A Weighted Mean Value Analysis to Identify Biological Pathway Activity Changes during Poplar Seed Germination


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Conditions, Data Collection and Analysis
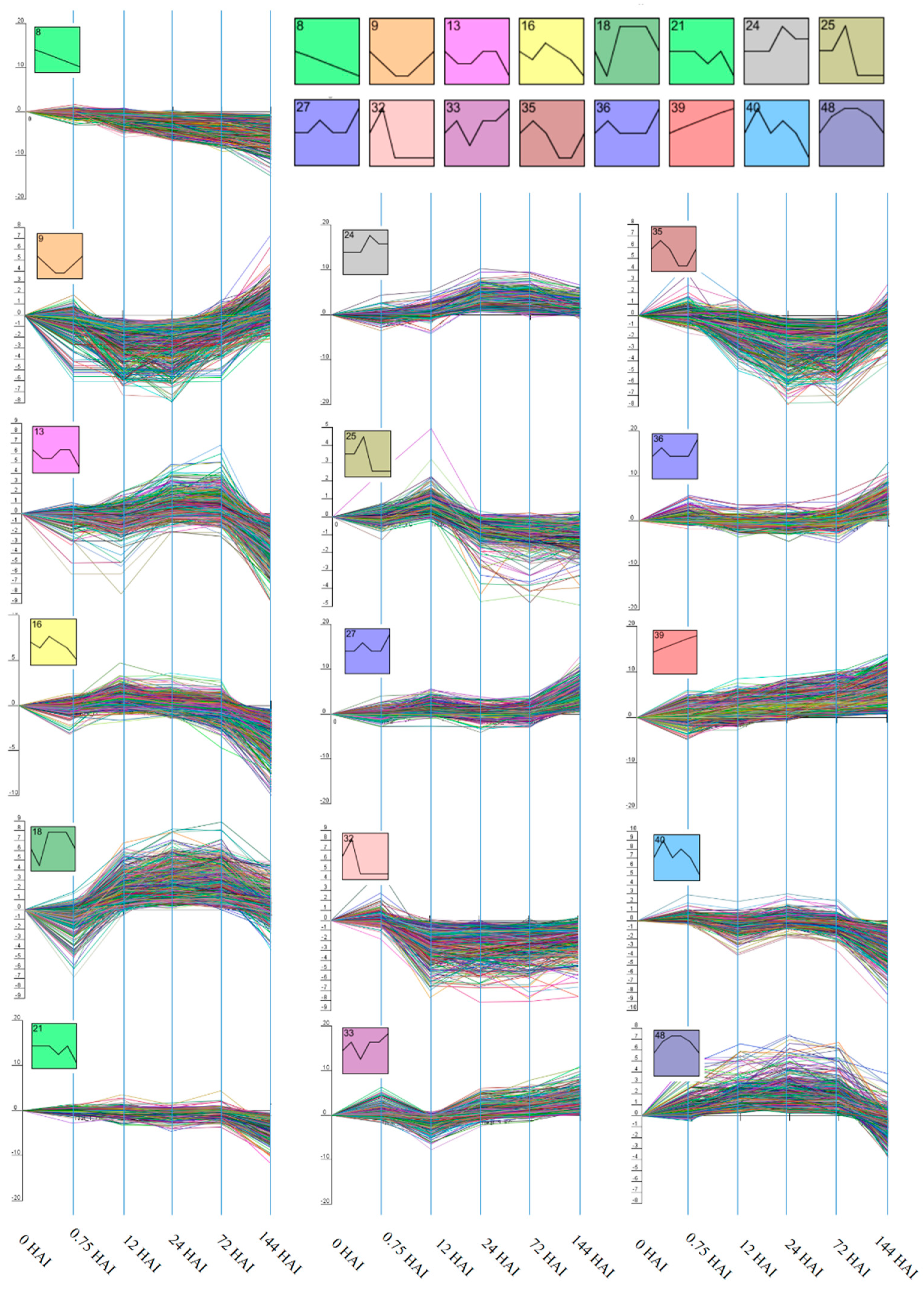
2.2. Clustering of Gene Expression Data
2.3. Gene Ontology (GO) Enrichment Analysis
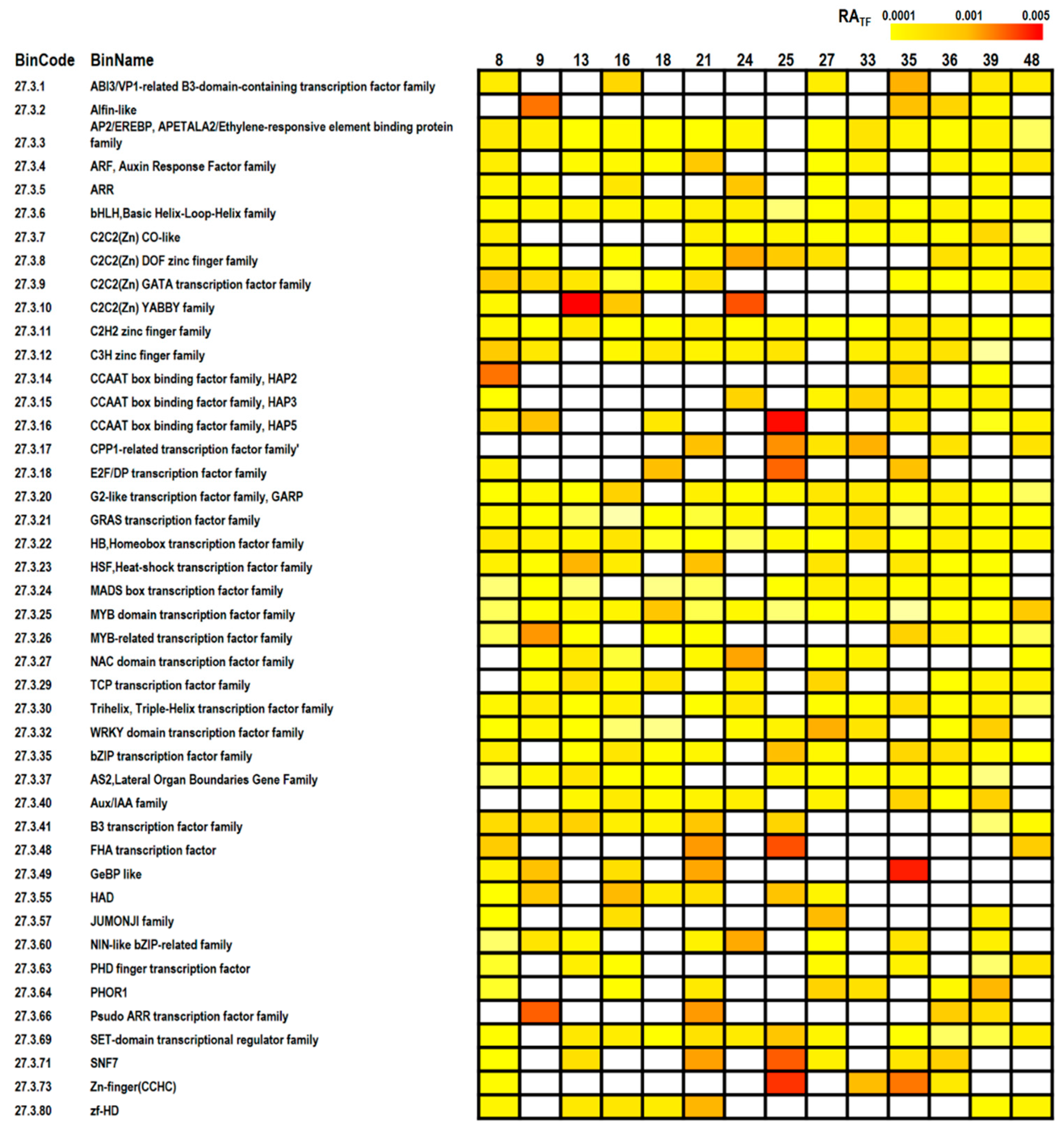
2.4. MapMan Analysis
2.5. Pathway Activity
3. Results
3.1. Cluster Analysis of DEGs
3.2. GO Enrichment Analysis
3.3. Pathway Analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Nonogaki, H.; Bassel, G.W.; Bewley, J.D. Germination-Still a mystery. Plant Sci. 2010, 179, 574–581. [Google Scholar] [CrossRef]
- Kamran, M.; Imran, Q.; Khatoon, A.; Lee, I.J.; Rehman, S. Effect of plant extracted smoke and reversion of abscisic acid stress on lettuce. Pak. J. Bot. 2013, 45, 1541–1549. [Google Scholar]
- Nambara, E.; Nonogaki, H. Seed biology in the 21st century: Perspectives and new directions. Plant Cell Physiol. 2012, 53, 1–4. [Google Scholar] [CrossRef]
- Bewley, J.D. Seed germination and dormancy. Plant Cell 1997, 9, 1055. [Google Scholar] [CrossRef]
- Bove, J.; Jullien, M.; Grappin, P. Functional genomics in the study of seed germination. Genome Biol. 2001, 3, 1001–1002. [Google Scholar] [CrossRef]
- Manz, B.; Müller, K.; Kucera, B.; Volke, F.; Leubner-Metzger, G. Water uptake and distribution in germinating tobacco seeds investigated in vivo by nuclear magnetic resonance imaging. Plant Physiol. 2005, 138, 1538–1551. [Google Scholar] [CrossRef]
- Finch-Savage, W.E.; Leubner-Metzger, G. Seed dormancy and the control of germination. New Phytol. 2006, 171, 501–523. [Google Scholar] [CrossRef]
- Girke, T.; Todd, J.; Ruuska, S.; White, J.; Benning, C.; Ohlrogge, J. Microarray analysis of developing Arabidopsis seeds. Plant Physiol. 2000, 124, 1570–1581. [Google Scholar] [CrossRef]
- Howell, K.A.; Narsai, R.; Carroll, A.; Ivanova, A.; Lohse, M.; Usadel, B.; Millar, A.H.; Whelan, J. Mapping metabolic and transcript temporal switches during germination in rice highlights specific transcription factors and the role of RNA instability in the germination process. Plant Physiol. 2009, 149, 961–980. [Google Scholar] [CrossRef]
- Le, B.H.; Cheng, C.; Bui, A.Q.; Wagmaister, J.A.; Henry, K.F.; Pelletier, J.; Kwong, L.; Belmonte, M.; Kirkbride, R.; Horvath, S. Global analysis of gene activity during Arabidopsis seed development and identification of seed-specific transcription factors. Proc. Natl. Acad. Sci. USA 2010, 107, 8063–8070. [Google Scholar] [CrossRef]
- Gao, J.; Yu, X.; Ma, F.; Li, J. RNA-seq analysis of transcriptome and glucosinolate metabolism in seeds and sprouts of broccoli (Brassica oleracea var. italic). PLoS ONE 2014, 9, e88804. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, W.Q.; Liu, S.J.; Moller, I.M.; Song, S.Q. Proteome Analysis of Poplar Seed Vigor. PLoS ONE 2015, 10, e0132509. [Google Scholar] [CrossRef]
- Qu, C.; Zuo, Z.; Cao, L.; Huang, J.; Sun, X.; Zhang, P.; Yang, C.; Li, L.; Xu, Z.; Liu, G. Comprehensive Dissection of Transcript and Metabolite Shifts during Seed Germination and Post-Germination Stages in Poplar. BMC Plant Biol. 2019, 19, 279. [Google Scholar] [CrossRef]
- Zhang, C.; Luo, W.; Li, Y.; Zhang, X.; Bai, X.; Niu, Z.; Zhang, X.; Li, Z.; Wan, D. Transcriptomic Analysis of Seed Germination Under Salt Stress in Two Desert Sister Species (Populus euphratica and P. pruinosa). Front. Genet. 2019, 10, 231. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Arxiv Prepr. Arxiv 2013, 1303, 3997. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. Edger: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef]
- Ernst, J.; Bar-Joseph, Z. STEM: A tool for the analysis of short time series gene expression data. BMC Bioinform. 2006, 7, 191. [Google Scholar] [CrossRef]
- Du, Z.; Zhou, X.; Ling, Y.; Zhang, Z.; Su, Z. Agrigo: A GO analysis toolkit for the agricultural community. Nucleic Acids Res. 2010, 64–70. [Google Scholar] [CrossRef]
- Thimm, O.; Bläsing, O.; Gibon, Y.; Nagel, A.; Meyer, S.; Krüger, P.; Selbig, J.; Müller, L.A.; Rhee, S.Y.; Stitt, M. Mapman: A user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 2004, 37, 914–939. [Google Scholar] [CrossRef]
- Ernst, J.; Nau, G.J.; Bar-Joseph, Z. Clustering short time series gene expression data. Bioinformatics 2005, 21, 59–168. [Google Scholar] [CrossRef]
- Balazadeh, S.; Schildhauer, J.; Araujo, W.L.; Munne-Bosch, S.; Fernie, A.R.; Proost, S.; Humbeck, K.; Mueller-Roeber, B. Reversal of senescence by N resupply to N-starved Arabidopsis thaliana: Transcriptomic and metabolomic consequences. J. Exp. Bot. 2014, 65, 3975–3992. [Google Scholar] [CrossRef]
- Swamy, P.; Sandhyarani, C. Contribution of the pentose phosphate pathway and glycolytic pathway to dormancy breakage and germination of peanut (Arachis hypogaea L.) seeds. J. Exp. Bot. 1986, 37, 80–88. [Google Scholar] [CrossRef]
- Gahan, P.B.; Dawson, A.L.; Black, M.; Chapman, J.M. Localization of Glucose-6-phosphate Dehydrogenase Activity in Seeds and its Possible Involvement in Dormancy Breakage. Ann. Bot. 1986, 57, 791–799. [Google Scholar] [CrossRef]
- Muscolo, A.; Panuccio, M.; Sidari, M. The effect of phenols on respiratory enzymes in seed germination. Plant Growth Regul. 2001, 35, 31–35. [Google Scholar] [CrossRef]
- Hare, P.D.; Cress, W.A.; van Staden, J. A regulatory role for proline metabolism in stimulating Arabidopsis thaliana seed germination. Plant Growth Regul. 2003, 39, 41–50. [Google Scholar] [CrossRef]
- Dekkers, B.J.W.; Pearce, S.; van Bolderen-Veldkamp, R.P.; Marshall, A.; Widera, P.; Gilbert, J.; Drost, H.G.; Bassel, G.W.; Muller, K.; King, J.R.; et al. Transcriptional Dynamics of Two Seed Compartments with Opposing Roles in Arabidopsis Seed Germination. Plant Physiol. 2013, 163, 205–215. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qu, C.; Zhang, Y.; Chen, J.; Zhang, S.; Yu, J.; Yang, C.; Zhang, X.; Xu, Z.-R.; Liu, G.-J. A Weighted Mean Value Analysis to Identify Biological Pathway Activity Changes during Poplar Seed Germination. Forests 2019, 10, 664. https://doi.org/10.3390/f10080664
Qu C, Zhang Y, Chen J, Zhang S, Yu J, Yang C, Zhang X, Xu Z-R, Liu G-J. A Weighted Mean Value Analysis to Identify Biological Pathway Activity Changes during Poplar Seed Germination. Forests. 2019; 10(8):664. https://doi.org/10.3390/f10080664
Chicago/Turabian StyleQu, Chunpu, Yuqing Zhang, Jinyuan Chen, Shuang Zhang, Jiajie Yu, Chengjun Yang, Xiuli Zhang, Zhi-Ru Xu, and Guan-Jun Liu. 2019. "A Weighted Mean Value Analysis to Identify Biological Pathway Activity Changes during Poplar Seed Germination" Forests 10, no. 8: 664. https://doi.org/10.3390/f10080664
APA StyleQu, C., Zhang, Y., Chen, J., Zhang, S., Yu, J., Yang, C., Zhang, X., Xu, Z.-R., & Liu, G.-J. (2019). A Weighted Mean Value Analysis to Identify Biological Pathway Activity Changes during Poplar Seed Germination. Forests, 10(8), 664. https://doi.org/10.3390/f10080664