Marker-Trait Associations for Tolerance to Ash Dieback in Common Ash (Fraxinus excelsior L.)

 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials and Phenotyping
2.2. DNA Extraction
2.3. Selection of the Gene Models and Primer Design
2.4. Multiplex PCR and Library Preparation
2.5. Bioinformatic Analysis, Demultiplexing and Variant Calling
2.6. Population Structure Analysis
2.7. Association Analysis
2.8. Prediction of Functional Effect of Non-Synonymous SNP and Secondary Structure
3. Results
3.1. Generation of SNP Markers
3.2. Low Levels of Differentiation between Material Selected for Disease Tolerance Phenotype and Susceptible Wild Population
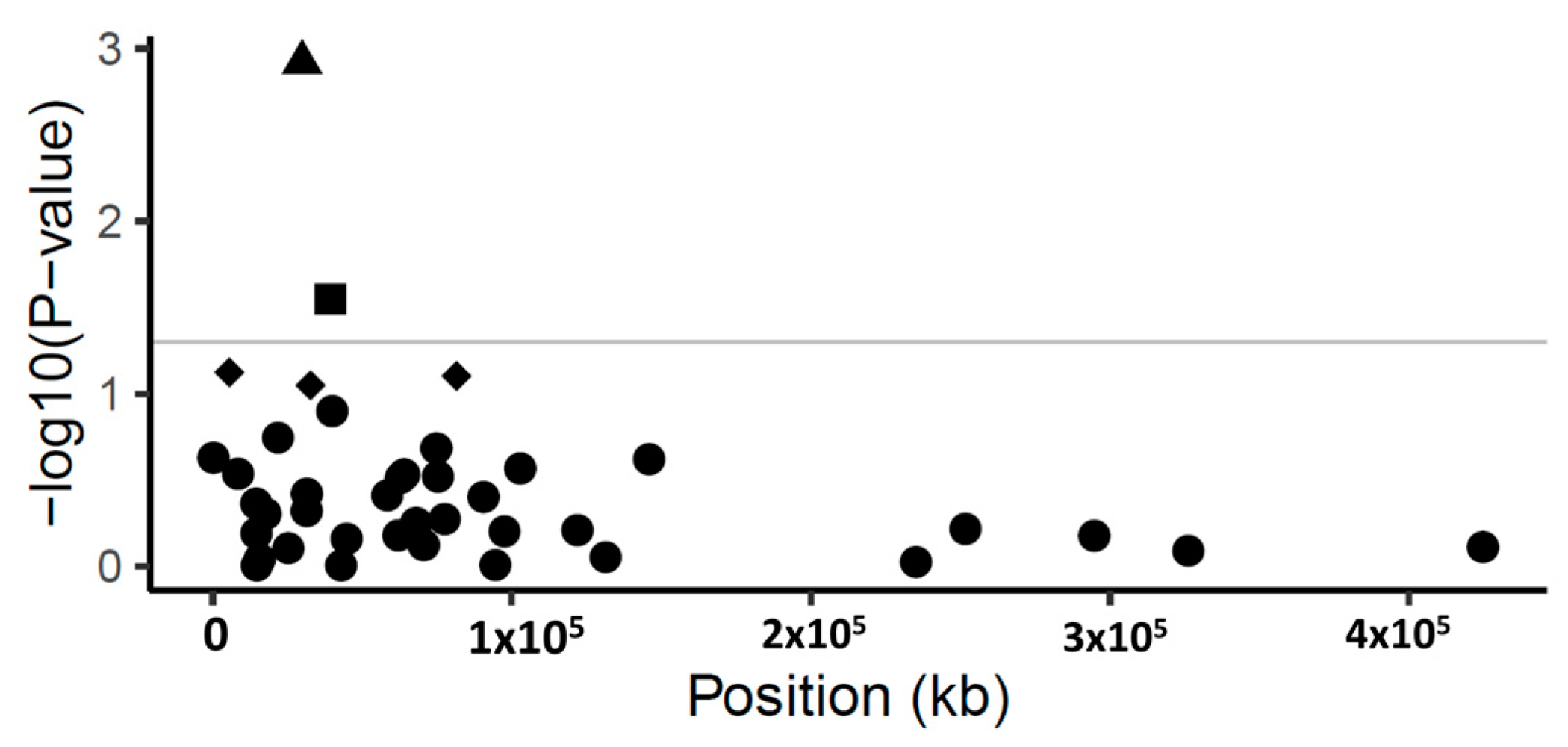
3.3. Marker-Traits Association Identifies Two Scaffolds Associated with the Tolerance Phenotype
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- FRAXIGEN. Ash species in Europe: Biological characteristics and practical guidelines for sustainable use. Folia Oecologica 2005, 33, 137. [Google Scholar]
- Baral, H.O.; Queloz, V.; Hosoya, T. Hymenoscyphus fraxineus, the correct scientific name for the fungus causing ash dieback in Europe. IMA Fungus 2014, 5, 79–80. [Google Scholar] [CrossRef] [PubMed]
- Enderle, R.; Stenlid, J.; Vasaitis, R. An overview of ash (Fraxinus spp.) and the ash dieback disease in Europe. CAB Rev. 2019, 14, 1–12. [Google Scholar] [CrossRef]
- Pautasso, M.; Aas, G.; Queloz, V.; Holdenrieder, O. European ash (Fraxinus excelsior) dieback–A conservation biology challenge. Biol. Conserv. 2013, 158, 37–49. [Google Scholar] [CrossRef]
- Coker, T.L.; Rozsypálek, J.; Edwards, A.; Harwood, T.P.; Butfoy, L.; Buggs, R.J. Estimating mortality rates of European ash (Fraxinus excelsior) under the ash dieback (Hymenoscyphus fraxineus) epidemic. Plants People Planet 2019, 1, 48–58. [Google Scholar] [CrossRef]
- Pautasso, M.; Schlegel, M.; Holdenrieder, O. Forest Health in a Changing World. Microb. Ecol. 2015, 69, 826–842. [Google Scholar] [CrossRef]
- Barklund, P. Askdöd grasserar över Syd-och Mellansverige. SkogsEko 2005, 3, 11–13. [Google Scholar]
- Cleary, M.; Nguyen, D.; Stener, L.; Stenlid, J.; Skovsgaard, J. Ash and ash dieback in Sweden: A review of disease history, current status, pathogen and host dynamics, host tolerance and management options in forests and landscapes. Dieback Eur. Ash (Fraxinus spp.) Conseq. Guidel. Sustain. Manag. 2017, 195–208. [Google Scholar]
- Stenlid, J.; Oliva, J.; Boberg, J.B.; Hopkins, A.J. Emerging diseases in European forest ecosystems and responses in society. Forests 2011, 2, 486–504. [Google Scholar] [CrossRef]
- Hultberg, T.; Sandström, J.; Felton, A.; Öhman, K.; Rönnberg, J.; Witzell, J.; Cleary, M. Ash dieback risks an extinction cascade. Biol. Conserv. 2020, 244, 108516. [Google Scholar] [CrossRef]
- Jönsson, M.T.; Thor, G. Estimating coextinction risks from epidemic tree death: Affiliate lichen communities among diseased host tree populations of Fraxinus excelsior. PLoS ONE 2012, 7, e45701. [Google Scholar] [CrossRef] [PubMed]
- Pliura, A.; Bakys, R.; Suchockas, V.; Marčiulyniene, D.; Gustiene, V.; Verbyla, V.; Lygis, V. Ash dieback in Lithuania: Disease history, research on impact and genetic variation in disease resistance, tree breeding and options for forest management. Dieback Eur. Ash (Fraxinus spp.) Conseq. Guidel. Sustain. Manag. 2017, 150–165. [Google Scholar]
- Kjær, E.D.; McKinney, L.V.; Nielsen, L.R.; Hansen, L.N.; Hansen, J.K. Adaptive potential of ash (Fraxinus excelsior) populations against the novel emerging pathogen Hymenoscyphus pseudoalbidus. Evol. Appl. 2012, 5, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Lobo, A.; Hansen, J.K.; McKinney, L.V.; Nielsen, L.R.; Kjær, E.D. Genetic variation in dieback resistance: Growth and survival of Fraxinus excelsior under the influence of Hymenoscyphus pseudoalbidus. Scand. J. Forest Res. 2014, 29, 519–526. [Google Scholar] [CrossRef]
- Harper, A.L.; McKinney, L.V.; Nielsen, L.R.; Havlickova, L.; Li, Y.; Trick, M.; Fraser, F.; Wang, L.; Fellgett, A.; Sollars, E.S.; et al. Molecular markers for tolerance of European ash (Fraxinus excelsior) to dieback disease identified using Associative Transcriptomics. Sci. Rep. 2016, 6, 19335. [Google Scholar] [CrossRef]
- McKinney, L.V.; Nielsen, L.R.; Hansen, J.K.; Kjær, E.D. Presence of natural genetic resistance in Fraxinus excelsior (Oleraceae) to Chalara fraxinea (Ascomycota): An emerging infectious disease. Heredity 2011, 106, 788. [Google Scholar] [CrossRef]
- Stener, L.G. Clonal differences in susceptibility to the dieback of Fraxinus excelsior in southern Sweden. Scand. J. For. Res. 2013, 28, 205–216. [Google Scholar] [CrossRef]
- Kjær, E.D.; McKinney, L.V.; Hansen, L.N.; Olrik, D.C.; Lobo, A.; Thomsen, I.M.; Hansen, J.K.; Nielsen, L.R. Genetics of ash dieback resistance in a restoration context–experiences from Denmark. Dieback Eur. Ash 2017, 106–114. [Google Scholar]
- Lobo, A.; McKinney, L.V.; Hansen, J.K.; Kjær, E.D.; Nielsen, L.R. Genetic variation in dieback resistance in Fraxinus excelsior confirmed by progeny inoculation assay. For. Pathol. 2015, 45, 379–387. [Google Scholar] [CrossRef]
- McKinney, L.V.; Thomsen, I.M.; Kjær, E.D.; Nielsen, L.R. Genetic resistance to Hymenoscyphus pseudoalbidus limits fungal growth and symptom occurrence in Fraxinus excelsior. For. Pathol. 2012, 42, 69–74. [Google Scholar] [CrossRef]
- McKinney, L.V.; Nielsen, L.R.; Collinge, D.B.; Thomsen, I.M.; Hansen, J.K.; Kjær, E.D. The ash dieback crisis: Genetic variation in resistance can prove a long-term solution. Plant Pathol. 2014, 63, 485–499. [Google Scholar] [CrossRef]
- Sollars, E.S.; Harper, A.L.; Kelly, L.J.; Sambles, C.M.; Ramirez-Gonzalez, R.H.; Swarbreck, D.; Kaithakottil, G.; Cooper, E.D.; Uauy, C.; Havlickova, L.; et al. Genome sequence and genetic diversity of European ash trees. Nature 2017, 541, 212. [Google Scholar] [CrossRef] [PubMed]
- Pliura, A.; Lygis, V.; Suchockas, V.; Bartkevicius, E. Performance of twenty four European Fraxinus excelsior populations in three Lithuanian progeny trials with a special emphasis on resistance to Chalara fraxinea. Balt. For. 2011, 17, 17–34. [Google Scholar]
- Neale, D.B.; Savolainen, O. Association genetics of complex traits in conifers. Trends Plant Sci. 2004, 9, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Menkis, A.; Bakys, R.; Stein Åslund, M.; Davydenko, K.; Elfstrand, M.; Stenlid, J.; Vasaitis, R. Identifying Fraxinus excelsior tolerant to ash dieback: Visual field monitoring versus a molecular marker. For. Pathol. 2019, e12572. [Google Scholar] [CrossRef]
- Stocks, J.J.; Metheringham, C.L.; Plumb, W.J.; Lee, S.J.; Kelly, L.J.; Nichols, R.A.; Buggs, R.J.A. Genomic basis of European ash tree resistance to ash dieback fungus. Nat. Ecol. Evol. 2019, 3, 1686–1696. [Google Scholar] [CrossRef]
- Baird, N.A.; Etter, P.D.; Atwood, T.S.; Currey, M.C.; Shiver, A.L.; Lewis, Z.A.; Selker, E.U.; Cresko, W.A.; Johnson, E.A. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS ONE 2008, 3, e3376. [Google Scholar] [CrossRef]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Dumont, T.; Pope, B.J.; Hammet, F.; Southey, M.C.; Park, D.J. A high-plex PCR approach for massively parallel sequencing. Biotechniques 2013, 55, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Marchese, A.; Marra, F.P.; Caruso, T.; Mhelembe, K.; Costa, F.; Fretto, S.; Sargent, D.J. The first high-density sequence characterized SNP-based linkage map of olive (‘Olea europaea’L. subsp. ‘europaea’) developed using genotyping by sequencing. Aust. J. Crop Sci. 2016, 10, 857. [Google Scholar] [CrossRef]
- Wu, D.; Koch, J.; Coggeshall, M.; Carlson, J. The first genetic linkage map for Fraxinus pennsylvanica and syntenic relationships with four related species. Plant Mol. Biol. 2019, 99, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Zhigunov, A.V.; Ulianich, P.S.; Lebedeva, M.V.; Chang, P.L.; Nuzhdin, S.V.; Potokina, E.K. Development of F1 hybrid population and the high-density linkage map for European aspen (Populus tremula L.) using RADseq technology. BMC Plant Biol. 2017, 17, 180. [Google Scholar] [CrossRef] [PubMed]
- Grattapaglia, D.; de Alencar, S.; Pappas, G. Genome-wide genotyping and SNP discovery by ultra-deep Restriction-Associated DNA (RAD) tag sequencing of pooled samples of E. grandis and E. globulus. BMC Proc. BioMed Cent. 2011, 5, 45. [Google Scholar] [CrossRef]
- Konar, A.; Choudhury, O.; Bullis, R.; Fiedler, L.; Kruser, J.M.; Stephens, M.T.; Gailing, O.; Schlarbaum, S.; Coggeshall, M.V.; Staton, M.E.; et al. High-quality genetic mapping with ddRADseq in the non-model tree Quercus rubra. BMC Genom. 2017, 18, 417. [Google Scholar] [CrossRef] [PubMed]
- Kirisits, T.; Freinschlag, C. Ash dieback caused by Hymenoscyphus pseudoalbidus in a seed plantation of Fraxinus excelsior in Austria. J. Agric. Ext. Rural. Dev. 2012, 4, 184–191. [Google Scholar] [CrossRef]
- Chang, S.; Puryear, J.; Cairney, J. A simple and efficient method for isolating RNA from pine trees. Plant Mol. Biol. Report. 1993, 11, 113–116. [Google Scholar] [CrossRef]
- You, F.M.; Huo, N.; Gu, Y.Q.; Luo, M.-C.; Ma, Y.; Hane, D.; Lazo, G.R.; Dvorak, J.; Anderson, O.D. BatchPrimer3: A high throughput web application for PCR and sequencing primer design. BMC Bioinform. 2008, 9, 253. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Li, H.; Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Eprint Arxiv 2013, arXiv:1303.13033997. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [PubMed]
- Earl, D.A. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. In Nucleic Acids Symposium Series; Information Retrieval Ltd.: London, UK, 1999; pp. 95–98. [Google Scholar]
- Marchler-Bauer, A.; Derbyshire, M.K.; Gonzales, N.R.; Lu, S.; Chitsaz, F.; Geer, L.Y.; Geer, R.C.; He, J.; Gwadz, M.; Hurwitz, D.I.; et al. CDD: NCBI’s conserved domain database. Nucleic Acids Res. 2014, 43, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef]
- Buchan, D.W.; Jones, D.T. The PSIPRED protein analysis workbench: 20 years on. Nucleic Acids Res. 2019, 47, 402–407. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, Y. I-TASSER server: New development for protein structure and function predictions. Nucleic Acids Res. 2015, 43, 174–181. [Google Scholar] [CrossRef]
- Sniezko, R.A.; Koch, J. Breeding trees resistant to insects and diseases: Putting theory into application. Biol. Invasions 2017, 19, 3377–3400. [Google Scholar] [CrossRef]
- Namkoong, G. Introduction to Quantitative Genetics in Forestry; United States Forest Service: Washington, DC, USA, 1979. [Google Scholar]
- Hall, D.; Hallingbäck, H.R.; Wu, H.X. Estimation of number and size of QTL effects in forest tree traits. Tree Genet. Genomes 2016, 12, 110. [Google Scholar] [CrossRef]
- Nemesio-Gorriz, M.; Hammerbacher, A.; Ihrmark, K.; Källman, T.; Olson, Å.; Lascoux, M.; Stenlid, J.; Gershenzon, J.; Elfstrand, M. Different alleles of a gene encoding leucoanthocyanidin reductase (PaLAR3) influence resistance against the fungus Heterobasidion parviporum in Picea abies. Plant Physiol. 2016, 171, 2671–2681. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Deng, D.; Wang, Z.; Duan, C.; Wu, X.; Wang, X.; Zong, X.; Zhu, Z. A novel er1 allele and the development and validation of its functional marker for breeding pea (Pisum sativum L.) resistance to powdery mildew. Theor. Appl. Genet. 2016, 129, 909–919. [Google Scholar] [CrossRef] [PubMed]
- Antão, C.M.; Malcata, F.X. Plant serine proteases: Biochemical, physiological and molecular features. Plant Physiol. Biochem. 2005, 43, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, A.; Monteiro, F.; Sebastiana, M. Subtilisin-like proteases in plant–pathogen recognition and immune priming: A perspective. Front. Plant Sci. 2014, 5, 739. [Google Scholar] [CrossRef] [PubMed]
- Laplaze, L.; Ribeiro, A.; Franche, C.; Duhoux, E.; Auguy, F.; Bogusz, D.; Pawlowski, K. Characterization of a Casuarina glauca nodule-specific subtilisin-like protease gene, a homolog of Alnus glutinosa ag12. Mol. Plant Microbe Interact. 2000, 13, 113–117. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tornero, P.; Conejero, V.; Vera, P. Identification of a new pathogen-induced member of the subtilisin-like processing protease family from plants. J. Biol. Chem. 1997, 272, 14412–14419. [Google Scholar] [CrossRef]
- Duan, X.; Zhang, Z.; Wang, J.; Zuo, K. Characterization of a novel cotton subtilase gene GbSBT1 in response to extracellular stimulations and its role in Verticillium resistance. PLoS ONE 2016, 11, e0153988. [Google Scholar] [CrossRef]
- Semizer-Cuming, D.; Finkeldey, R.; Nielsen, L.R.; Kjær, E.D. Negative correlation between ash dieback susceptibility and reproductive success: Good news for European ash forests. Ann. For. Sci. 2019, 76, 16. [Google Scholar] [CrossRef]
- Aitken, S.N.; Yeaman, S.; Holliday, J.A.; Wang, T.; Curtis-McLane, S. Adaptation, migration or extirpation: Climate change outcomes for tree populations. Evol. Appl. 2008, 1, 95–111. [Google Scholar] [CrossRef]
- Namkoong, G. Maintaining genetic diversity in breeding for resistance in forest trees. Annu. Rev. Phytopathol. 1991, 29, 325–342. [Google Scholar] [CrossRef]


{kind=link}
| Type | Pairwise FST |
|---|---|
| Tolerant vs. Susceptible genotypes | 0.0220 |
| Selected for tolerance vs. other materials | 0.0217 |
| Selected for tolerance vs. Uppland | 0.0228 |
| Uppland vs. Öland | 0.0187 |
| Marker a | Scaffold b | Variant c | Gene Model d | Gene Model CDS Length (Bp) | Position e | p-Value | FDR Adj p-Value f | PVE% g | SNP Feature h | Annotation |
|---|---|---|---|---|---|---|---|---|---|---|
| SCONTIG5992_29927 | Scaffold 5992 | A/G | FRAEX38873_v2_000299890.1 | 2349 | 29,927 | 0.001 | 0.048 | 5.4 | non-synonymous | Peptidase S8, subtilisin-related|Peptidase S8/S53 domain |
| SCONTIG6368_39377 | Scaffold 6368 | C/G | FRAEX38873_v2_000311990.1 | 2601 | 39,377 | 0.028 | 0.568 | 3.0 | non-synonymous | Leucine-rich repeat |
| SCONTIG2549_5550 | Scaffold 2549 | T/A | FRAEX38873_v2_000132480.1 | 2571 | 5550 | 0.075 | 1.000 | 2.2 | synonymous | Pentatricopeptide repeat |
| SCONTIG8553_81512 | Scaffold 8553 | A/G | FRAEX38873_v2_000368890.1 | 3237 | 81,512 | 0.079 | 0.790 | 2.1 | non-synonymous | unknown |
| SCONTIG874_32729 | Scaffold 874 | A/C | FRAEX38873_v2_000373260.1 | 1464 | 32,729 | 0.089 | 0.716 | 2.1 | synonymous | cytochrome p450 94a1-like |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chaudhary, R.; Rönneburg, T.; Stein Åslund, M.; Lundén, K.; Durling, M.B.; Ihrmark, K.; Menkis, A.; Stener, L.-G.; Elfstrand, M.; Cleary, M.; et al. Marker-Trait Associations for Tolerance to Ash Dieback in Common Ash (Fraxinus excelsior L.). Forests 2020, 11, 1083. https://doi.org/10.3390/f11101083
Chaudhary R, Rönneburg T, Stein Åslund M, Lundén K, Durling MB, Ihrmark K, Menkis A, Stener L-G, Elfstrand M, Cleary M, et al. Marker-Trait Associations for Tolerance to Ash Dieback in Common Ash (Fraxinus excelsior L.). Forests. 2020; 11(10):1083. https://doi.org/10.3390/f11101083
Chicago/Turabian StyleChaudhary, Rajiv, Tilman Rönneburg, Matilda Stein Åslund, Karl Lundén, Mikael Brandström Durling, Katarina Ihrmark, Audrius Menkis, Lars-Göran Stener, Malin Elfstrand, Michelle Cleary, and et al. 2020. "Marker-Trait Associations for Tolerance to Ash Dieback in Common Ash (Fraxinus excelsior L.)" Forests 11, no. 10: 1083. https://doi.org/10.3390/f11101083
APA StyleChaudhary, R., Rönneburg, T., Stein Åslund, M., Lundén, K., Durling, M. B., Ihrmark, K., Menkis, A., Stener, L.-G., Elfstrand, M., Cleary, M., & Stenlid, J. (2020). Marker-Trait Associations for Tolerance to Ash Dieback in Common Ash (Fraxinus excelsior L.). Forests, 11(10), 1083. https://doi.org/10.3390/f11101083