Transcriptomic Profiling of Cryptomeria fortunei Hooibrenk Vascular Cambium Identifies Candidate Genes Involved in Phenylpropanoid Metabolism

 , , ,
, , ,
Abstract
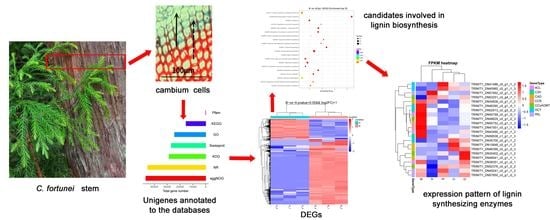
1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. RNA Extraction and Transcriptome Sequencing
2.3. Assembly and Functional Annotation
2.4. Differential Expression Analysis
2.5. Verification of Gene Expression Using qRT-PCR
2.6. Determination of Lignin Content
3. Results
3.1. Statistics of Transcriptome Sequencing Results and De Novo Assembly
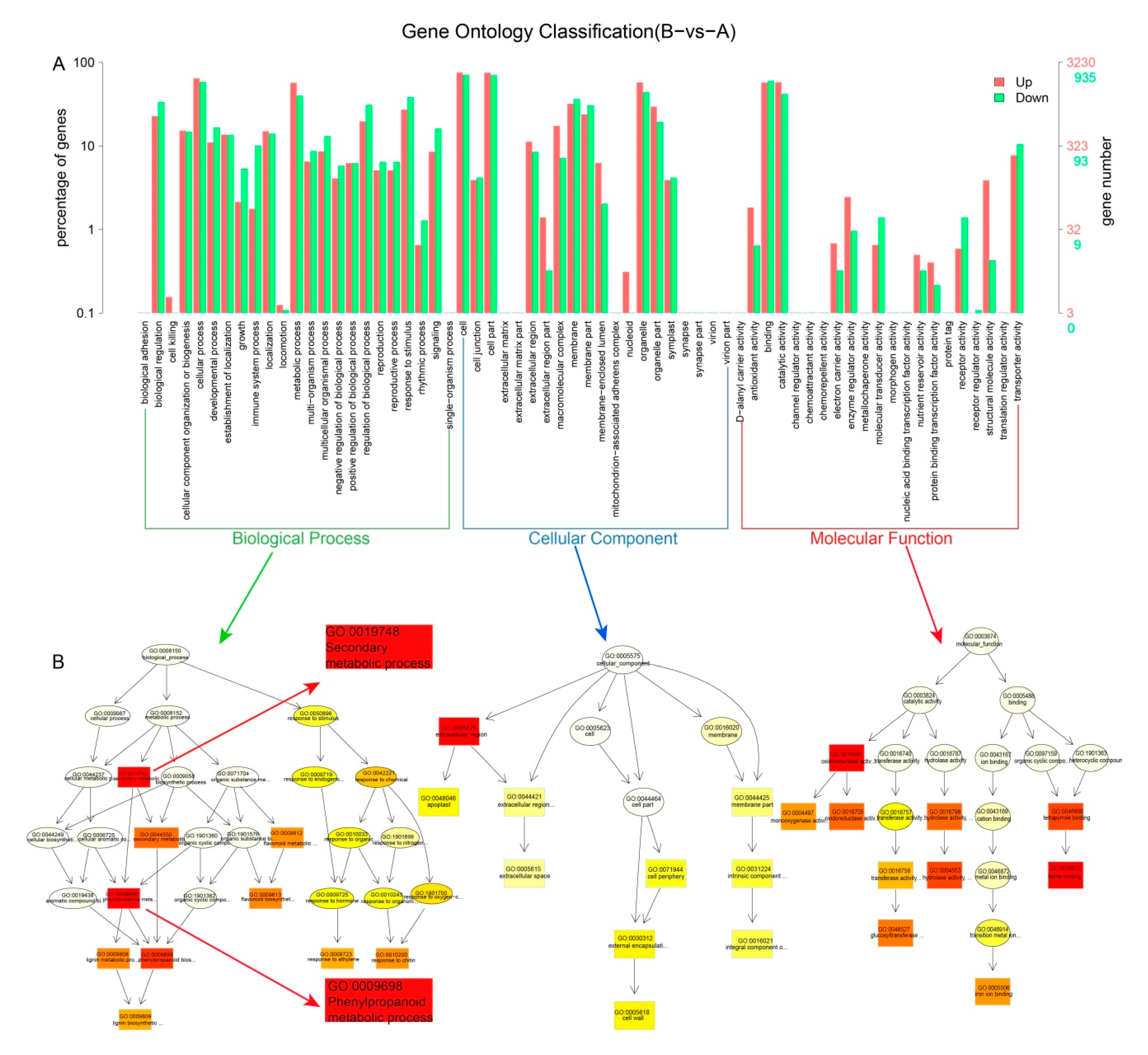
3.2. Functional Annotation and Classification of All Unigenes
3.3. Identification of DEGs and the GO and KEGG Enrichment Analysis
3.4. Identification of Candidate Genes Involved in Phenylpropanoid Metabolism
3.5. Quantitative Real-Time PCR Validation of Candidate Genes Involved in Phenylpropanoid Biosynthesis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhao, H.; Mo, J.X.; Hua, H.; Guo, Z.H.; Xu, J. Meiosis process and abnormal behavior of pollen mother cells in Cryptomeria fortunei. J. Nanjing For. Univ. (Nat. Sci. Ed.) 2019, 43, 45–50. (In Chinese) [Google Scholar]
- Růžička, K.; Ursache, R.; Hejátko, J.; Helariutta, Y. Xylem development—From the cradle to the grave. New Phytol. 2015, 207, 519–535. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Zhang, L.; Lei, L.; Cheng, Z.S.; Sun, S.W.; Lai, M. Analysis of SSR information in transcriptome and development of EST-SSR molecular markers in Pinus elliottii Engelm. J. Nanjing For. Univ. (Nat. Sci. Ed.) 2020, 44, 75–83. (In Chinese) [Google Scholar]
- Fagard, M.; Höfte, H.; Vernhettes, S. Cell wall mutants. Plant Physiol. Biochem. 2000, 38, 15–25. [Google Scholar] [CrossRef]
- Soler, M.; Plasencia, A.; Larbat, R.; Pouzet, C.; Jauneau, A.; Rivas, S.; Pesquet, E.; Lapierre, C.; Truchet, I.; Grima-Pettenati, J. The Eucalyptus linker histone variant EgH1.3 cooperates with the transcription factor EgMYB1 to control lignin biosynthesis during wood formation. New Phytol. 2016, 213, 287–299. [Google Scholar] [CrossRef]
- Shi, R.; Sun, Y.-H.; Li, Q.Z.; Heber, S.; Sederoff, R.; Chiang, V.L. Towards a systems approach for lignin biosynthesis in Populus trichocarpa: Transcript abundance and specificity of the monolignol biosynthetic genes. Plant Cell Physiol. 2009, 51, 144–163. [Google Scholar] [CrossRef]
- Jokipii-Lukkari, S.; Delhomme, N.; Schiffthaler, B.; Mannapperuma, C.; Prestele, J.; Nilsson, O.; Street, R.N.; Tuominen, H. Transcriptional Roadmap to Seasonal Variation in Wood Formation of Norway Spruce. Plant Physiol. 2018, 176, 2851–2870. [Google Scholar] [CrossRef]
- Boerjan, W.; Ralph, J.; Baucher, M. Ligninbiosynthesis. Annu. Rev. Plant Boil. 2003, 54, 519–546. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Cheng, X.F.; Leshkevich, J.; Umezawa, T.; Harding, S.A.; Chiang, V.L. the last step of syringyl monolignol biosynthesis in angiosperms is regulated by a novel gene encoding sinapyl alcohol dehydrogenase. Plant Cell 2001, 13, 1567–1586. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-H.; Chiang, V.L. Molecular cloning of 4-coumarate:coenzyme A ligase in loblolly pine and the roles of this enzyme in the biosynthesis of lignin in compression wood. Plant Physiol. 1997, 113, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Hua, H.; Xu, J.; Mo, J.; Zhao, H.; Yang, J. Cloning and Functional Analysis of Lignin Biosynthesis Genes Cf4CL and CfCCoAOMT in Cryptomeria fortunei. Genes 2019, 10, 619. [Google Scholar] [CrossRef]
- Qiu, Z.; Wan, L.; Chen, T.; Wan, Y.; He, X.; Lu, S.; Wang, Y.; Lin, J. The regulation of cambial activity in Chinese fir (Cunninghamia lanceolata) involves extensive transcriptome remodeling. New Phytol. 2013, 199, 708–719. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Jaroszewski, L.; Godzik, A. Clustering of highly homologous sequences to reduce the size of large protein databases. Bioinformatics 2001, 17, 282–283. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2014, 12, 59–60. [Google Scholar] [CrossRef]
- Mistry, J.; Finn, R.D.; Eddy, S.R.; Bateman, A.; Punta, M. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 2013, 41, e121. [Google Scholar] [CrossRef]
- Roberts, A.; Trapnell, C.; Donaghey, J.; Rinn, J.L.; Pachter, L. Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Boil. 2011, 12, R22. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Roberts, A. Ambiguous Fragment Assignment for High-Throughput Sequencing Experiments. Ph.D. Thesis, University of California, Berkeley, CA, USA, 2013. [Google Scholar]
- Anders, S.; Huber, W. Differential Expression of RNA-Seq Data at the Gene Level–the DESeq Package; European Molecular Biology Laboratory (EMBL): Heidelberg, Germany, 2012. [Google Scholar]
- Wu, Y.; Guo, J.; Zhou, Q.; Xin, Y.; Wang, G.; Xu, L.-A. De novo transcriptome analysis revealed genes involved in flavonoid biosynthesis, transport and regulation in Ginkgo biloba. Ind. Crop. Prod. 2018, 124, 226–235. [Google Scholar] [CrossRef]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2009, 26, 136–138. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Rodrigues, J.; Faix, O.; Pereira, H. Improvement of the acetylbromide method for lignin determination within large scale screening programmes. Holz als Roh-und Werkst. 1999, 57, 341–345. [Google Scholar] [CrossRef]
- Cao, Y.T. Transcriptome Sequencing and Profiling of Expression in Vascular Cambium of Cryptomeria fortunei. Master’ s Thesis, Nanjing Forestry University, Nanjing, China, 2016. (In Chinese). [Google Scholar]
- Alexa, A.; Rahnenfuhrer, J. Enrichment Analysis for Gene Ontology; R Package Version 2.8; Bioconductor: Buffalo, NY, USA, 2010. [Google Scholar]
- Mo, J.; Xu, J.; Cao, Y.; Yang, L.; Yin, T.; Hua, H.; Zhao, H.; Guo, Z.; Yang, J.; Shi, J. Pinus massoniana Introgression Hybrids Display Differential Expression of Reproductive Genes. Forests 2019, 10, 230. [Google Scholar] [CrossRef]
- Huang, H.; Xu, L.-L.; Tong, Z.; Lin, E.; Liu, Q.; Cheng, L.J.; Zhu, M.-Y. De novo characterization of the Chinese fir (Cunninghamia lanceolata) transcriptome and analysis of candidate genes involved in cellulose and lignin biosynthesis. BMC Genom. 2012, 13, 648. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.-Y.; Yang, H.; Wei, C.; Yu, O.; Zhang, Z.; Jiang, C.; Sun, J.; Li, Y.; Chen, Q.; Xia, T.; et al. Deep sequencing of the Camellia sinensis transcriptome revealed candidate genes for major metabolic pathways of tea-specific compounds. BMC Genom. 2011, 12, 131. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Mao, Y.; Kong, F.; Yang, G.; Ma, F.; Wang, L. Profiling of the transcriptome of Porphyra yezoensis with Solexa sequencing technology. Chin. Sci. Bull. 2011, 56, 2119–2130. [Google Scholar] [CrossRef]
- Mishima, K.; Fujiwara, T.; Iki, T.; Kuroda, K.; Yamashita, K.; Tamura, M.; Fujisawa, Y.; Watanabe, A. Transcriptome sequencing and profiling of expressed genes in cambial zone and differentiating xylem of Japanese cedar (Cryptomeria japonica). BMC Genom. 2014, 15, 219. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.H.; Xiong, Z.H.; Wu, W.L.; Li, W.L. CAD enzyme activity and gene expression in connection with lignin synthesis during fruit development and ripening process of blackberry. J. Nanjing For. Univ. (Nat. Sci. Ed.) 2018, 42, 141–148. (In Chinese) [Google Scholar]
- Douglas, C.J. Phenylpropanoid metabolism and lignin biosynthesis: From weeds to trees. Trends Plant Sci. 1996, 1, 171–178. [Google Scholar] [CrossRef]
- Heide, O.M. Growth and dormancy in norway spruce ecotypes (Picea abies) i. interaction of photoperiod and temperature. Physiol. Plant. 1974, 30, 1–12. [Google Scholar] [CrossRef]
- Sato, S.; Yoshida, M.; Ashizaki, Y.; Yamamoto, H. Diurnal periodicity of the expression of genes involved in monolignol biosynthesis in differentiating xylem of cryptomeria japonica. Am. J. Plant Sci. 2016, 7, 2457–2469. [Google Scholar] [CrossRef]
- Zhong, R.; Ye, Z. Transcriptional regulation of lignin biosynthesis. Plant Signal. Behav. 2009, 4, 1028–1034. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.Z.; Zheng, J.; Chen, J.H.; Shi, J.S. Cloning transcription factor ClMYB4 involving in secondary cell wall biosynthesis from Cunninghamia lanceolata (Lamb) Hook and expressing in E. coli. Mol. Plant Breed. 2012, 10, 512–519. (In Chinese) [Google Scholar]
- Cassan-Wang, H.; Goué, N.; Saidi, M.N.; Legay, S.; Sivadon, P.; Goffner, D.; Grima-Pettenati, J. Identification of novel transcription factors regulating secondary cell wall formation in Arabidopsis. Front. Plant Sci. 2013, 4, 189. [Google Scholar] [CrossRef]
- Movahedi, A.; Zhang, J.; Yin, T.; Zhuge, Q. Functional Analysis of Two Orthologous NAC Genes, CarNAC3, and CarNAC6 from Cicer arietinum, Involved in Abiotic Stresses in Poplar. Plant Mol. Boil. Rep. 2015, 33, 1539–1551. [Google Scholar] [CrossRef]
- Wang, J.; Xu, M.; Gu, Y.; Xu, L. Differentially expressed gene analysis of Tamarix chinensis provides insights into NaCl-stress response. Trees 2016, 31, 645–658. [Google Scholar] [CrossRef]
- Liu, X.; Yu, W.; Zhang, X.; Wang, G.; Cao, F.-L.; Cheng, H. Identification and expression analysis under abiotic stress of the R2R3-MYB genes in Ginkgo biloba L. Physiol. Mol. Boil. Plants 2017, 23, 503–516. [Google Scholar] [CrossRef]
- Wang, Y.; Qiu, W.M.; Li, H.; He, X.L.; Liu, M.Y.; Han, X.J.; Qu, T.B.; Zhuo, R.Y. Research on the response of SaWRKY7 gene to cadmium stress in Sedum alfredii Hance. J. Nanjing For. Univ. (Nat. Sci. Ed.) 2019, 43, 59–66. (In Chinese) [Google Scholar]
- Tian, X.Y.; Zhou, J.; Wang, B.S.; He, K.Y.; He, X.D. Cloning and expression pattern analysis of NAC genes in Salix. J. Nanjing For. Univ. (Nat. Sci. Ed.) 2020, 44, 119–124. (In Chinese) [Google Scholar]
- Yang, C.; Xu, M.; Xuan, L.; Jiang, X.; Huang, M. Identification and expression analysis of twenty ARF genes in Populus. Gene 2014, 544, 134–144. [Google Scholar] [CrossRef]
- Bi, C.; Xu, Y.; Ye, Q.; Yin, T.; Ye, N. Genome-wide identification and characterization of WRKY gene family in Salix suchowensis. PeerJ 2016, 4, e2437. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Cheng, T.; Lu, M.; Liu, G.; Li, M.; Shi, J.; Lu, Y.; Laux, T.; Chen, J. Expansion and Functional Divergence of AP2 Group Genes in Spermatophytes Determined by Molecular Evolution and Arabidopsis Mutant Analysis. Front. Plant Sci. 2016, 7, 1585. [Google Scholar] [CrossRef] [PubMed]
- Mo, Z.H.; Li, F.D.; Su, W.C.; Cao, F.; Peng, F.R.; Li, Y.R. Cloning and expression analysis of CiMYB46 in the graft healing process of Carya illinoinensis. J. Nanjing For. Univ. (Nat. Sci. Ed.) 2019, 43, 156–162. (In Chinese) [Google Scholar]
- Chen, G.; Wang, L.; Li, L.; Li, Y.; Li, H.; Ding, W.; Shi, T.; Yang, X.; Wang, L. Genome-Wide Identification of the Auxin Response Factor (ARF) Gene Family and Their Expression Analysis during Flower Development of Osmanthus fragrans. Forests 2020, 11, 245. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw_Reads | Clean_Reads | Clean_Bases | Valid_Bases (%) | Q30 1 (%) | GC 2 (%) |
|---|---|---|---|---|---|---|
| A_1 | 46,571,826 | 45,283,706 | 6,697,931,982 | 95.88 | 93.64 | 43.53 |
| A_2 | 53,925,378 | 52,724,482 | 7,839,827,405 | 96.92 | 93.96 | 43.81 |
| A_3 | 53,242,134 | 51,808,658 | 7,669,074,710 | 96.03 | 93.55 | 43.83 |
| B_1 | 46,927,200 | 45,482,626 | 6,725,348,970 | 95.54 | 93.11 | 44.00 |
| B_2 | 52,320,204 | 50,588,050 | 7,464,083,436 | 95.11 | 92.91 | 43.84 |
| B_3 | 47,901,258 | 46,465,580 | 6,850,549,711 | 95.34 | 92.33 | 44.10 |
| C_1 | 48,353,956 | 47,494,412 | 7,067,068,800 | 97.44 | 94.45 | 44.15 |
| C_2 | 49,948,000 | 49,090,122 | 7,312,542,131 | 97.60 | 94.33 | 44.56 |
| C_3 | 48,035,404 | 47,186,328 | 7,004,509,885 | 97.21 | 94.30 | 44.44 |
| D_1 | 46,718,046 | 45,434,522 | 6,720,860,268 | 95.91 | 93.01 | 44.09 |
| D_2 | 48,454,586 | 47,792,530 | 7,068,774,362 | 97.26 | 94.85 | 44.03 |
| D_3 | 43,553,002 | 42,426,072 | 6,280,307,490 | 96.13 | 92.99 | 43.71 |
| E_1 | 47,230,764 | 46,004,016 | 6,795,292,538 | 95.92 | 93.20 | 44.04 |
| E_2 | 47,250,800 | 45,758,216 | 6,745,283,335 | 95.17 | 92.51 | 44.04 |
| E_3 | 44,020,258 | 43,396,072 | 6,443,702,336 | 97.59 | 94.89 | 44.03 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, J.; Guo, Z.; Zhang, Y.; Mo, J.; Cui, J.; Hu, H.; He, Y.; Xu, J. Transcriptomic Profiling of Cryptomeria fortunei Hooibrenk Vascular Cambium Identifies Candidate Genes Involved in Phenylpropanoid Metabolism. Forests 2020, 11, 766. https://doi.org/10.3390/f11070766
Yang J, Guo Z, Zhang Y, Mo J, Cui J, Hu H, He Y, Xu J. Transcriptomic Profiling of Cryptomeria fortunei Hooibrenk Vascular Cambium Identifies Candidate Genes Involved in Phenylpropanoid Metabolism. Forests. 2020; 11(7):766. https://doi.org/10.3390/f11070766
Chicago/Turabian StyleYang, Junjie, Zhenhao Guo, Yingting Zhang, Jiaxing Mo, Jiebing Cui, Hailiang Hu, Yunya He, and Jin Xu. 2020. "Transcriptomic Profiling of Cryptomeria fortunei Hooibrenk Vascular Cambium Identifies Candidate Genes Involved in Phenylpropanoid Metabolism" Forests 11, no. 7: 766. https://doi.org/10.3390/f11070766
APA StyleYang, J., Guo, Z., Zhang, Y., Mo, J., Cui, J., Hu, H., He, Y., & Xu, J. (2020). Transcriptomic Profiling of Cryptomeria fortunei Hooibrenk Vascular Cambium Identifies Candidate Genes Involved in Phenylpropanoid Metabolism. Forests, 11(7), 766. https://doi.org/10.3390/f11070766