Comparative Proteomic Analysis of Leaves at Different Ages in Allotriploid Populus


Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Protein Extraction, Digestion, and Desalting
2.3. LC-MS/MS Analysis of Digested Peptides
2.4. Protein Identification
2.5. Analysis of Differentially Accumulated Proteins (DAPs)
2.6. Protein Annotation and Enrichment Analysis
2.7. Protein-Protein Interaction (PPI) Network Construction
2.8. RNA-Seq Data Analysis
3. Results
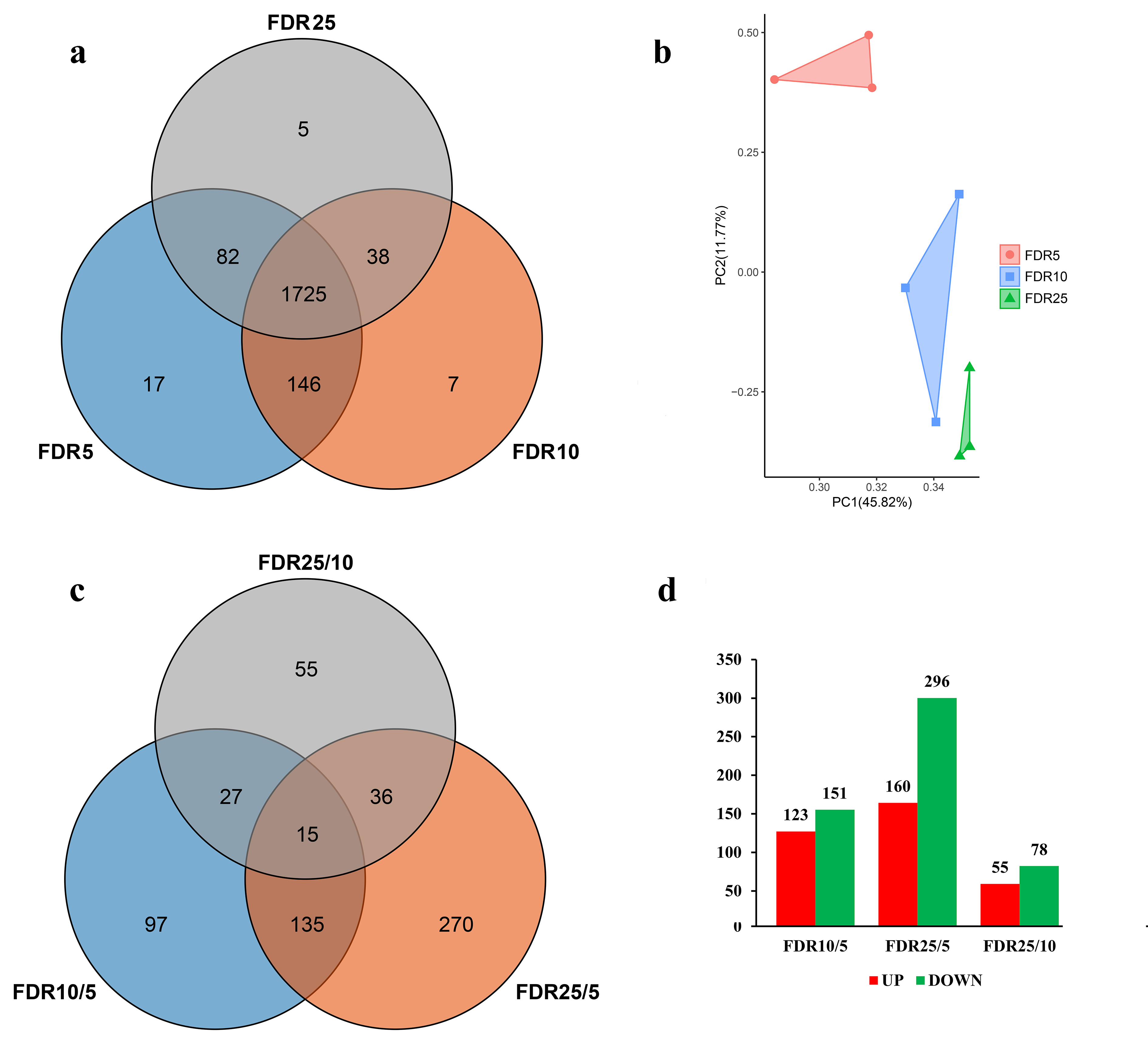
3.1. Overview of Proteome during Leaf Development
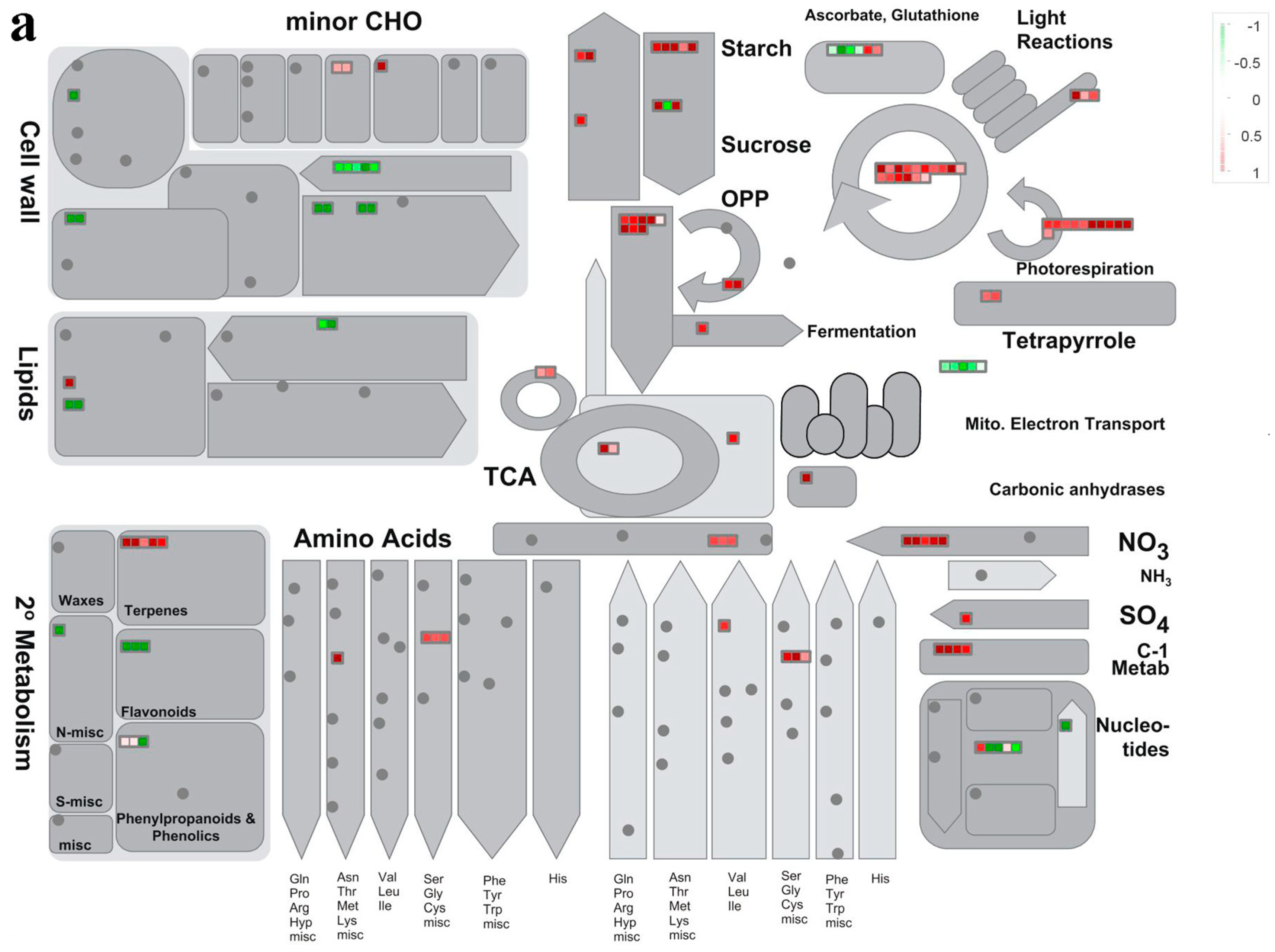
3.2. Cellular Processes Reflected by DAPs
3.3. Accumulation Patterns of DAPs
3.4. KEGG Enrichment Analysis
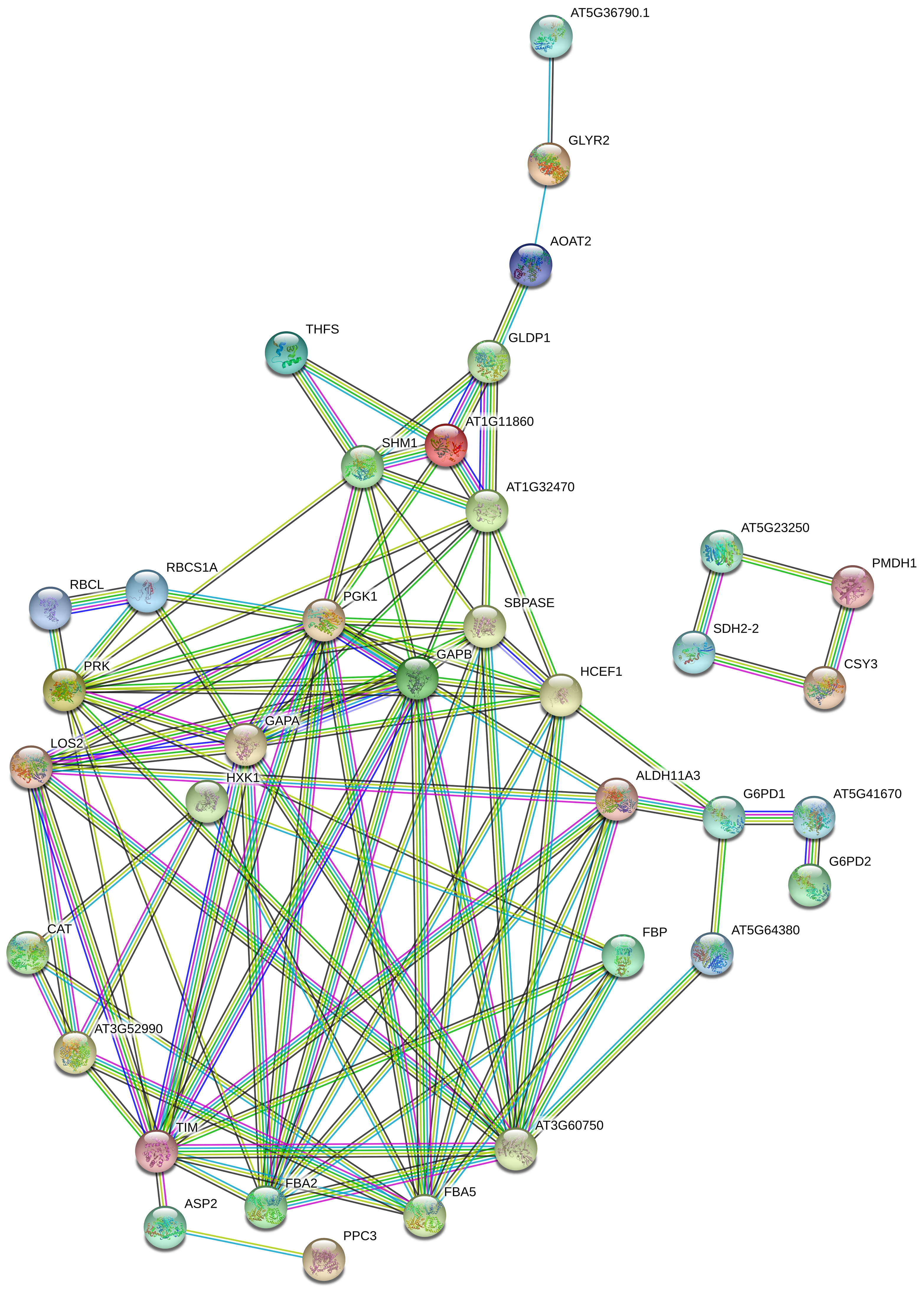
3.5. PPI Network Construction for DAPs Involved in Carbon Metabolism
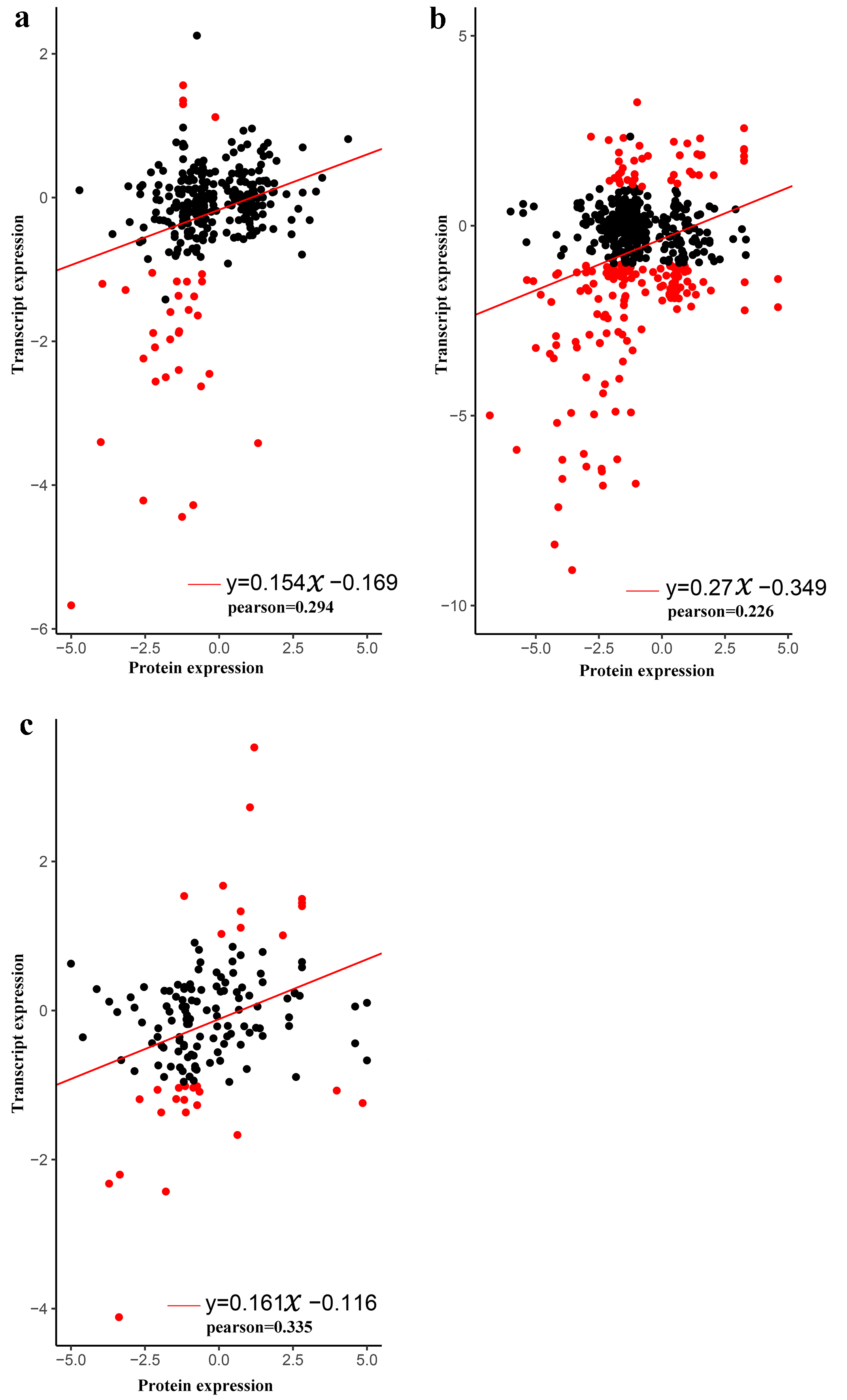
3.6. Transcriptome-Proteome Associated Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Szymanski, J.; Levin, Y.; Savidor, A.; Breitel, D.; Chappell-Maor, L.; Heinig, U.; Topfer, N.; Aharoni, A. Label-free deep shotgun proteomics reveals protein dynamics during tomato fruit tissues development. Plant J. 2017, 90, 396–417. [Google Scholar] [CrossRef]
- Jensen, O.N. Modification-specific proteomics: Characterization of post-translational modifications by mass spectrometry. Curr. Opin. Chem. Biol. 2004, 8, 33–41. [Google Scholar] [CrossRef]
- Kaufmann, K.; Smaczniak, C.; de Vries, S.; Angenent, G.C.; Karlova, R. Proteomics insights into plant signaling and development. Proteomics 2011, 11, 744–755. [Google Scholar] [CrossRef] [PubMed]
- Hossain, Z.; Nouri, M.Z.; Komatsu, S. Plant cell organelle proteomics in response to abiotic stress. J. Proteome Res. 2012, 11, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Pflieger, D.; Bigeard, J.; Hirt, H. Isolation and characterization of plant protein complexes by mass spectrometry. Proteomics 2011, 11, 1824–1833. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Lu, S.; Liu, K.; Wang, S.; Huang, L.; Guo, L. Proteomics: A powerful tool to study plant responses to biotic stress. Plant Methods 2019, 15, 135. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Han, B.; Wang, T.; Chen, S.; Li, H.; Zhang, Y.; Dai, S. Mechanisms of plant salt response: Insights from proteomics. J. Proteome Res. 2012, 11, 49–67. [Google Scholar] [CrossRef] [PubMed]
- Mitprasat, M.; Roytrakul, S.; Jiemsup, S.; Boonseng, O.; Yokthongwattana, K. Leaf proteomic analysis in cassava (Manihot esculenta, Crantz) during plant development, from planting of stem cutting to storage root formation. Planta 2011, 233, 1209–1221. [Google Scholar] [CrossRef]
- Liang, C.; Tian, J.; Liao, H. Proteomics dissection of plant responses to mineral nutrient deficiency. Proteomics 2013, 13, 624–636. [Google Scholar] [CrossRef]
- Soltis, D.E.; Misra, B.B.; Shan, S.; Chen, S.; Soltis, P.S. Polyploidy and the proteome. Biochim. Biophys. Acta 2016, 1864, 896–907. [Google Scholar] [CrossRef]
- Jansson, S.; Douglas, C.J. Populus: A model system for plant biology. Annu. Rev. Plant Biol. 2007, 58, 435–458. [Google Scholar] [CrossRef] [PubMed]
- Plomion, C.; Lalanne, C.; Claverol, S.; Meddour, H.; Kohler, A.; Bogeat-Triboulot, M.B.; Barre, A.; Le Provost, G.; Dumazet, H.; Jacob, D.; et al. Mapping the proteome of poplar and application to the discovery of drought-stress responsive proteins. Proteomics 2006, 6, 6509–6527. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, U.C.; Hurst, G.B.; Lankford, P.K.; Ranjan, P.; Pelletier, D.A. Shotgun proteome profile of Populus developing xylem. Proteomics 2009, 9, 4871–4880. [Google Scholar] [CrossRef] [PubMed]
- Hurst, G.; Yang, X.H.; Tschaplinski, T.; Tuskan, G.; Lankford, P.; Shah, M.; Jawdy, S.; Gunter, L.; Engle, N. Proteomics of Leaf Tissues from Populus. Proteom. 2010, 3, 4. [Google Scholar]
- Liu, J.; Hai, G.; Wang, C.; Cao, S.; Xu, W.; Jia, Z.; Yang, C.; Wang, J.P.; Dai, S.; Cheng, Y. Comparative proteomic analysis of Populus trichocarpa early stem from primary to secondary growth. J. Proteom. 2015, 126, 94–108. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, X.; Zhang, J.; Fan, W.; Lu, M.; Hu, J. Proteomic Analysis and Identification of Possible Allergenic Proteins in Mature Pollen of Populus tomentosa. Int. J. Mol. Sci. 2018, 19, 250. [Google Scholar] [CrossRef]
- Leijon, F.; Melzer, M.; Zhou, Q.; Srivastava, V.; Bulone, V. Proteomic Analysis of Plasmodesmata From Populus Cell Suspension Cultures in Relation With Callose Biosynthesis. Front. Plant Sci. 2018, 9, 1681. [Google Scholar] [CrossRef]
- Bonhomme, L.; Monclus, R.; Vincent, D.; Carpin, S.; Lomenech, A.M.; Plomion, C.; Brignolas, F.; Morabito, D. Leaf proteome analysis of eight Populus x euramericana genotypes: Genetic variation in drought response and in water-use efficiency involves photosynthesis-related proteins. Proteomics 2009, 9, 4121–4142. [Google Scholar] [CrossRef]
- Zhang, Y.; Feng, L.; Jiang, H.; Zhang, Y.; Zhang, S. Different Proteome Profiles between Male and Female Populus cathayana Exposed to UV-B Radiation. Front. Plant Sci. 2017, 8, 320. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Chen, F.; Peng, S.; Ma, W.; Korpelainen, H.; Li, C. Comparative physiological, ultrastructural and proteomic analyses reveal sexual differences in the responses of Populus cathayana under drought stress. Proteomics 2010, 10, 2661–2677. [Google Scholar] [CrossRef]
- Zhang, S.; Feng, L.; Jiang, H.; Ma, W.; Korpelainen, H.; Li, C. Biochemical and proteomic analyses reveal that Populus cathayana males and females have different metabolic activities under chilling stress. J. Proteome Res. 2012, 11, 5815–5826. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Wu, F.; Kang, X. Genetic control of fiber properties and growth in triploid hybrid clones of Populus tomentosa. Scand. J. Forest Res. 2013, 28, 621–630. [Google Scholar] [CrossRef]
- Liao, T.; Cheng, S.; Zhu, X.; Min, Y.; Kang, X. Effects of triploid status on growth, photosynthesis, and leaf area in Populus. Trees 2016, 30, 1137–1147. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Y.; Suo, Y.; Min, Y.; Kang, X. Proteomic Changes Between Populus Allotriploids and Diploids Revealed Using an iTRAQ-based Quantitative Approach. Curr. Proteom. 2017, 14, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Du, K.; Liao, T.; Ren, Y.; Geng, X.; Kang, X. Molecular Mechanism of Vegetative Growth Advantage in Allotriploid Populus. Int. J. Mol. Sci. 2020, 21, 441. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote) omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]
- Usadel, B.; Nagel, A.; Thimm, O.; Redestig, H.; Blaesing, O.E.; Palacios-Rojas, N.; Selbig, J.; Hannemann, J.; Piques, M.C.; Steinhauser, D.; et al. Extension of the visualization tool MapMan to allow statistical analysis of arrays, display of corresponding genes, and comparison with known responses. Plant Physiol. 2005, 138, 1195–1204. [Google Scholar] [CrossRef]
- Wang, K.; Singh, D.; Zeng, Z.; Coleman, S.J.; Huang, Y.; Savich, G.L.; He, X.; Mieczkowski, P.; Grimm, S.A.; Perou, C.M.; et al. MapSplice: Accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res. 2010, 38, e178. [Google Scholar] [CrossRef]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef]
- Shalit, T.; Elinger, D.; Savidor, A.; Gabashvili, A.; Levin, Y. MS1-based label-free proteomics using a quadrupole orbitrap mass spectrometer. J. Proteome Res. 2015, 14, 1979–1986. [Google Scholar] [CrossRef] [PubMed]
- Wilson, K.B.; Hanson, P.J.; Baldocchi, D.D. Spatial and seasonal variability of photosynthetic parameters and their relationship to leaf nitrogen in a deciduous forest. Tree Physiol. 2000, 20, 565–578. [Google Scholar] [CrossRef]
- Hanba, Y.T.; Miyazawa, S.-I.; Kogami, H.; Terashima, I. Effects of leaf age on internal CO2 transfer conductance and photosynthesis in tree species having different types of shoot phenology. Funct. Plant Biol. 2001, 28, 1075. [Google Scholar] [CrossRef]
- Shin-Ichi, M.; Shigenari, S.; Ichiro, T. Slow leaf development of evergreen broad-leaved tree species in Japanese warm temperate forests. Ann. Bot. 1998, 6, 859–869. [Google Scholar]
- Miyazawa, S.I.; Terashima, I. Slow development of leaf photosynthesis in an evergreen broad-leaved tree, castanopsis sieboldii: Relationships between leaf anatomical characteristics and photosynthetic rate. Plant Cell Environ. 2010, 24, 279–291. [Google Scholar] [CrossRef]
- Niinemets, Ü.; García-Plazaola, J.I.; Tosens, T.; Flexas, J.; Loreto, F.; Medrano, H. Photosynthesis during leaf development and ageing. In Terrestrial Photosynthesis in a Changing Environment; Cambridge University Press: Cambridge, UK, 2012; pp. 353–372. [Google Scholar]
- Tosens, T.; Niinemets, U.; Vislap, V.; Eichelmann, H.; Castro Diez, P. Developmental changes in mesophyll diffusion conductance and photosynthetic capacity under different light and water availabilities in Populus tremula: How structure constrains function. Plant Cell Environ. 2012, 35, 839–856. [Google Scholar] [CrossRef]
- Lim, P.O.; Kim, H.J.; GilNam, H. Leaf senescence. Annu. Rev. Plant Biol. 2007, 58, 115–136. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, Y.; Zou, D.; Zhao, Y.; Wang, Y.; Zhang, Y.; Xia, X.; Luo, J.; Guo, H.; Zhang, Z. LSD 3.0: A comprehensive resource for the leaf senescence research community. Nucleic Acids Res. 2020, 48, D1069–D1075. [Google Scholar] [CrossRef]
- Cho, M.-H.; Park, H.L.; Hahn, T.-R. Engineering leaf carbon metabolism to improve plant productivity. Plant Biotechnol. Rep. 2014, 9, 1–10. [Google Scholar] [CrossRef]
- Ebenhoh, O.; Spelberg, S. The importance of the photosynthetic Gibbs effect in the elucidation of the Calvin-Benson-Bassham cycle. Biochem. Soc. Trans. 2018, 46, 131–140. [Google Scholar] [CrossRef]
- Hagemann, M.; Bauwe, H. Photorespiration and the potential to improve photosynthesis. Curr. Opin. Chem. Biol. 2016, 35, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Fernie, A.R. On the role of the tricarboxylic acid cycle in plant productivity. J. Integr. Plant Biol. 2018, 60, 1199–1216. [Google Scholar] [CrossRef] [PubMed]
- Fernie, A.R.; Carrari, F.; Sweetlove, L.J. Respiratory metabolism: Glycolysis, the TCA cycle and mitochondrial electron transport. Curr. Opin. Plant Biol. 2004, 7, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Andriotis, V.M.E.; Smith, A.M. The plastidial pentose phosphate pathway is essential for postglobular embryo development in Arabidopsis. Proc. Natl Acad. Sci. USA 2019, 116, 15297–15306. [Google Scholar] [CrossRef] [PubMed]
- Khare, S.; Singh, N.B.; Singh, A.; Hussain, I.; Amist, N. Plant secondary metabolites synthesis and their regulations under biotic and abiotic constraints. J. Plant Biol. 2020, 63, 203–216. [Google Scholar] [CrossRef]
- Perchlik, M.; Tegeder, M. Leaf amino acid supply affects photosynthetic and plant nitrogen use efficiency under nitrogen stress. Plant Physiol. 2018, 178, 174–188. [Google Scholar] [CrossRef]
- Mary, C.W. Fatty acids in chloroplasts and leaves. J. Exp. Bot. 1958, 9, 254–261. [Google Scholar]
- Yang, Z.; Ohlrogge, J.B. Turnover of fatty acids during natural senescence of Arabidopsis, brachypodium, and switchgrass and in Arabidopsis β-oxidation mutants. Plant Physiol. 2009, 150, 1981–1989. [Google Scholar] [CrossRef]
- Griffin, T.J.; Gygi, S.P.; Ideker, T.; Rist, B.; Eng, J.; Hood, L.; Aebersold, R. Complementary profiling of gene expression at the transcriptome and proteome levels in Saccharomyces cerevisiae. Mol. Cell Proteom. 2002, 1, 323–333. [Google Scholar] [CrossRef]
- Lan, P.; Li, W.; Schmidt, W. Complementary proteome and transcriptome profiling in phosphate-deficient Arabidopsis roots reveals multiple levels of gene regulation. Mol. Cell Proteom. 2012, 11, 1156–1166. [Google Scholar] [CrossRef] [PubMed]
- Gygi, S.P.; Rochon, Y.; Franza, B.R.; Aebersold, R. Correlation between protein and mRNA abundance in yeast. Mol. Cell Biol. 1999, 19, 1720–1730. [Google Scholar] [CrossRef] [PubMed]
- Hajduch, M.; Hearne, L.B.; Miernyk, J.A.; Casteel, J.E.; Joshi, T.; Agrawal, G.K.; Song, Z.; Zhou, M.; Xu, D.; Thelen, J.J. Systems analysis of seed filling in Arabidopsis: Using general linear modeling to assess concordance of transcript and protein expression. Plant Physiol. 2010, 152, 2078–2087. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway ID | Pathway Description | False Discovery Rate | ||
|---|---|---|---|---|
| FDR10/5 | FDR25/5 | FDR25/5 | ||
| pop01200 | Carbon metabolism | 3.04 × 10−28 | 4.63 × 10−36 | 2.97 × 10−5 |
| pop00630 | Glyoxylate and dicarboxylate metabolism | 2.93 × 10−26 | 4 × 10−22 | 6.62 × 10−5 |
| pop01100 | Metabolic pathways | 2.93 × 10−26 | 4.2 × 10−42 | 2.58 × 10−16 |
| pop01110 | Biosynthesis of secondary metabolites | 4.45 × 10−20 | 2.52 × 10−24 | 0.000001 |
| pop00710 | Carbon fixation in photosynthetic organisms | 1.24 × 10−18 | 1.96 × 10−21 | |
| pop00010 | Glycolysis/Gluconeogenesis | 1.58 × 10−11 | 6.89 × 10−11 | |
| pop00260 | Glycine, serine and threonine metabolism | 2.4 × 10−11 | 7.06 × 10−6 | |
| pop03010 | Ribosome | 2.09 × 10−10 | 4.07 × 10−87 | |
| pop00030 | Pentose phosphate pathway | 1.54 × 10−7 | 1.22 × 10−9 | |
| pop01230 | Biosynthesis of amino acids | 4.01 × 107 | 1.57 × 10−12 | 0.00037 |
| pop00500 | Starch and sucrose metabolism | 5.42 × 10−7 | 1.72 × 10−7 | |
| pop00520 | Amino sugar and nucleotide sugar metabolism | 1.61 × 10−6 | 0.00061 | |
| pop00051 | Fructose and mannose metabolism | 2.84 × 10−6 | 0.00012 | |
| pop00910 | Nitrogen metabolism | 2.15 × 10−5 | 4.37 × 10−5 | 0.0064 |
| pop00620 | Pyruvate metabolism | 2.99 × 10−5 | 7.06 × 10−6 | |
| pop00250 | Alanine, aspartate and glutamate metabolism | 7.57 × 10−5 | 0.00018 | 0.012 |
| pop00052 | Galactose metabolism | 0.00021 | 0.00043 | |
| pop00220 | Arginine biosynthesis | 0.00027 | 0.00033 | 0.00048 |
| pop00670 | One carbon pool by folate | 0.00029 | 0.00019 | |
| pop00230 | Purine metabolism | 0.00053 | 0.028 | |
| pop00480 | Glutathione metabolism | 0.00064 | 0.00062 | |
| pop00460 | Cyanoamino acid metabolism | 0.00088 | 0.0013 | |
| pop04146 | Peroxisome | 0.0021 | 0.042 | |
| pop03050 | Proteasome | 0.0033 | 0.00026 | |
| pop00280 | Valine, leucine and isoleucine degradation | 0.0071 | 0.0292 | |
| pop00020 | Citrate cycle (TCA cycle) | 0.0104 | 1.13 × 10−5 | 0.0161 |
| pop01040 | Biosynthesis of unsaturated fatty acids | 0.0108 | 0.0056 | |
| pop00240 | Pyrimidine metabolism | 0.0151 | ||
| pop00062 | Fatty acid elongation | 0.0187 | ||
| pop00190 | Oxidative phosphorylation | 0.0187 | 3.12 × 10−7 | 0.0023 |
| pop01212 | Fatty acid metabolism | 0.0187 | 0.00026 | 0.0206 |
| pop04144 | Endocytosis | 0.0202 | 0.00081 | |
| pop00053 | Ascorbate and aldarate metabolism | 0.025 | ||
| pop00380 | Tryptophan metabolism | 0.0276 | ||
| pop00780 | Biotin metabolism | 0.00025 | ||
| pop01210 | 2-Oxocarboxylic acid metabolism | 0.00036 | 0.0161 | |
| pop00195 | Photosynthesis | 0.0025 | ||
| pop00071 | Fatty acid degradation | 0.0035 | ||
| pop00061 | Fatty acid biosynthesis | 0.0041 | ||
| pop00970 | Aminoacyl-tRNA biosynthesis | 0.0077 | 0.00013 | |
| pop00040 | Pentose and glucuronate interconversions | 0.009 | ||
| pop00450 | Selenocompound metabolism | 0.0101 | 0.00096 | |
| pop00270 | Cysteine and methionine metabolism | 0.0109 | 0.0152 | |
| pop00350 | Tyrosine metabolism | 0.0121 | ||
| pop00130 | Ubiquinone and other terpenoid-quinone biosynthesis | 0.0157 | ||
| pop00660 | C5-Branched dibasic acid metabolism | 0.0175 | ||
| pop00730 | Thiamine metabolism | 0.0239 | ||
| pop00400 | Phenylalanine, tyrosine and tryptophan biosynthesis | 0.034 | ||
| pop04145 | Phagosome | 0.0368 | ||
| pop00740 | Riboflavin metabolism | 0.0161 | ||
| pop00940 | Phenylpropanoid biosynthesis | 0.0161 | ||
| pop04070 | Phosphatidylinositol signaling system | 0.0161 | ||
| pop00562 | Inositol phosphate metabolism | 0.0167 | ||
| pop00591 | Linoleic acid metabolism | 0.0206 | ||
| pop00920 | Sulfur metabolism | 0.0444 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Wang, Y.; Wei, H.; Kang, X. Comparative Proteomic Analysis of Leaves at Different Ages in Allotriploid Populus. Forests 2020, 11, 1154. https://doi.org/10.3390/f11111154
Li J, Wang Y, Wei H, Kang X. Comparative Proteomic Analysis of Leaves at Different Ages in Allotriploid Populus. Forests. 2020; 11(11):1154. https://doi.org/10.3390/f11111154
Chicago/Turabian StyleLi, Jiang, Yi Wang, Hairong Wei, and Xiangyang Kang. 2020. "Comparative Proteomic Analysis of Leaves at Different Ages in Allotriploid Populus" Forests 11, no. 11: 1154. https://doi.org/10.3390/f11111154
APA StyleLi, J., Wang, Y., Wei, H., & Kang, X. (2020). Comparative Proteomic Analysis of Leaves at Different Ages in Allotriploid Populus. Forests, 11(11), 1154. https://doi.org/10.3390/f11111154