1. Introduction
Chinese elm (
Ulmus parvifolia), which is native to China, Japan and Korea, has become a widely distributed ornamental tree that is frequently planted on lawns, along streets and in parks [
1]. In China, the wild resources of
U. parvifolia are mainly located in the northern and eastern areas, exhibiting a wide range of adaptation. Within this area, Chinese elm is recognized as a drought, heat, and cold tolerant tree [
2,
3,
4]. Nevertheless, as global climate alteration will happen in the near future, it remains questionable to what degree the speed of future adaptation can keep up with the pace of climate change [
5]. Therefore, an in-depth understanding of the genetic diversity and the genetic regulation of adaptation in Chinese elms is essential. Revealing polymorphisms and genes that determine adaptation would provide the basis for breeding genetically improved germplasms that could be used in changing environments.
Genetic diversity is the maximum of genetic variation presented in the genetic makeup of a specific species [
6]. It is an important component of species biodiversity. Monitoring the genetic diversity of natural populations is of paramount importance, since it could shed light on the population structure, history, ecology, and adaptation of the species [
7]. Local adaptation occurs gradually over time, with relatively long generation times. During the adaptation process, alleles that are best fitted to the specific climate gradually prevail through positive selection [
8]. Those alleles, once identified, can give new insights into plant adaptive evolution, as well as be utilized for future molecular breeding.
Previous research on genetic diversity and local adaptation of plants has been conducted at the DNA-based molecular level, such as simple sequence repeat (SSR), inter-simple sequence repeat (ISSR), random amplified polymorphic DNA (RAPD), amplified fragment length polymorphism (AFLP), and single nucleotide polymorphism (SNP) [
7,
9,
10]. SNPs are genome sequence variations that occur when there is a single nucleotide change in the DNA sequence [
11]. SNPs are the most abundant and stable type of DNA variation in a genome, therefore, the density of SNP markers is much higher than any other molecular markers [
3]. Nowadays, reduced representation sequencing, such as genotyping-by-sequencing (GBS) and specific locus amplified fragment sequencing (SLAF-seq), has been used to quickly and efficiently identify numerous SNPs in plants [
12,
13]. As reduced representation sequencing can be performed without a reference genome, it has been tested on many kinds of trees, such as pecans [
14], Japanese conifers [
10], and
masson pine [
15].
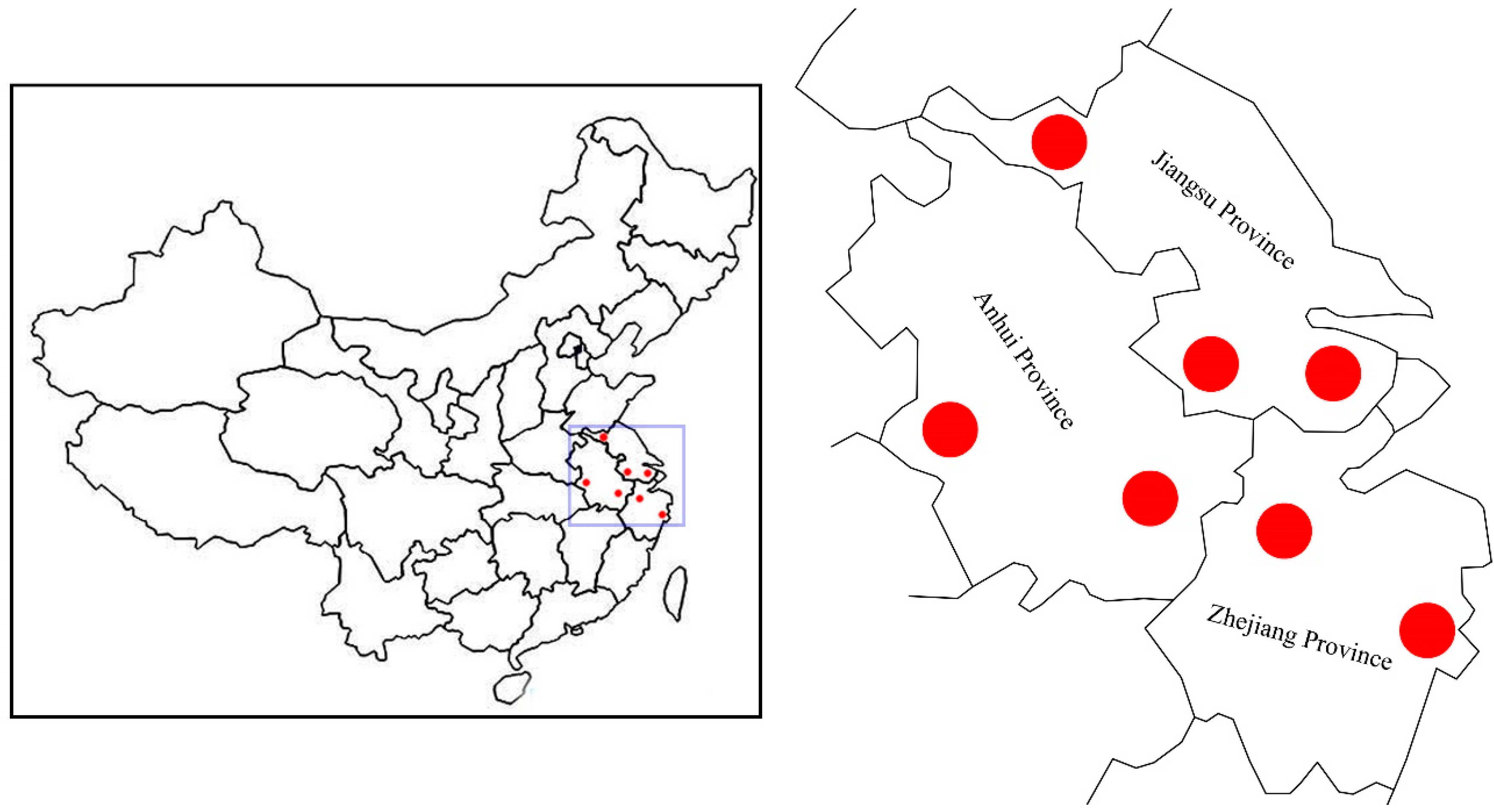
To date, reports regarding the genetic diversity and adaptive mechanisms in Chinese elms remain remarkably scant. In this study, we attempt to explore the genetic diversity of seven natural populations of Chinese elms in eastern China, and then identify the potential local adaptation genes based on SLAF-seq identified SNPs. Our results might help in the marker-assisted breeding of Chinese elms in the future.
4. Discussion
The present study is the first attempt to use SNPs derived from SLAF to assess the genetic diversity and explore the adaptation mechanisms of Chinese elms. In recent years, SLAF-seq technology has become a low-cost technique to effectively develop reliable SNP and InDel markers for genome-wide association analysis and high-density genetic map construction [
25,
26]. Our study identified a total of 4,138,972 SNPs and selected 457,888 SNPs with MAF > 5% and a missing rate < 0.2 for further analysis. The number of molecular markers was dramatically larger than that in previous reports on elm species [
27,
28], which facilitates precise genetic analysis.
Heterozygosity is an important measure of overall genetic diversity [
25]. In our study, the Ho and He values ranged from 0.1483 to 0.1822 (an average of 0.1599) and 0.3236 to 0.3427 (an average of 0.3315), respectively (
Table 3). These values were lower than the results observed in other trees [
25,
29]. A relative lower level of genetic heterozygosity for the Chinese elms might be due to the existence of spatial isolation in different groups, hindering the gene communication between individuals to some extent. The index, PIC, measures the degree of informativeness of a genetic marker, with values ranging from 0 to 1 [
29]. A locus with a PIC value of 0 is undesirable [
30]. When PIC < 0.25, it indicates a low polymorphism, and 0.25 < PIC < 0.50 represents a median polymorphism. In contrast, PIC > 0.50 is indicative of high polymorphism [
31]. According to this criteria, as the PIC values were between 0.2632 to 0.2761 (
Table 3), the tested seven populations in our study possessed medium genetic diversity in terms of PIC. The inter-individual fixation index (FIS) measures the deviation of genotype frequencies from Hardy–Weinberg proportions within each population. A negative FIS indicates heterozygote excess (outbreeding), while a positive value reflects a deficiency in heterozygosity (inbreeding) [
32]. In our study, the FY population presented a negative FIS (−0.03849) (
Table 3), suggesting a slight excess of heterozygotes. The other six populations exhibited a positive FIS (
Table 3). All the populations were not statistically significantly deviated from the Hardy–Weinberg equilibrium (
p > 0.05), indicating a relatively random mating for these populations. Overall, the FY population displayed higher genetic diversity than the other six populations, which was supported by the larger Ne, Ho, He, H, I, and PIC values (
Table 3).
Outcrossing woody plants tend to possess low levels of genetic differentiation among populations [
33]. In the current study, differentiation among populations was estimated by Fst values. Fst > 0.25 signifies a great genetic differentiation, 0.25 > Fst > 0.15 indicates a moderate genetic differentiation, 0.15 > Fst > 0.05 means a small genetic differentiation, and Fst < 0.05 represents negligible genetic differentiation [
34]. Based on this standard, a low genetic differentiation was found among the studied populations (Fst values ranging from 0.00712 to 0.09106) (
Table 4). Additionally, AMOVA analysis (
Table 5) also indicated a low percentage of variation (4.24%) among populations. Similar results could be found in other trees [
7,
35].
Investigating the population structure of tested individuals is the premise for association analysis, since the presence of the population structure could affect the validity of association results [
36,
37,
38]. In our study, the optimal K value of the seven populations was 1 (
Figure 2), indicating no population structure existed in the studied groups. The geographic boundaries had a weak effect on the genetic structure of Chinese elms. The existence of population structure might cause correlations between unlinked locis, and would usually result in increased false associations, the weak population structure of Chinese elm in our study would be conducive to subsequent association analysis.
Natural selection has an important impact on shaping the genetic variation of a population, and therefore promotes local adaptation [
39]. In this research, based on the identified SNP markers, an association study was used to uncover the hidden genetic basis of local adaptation. The associated SNP markers were blasted against public databases for putative genes. We found that the genes of
DEAD-box helicase and
V-type proton ATPase seemed to be candidates for adaptation to altitude.
DEAD-box helicase is involved in nucleic acid metabolism functions, such as transcription, translation, replication, repair, recombination, ribosome biogenesis and splicing, which control plant grow and development [
40].
V-type ATPase, as a transporter, is essential for energy metabolism and maintenance of solute homeostasis, which makes it indispensable for plant growth [
41,
42].
V-type proton ATPase has been shown to play a significant role in plant adaptation to stressful growth conditions [
42]. We deduced that variations in altitude would lead to a difference in plant growth according to the functions of
DEAD-box helicase and
V-type proton ATPase.
UDP-glycosyltransferase (
UGT) and
peroxisome biogenesis protein were associated with annual rainfall variable.
UGT belongs to the
glycosyltransferase (
GT) multigene family [
43]. In plants, GTs are a ubiquitous group of enzymes involved in the glycosylation process, and glycosylation leads to the formation of glycosylated secondary chemicals such as flavonols, anthocyanins, and plant hormones [
44,
45]. Glycosylated secondary products possess increased water solubility and molecule stability, which could change their biological activity [
44].
Peroxisome biogenesis protein might participate in the synthesis of peroxisomes, a metabolic organelle that exists in all eukaryotic cells [
46]. Peroxisomes contribute to resistance against oxidative stresses, β- and α-oxidation of fatty acids, and synthesis of ether lipids [
47,
48]. The products of
UGT and
Peroxisome biogenesis protein seemed to confer advantages for plants survival in rainy climate [
43]. It is reasonable that
UGT and
peroxisome biogenesis protein appeared as candidates for adaptation to rainfall climate.
Cysteine-rich receptor-like protein kinase (
CRK) was the putative gene that we found was associated with the annual average temperature variable. CRKs are critical signaling components that regulate plant developmental and defense processes. In
Arabidopsis, overexpression of a CRK gene confers drought tolerance without affecting plant growth [
49]. Considering that the temperature variable would be generally correlated with drought stress, it is possible that there may be a difference in drought-associated loci among populations. Identification of putative candidate genes correlated with the environment would reveal a primary insight into functional genes mediating local adaptation. However, further studies are required in the future to explain the accurate roles of those candidate genes in the adaptation processes of Chinese elms.

 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}