 ** | 74 |
| ycf2 | IR | 5115 F/6946 V | 1.000 ** | 7 | |
| cemA | LSC | 12302 T/12375A | 1.000 ** | 6 | |
| ndhB | IR | 13905 I/14140 E | 1.000 ** | 19 | |
| psbD | LSC | 20701 S | 1.000 ** | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, S.; Dong, X.; Yang, J.; Guo, C.; Cao, B.; Guo, Y.; Hu, G. Complete Chloroplast Genome of Clethra fargesii Franch., an Original Sympetalous Plant from Central China: Comparative Analysis, Adaptive Evolution, and Phylogenetic Relationships. Forests 2021, 12, 441. https://doi.org/10.3390/f12040441
Ding S, Dong X, Yang J, Guo C, Cao B, Guo Y, Hu G. Complete Chloroplast Genome of Clethra fargesii Franch., an Original Sympetalous Plant from Central China: Comparative Analysis, Adaptive Evolution, and Phylogenetic Relationships. Forests. 2021; 12(4):441. https://doi.org/10.3390/f12040441
Chicago/Turabian StyleDing, Shixiong, Xiang Dong, Jiaxin Yang, Chunce Guo, Binbin Cao, Yuan Guo, and Guangwan Hu. 2021. "Complete Chloroplast Genome of Clethra fargesii Franch., an Original Sympetalous Plant from Central China: Comparative Analysis, Adaptive Evolution, and Phylogenetic Relationships" Forests 12, no. 4: 441. https://doi.org/10.3390/f12040441
APA StyleDing, S., Dong, X., Yang, J., Guo, C., Cao, B., Guo, Y., & Hu, G. (2021). Complete Chloroplast Genome of Clethra fargesii Franch., an Original Sympetalous Plant from Central China: Comparative Analysis, Adaptive Evolution, and Phylogenetic Relationships. Forests, 12(4), 441. https://doi.org/10.3390/f12040441