DNA Methylation of Farnesyl Pyrophosphate Synthase, Squalene Synthase, and Squalene Epoxidase Gene Promoters and Effect on the Saponin Content of Eleutherococcus Senticosus

Abstract
:1. Introduction
2. Materials and Methods
2.1. Extraction of DNA and Total RNA from E. Senticosus and Synthesis of cDNA
2.2. Prediction of CpG Islands, and DNA Methylation Site Analysis of the FPS, SS, and SE Promoters of E. Senticosus
2.3. Analysis of FPS, SS, and SE Gene Expression of E. Senticosus
2.4. Determination of Total Saponin in E. Senticosus and Correlation Analysis
2.5. Preparation and Extraction of E. Senticosus Samples for Metabolite Analysis
2.6. UPLC Conditions
2.7. ESI-Q TRAP-MS/MS
2.8. Metabolome Data Analysis
3. Results
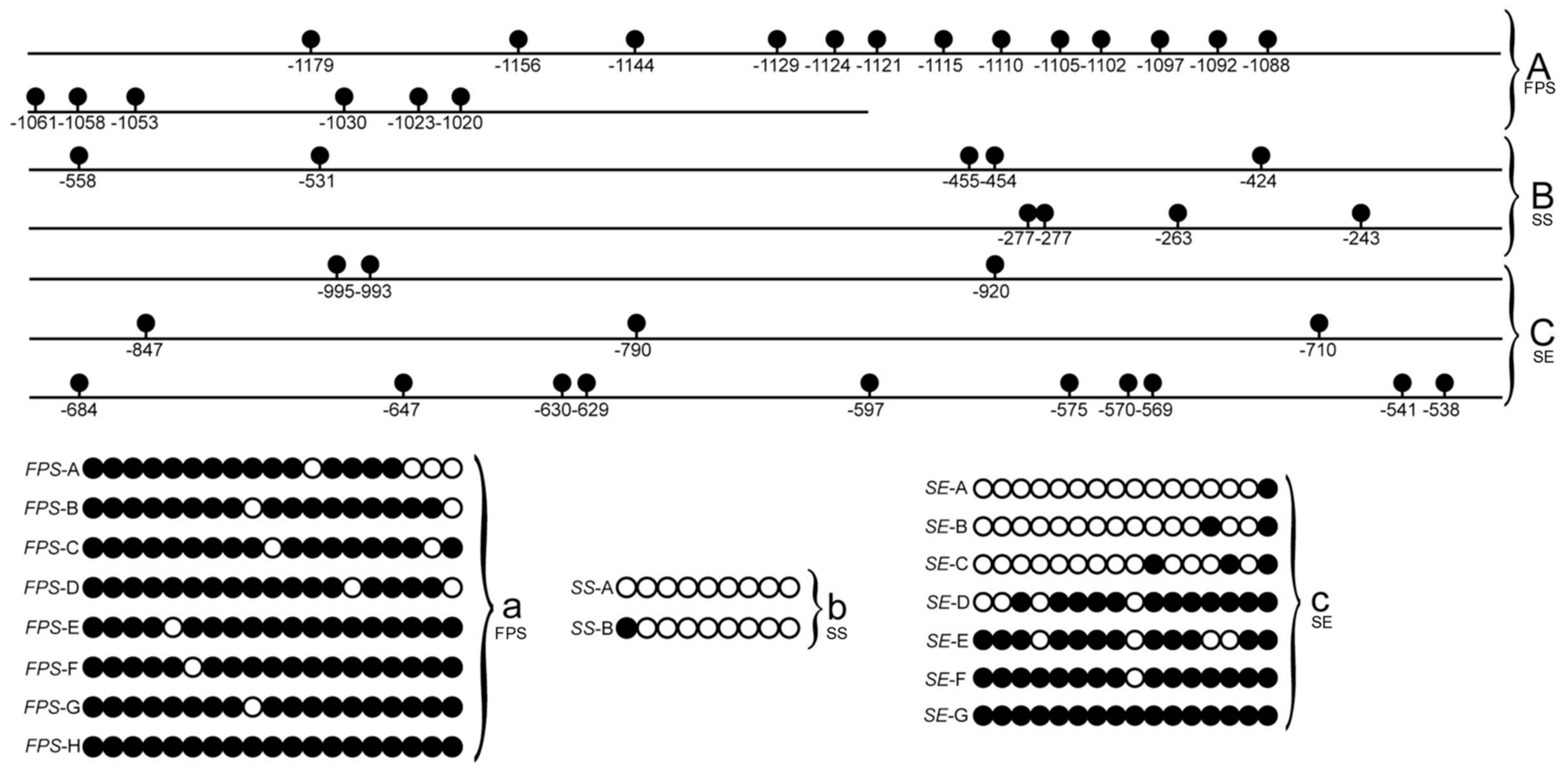
3.1. DNA Methylation Site Analysis of FPS, SS, and SE Gene Promoters of E. Senticosus
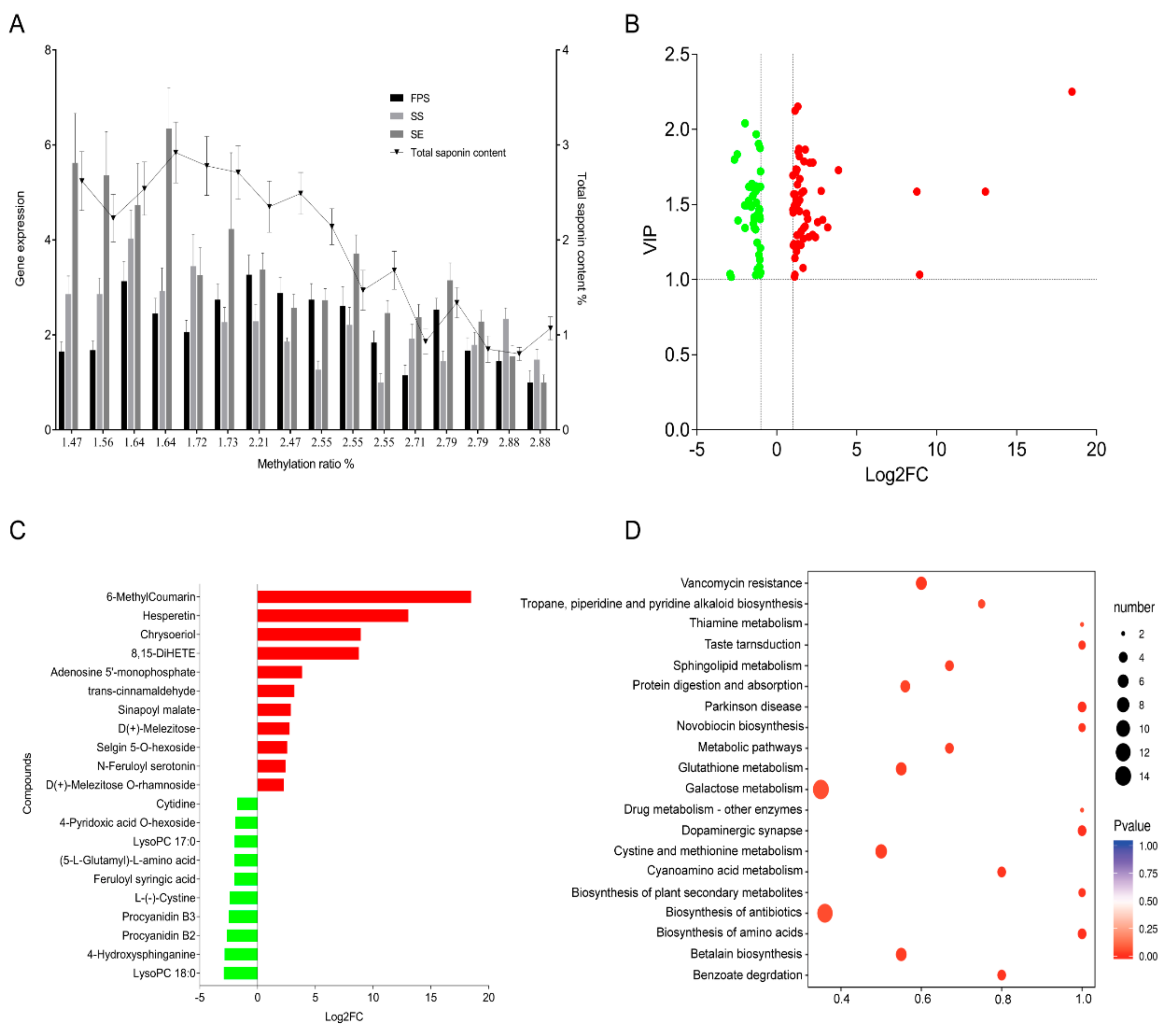
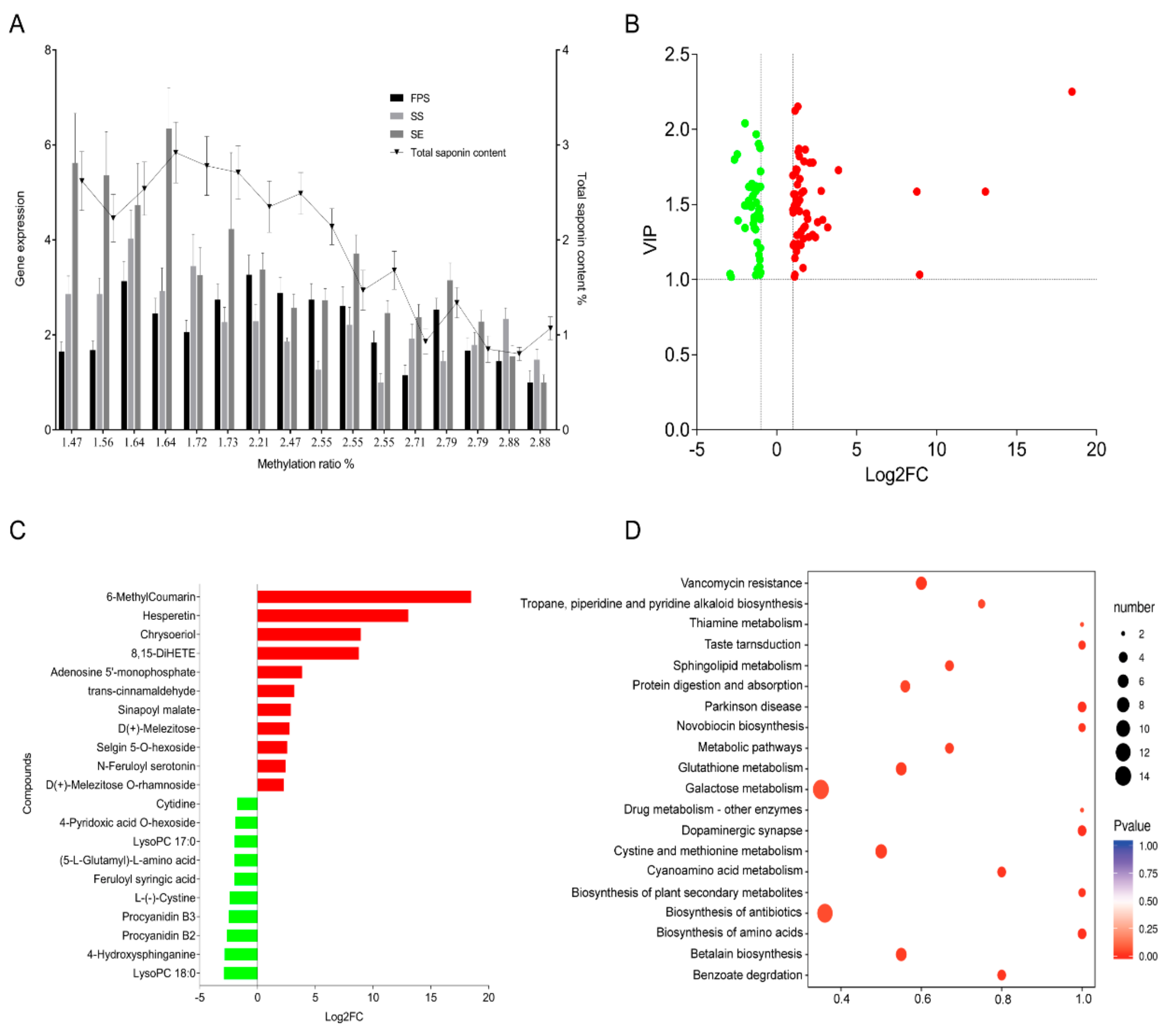
3.2. Effect of the DNA Methylation of FPS, SS, and SE Promoters of E. Senticosus on the Gene Expression and Saponin Content
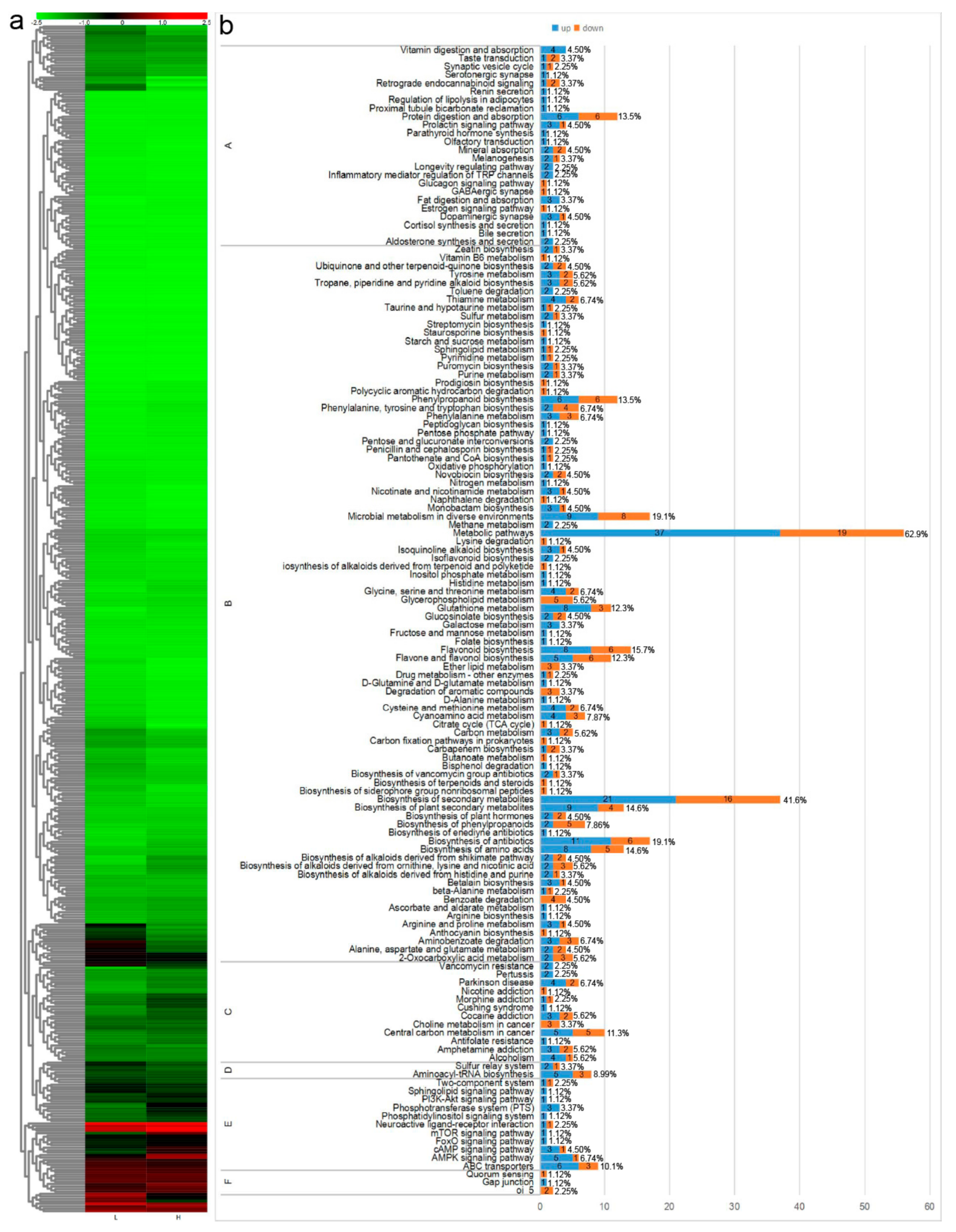
3.3. Screening of Differential Metabolites in the Leaves of E. Senticosus
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- The State Pharmacopoeia Commission of P.R. China. Pharmacopoeia of the People’s Republic of China; China Medical Science Press: Beijing, China, 2015; pp. 5–6. [Google Scholar]
- Huang, L.; Zhao, H.; Huang, B.; Zheng, C.; Peng, W.; Qin, L. Acanthopanax senticosus: Review of botany, chemistry and pharmacology. Pharmazie 2011, 66, 83–97. [Google Scholar] [PubMed]
- Murthy, H.N.; Kim, Y.S.; Georgiev, M.I.; Peak, K.Y. Biotechnological production of eleutherosides: Current state and perspectives. Appl. Microbiol. Biotechnol. 2014, 98, 7319–7329. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.D.; Park, H.W.; Hahn, Y.; Hur, C.G.; In, D.S.; Chung, H.J.; Liu, J.R.; Choi, D.W. Discovery of genes for ginsenoside biosynthesis by analysis of ginseng expressed sequence tags. Plant Cell Rep. 2003, 22, 224–230. [Google Scholar] [CrossRef] [PubMed]
- SzkoPińska, A.; Płochocka, D. Farnesyl diphosphate synthase; regulation of product specificity. Acta Biochim. Pol. 2005, 52, 45–55. [Google Scholar] [PubMed]
- Abe, I.; Rohmer, M.; Prestwich, G.D. Enzymatic cyclization of squalene and oxidosqualene to sterols and triterpenes. Chem. Rev. 1993, 93, 2189–2206. [Google Scholar] [CrossRef]
- Hwang, H.S.; Lee, H.; Choi, Y.E. Transcriptomic analysis of Siberian ginseng (Eleutherococcus senticosus) to discover genes involved in saponin biosynthesis. BMC Genom. 2015, 16, 180. [Google Scholar] [CrossRef]
- HaralamPidis, K.; Trojanowska, M.; Osbourn, A.E. Biosynthesis of triterpenoid saponins in plants. Adv. Biochem. Eng. Biotechnol. 2002, 75, 31–49. [Google Scholar]
- Pikaard, C.S.; Mittelsten Scheid, O. Epigenetic regulation in plants. Cold Spring Harb. Perspect. Biol. 2014, 6, a109315. [Google Scholar] [CrossRef]
- Nguyen, T.P.; Kyung, A.L.; Su, J.J.; Jung, K.C.; Young, H.K.; Jong, S.K. Capillary electrophoretic method for the determination of diterpenoid isomers in Acanthopanax species. J. Pharm. Biomed. Anal. 2006, 40, 56–61. [Google Scholar]
- PlantCARE, a Database of Plant Promoters and their Cis-Acting Regulatory Elements. Available online: http://bioinformatics.psb.ugent.be/webtools/plantcare/html/ (accessed on 20 November 2019).
- EMBOSS Programs < EMBL-EBI. Available online: https://www.ebi.ac.uk/Tools/emboss/ (accessed on 20 November 2019).
- MethPrimer-Design MSP/BSP Primers and Predict CpG Islands—Li Lab, PUMCH. Available online: http://www.urogene.org/cgi-bin/methprimer/methprimer.cgi (accessed on 20 November 2019).
- Singh, V.K.; Mangalam, A.K.; Dwivedi, S.; Naik, S. Primer premier: Program for design of degenerate primers from a protein sequence. Biotechniques 1998, 24, 318–319. [Google Scholar] [CrossRef]
- Naydenov, M.; Baev, V.; Apostolova, E.; Gospodinova, N.; Sablok, G.; Gozmanova, M.; Yahubyan, G. High-temperature effect on genes engaged in DNA methylation and affected by DNA methylation in Arabidopsis. Plant Physiol. Biochem. 2015, 87, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Fraga, C.G.; Clowers, B.H.; Moore, R.J.; Zink, E.M. Signature-discovery approach for sample matching of a nerve-agent precursor using liquid chromatography-mass spectrometry, XCMS, and chemometrics. Anal. Chem. 2010, 82, 4165–4173. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Susumu, G. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Arango, J.; Beltrán, J.; Nuñez, J.; Chavarriaga, P. Evidence of epigenetic mechanisms affecting carotenoids. Subcell. Biochem. 2016, 79, 295–307. [Google Scholar] [PubMed]
- Curradi, M.; Izzo, A.; Badaracco, G.; Landsberger, N. Molecular mechanisms of gene silencing mediated by DNA methylation. Mol. Cell. Biol. 2002, 22, 3157–3173. [Google Scholar] [CrossRef]
- Chujo, T.; Scott, B. Histone H3K9 and H3K27 methylation regulates fungal alkaloid biosynthesis in a fungal endophyte-plant symbiosis. Mol. Microbiol. 2014, 92, 413–434. [Google Scholar] [CrossRef]
- Li, L.Q.; Li, X.L.; Fu, C.H.; Zhao, C.F.; Yu, L.J. Sustainable use of Taxus media cell cultures through minimal growth conservation and manipulation of genome methylation. Process Biochem. 2013, 48, 525–531. [Google Scholar] [CrossRef]
- Dou, L.; Jia, X.; Wei, H.; Fan, S.; Wang, H.; Guo, Y.; Duan, S.; Pang, C.; Yu, S. Global analysis of DNA methylation in young (J1) and senescent (J2) Gossypium hirsutum L. cotyledons byMeDIP-Seq. PLoS ONE 2017, 12, e0179141. [Google Scholar] [CrossRef]
- Chan, S.W.; Henderson, I.R.; Jacobsen, S.E. Gardening the genome: DNA methylation in Arabidopsis thaliana. Nat. Rev. Genet. 2005, 6, 351–360. [Google Scholar] [CrossRef]
- Kuo, T.C.; Chen, C.H.; Chen, S.H.; Chun, M.J.; Huang, L.C.; Lin, C.Y.; Chen, C.Y.; Lo, H.F.; Jeng, S.T.; Chen, L.F. The effect of red light and far-red light conditions on secondary metabolism in Agarwood. BMC Plant Biol. 2015, 15, 139. [Google Scholar] [CrossRef]
- Da, K.; Nowak, J.; Flinn, B. Potato cytosine methylation and gene expression changes induced by a beneficial bacterial endophyte, Burkholderia phytofirmans strain PsJN. Plant Physiol. Biochem. 2012, 50, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Qin, L.; Xie, C.; Li, W.; Yuan, J.; Kong, L.; Yu, W.; Xia, G.; Liu, S. Induced and constitutive DNA methylation in a salinity-tolerant wheat introgression line. Plant Cell Physiol. 2014, 55, 1354–1365. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Wang, Z.; Jiang, C.; Wang, X.; Huang, L. Exploiting genes and functional diversity of chlorogenic acid and luteolin biosyntheses in Lonicera japonica and their substitutes. Gene 2014, 534, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Zha, L.; Liu, S.; Liu, J.; Jiang, C.; Yu, S.; Yuan, Y.; Yang, J.; Wang, Y.; Huang, L. DNA methylation influences chlorogenic acid biosynthesis in Lonicera japonica by mediating LjbZIP8 to regulate phenylalanine ammonia-lyase 2 expression. Front. Plant Sci. 2017, 8, 1178. [Google Scholar] [CrossRef]
- Kim, J.; Kim, J.H.; Richards, E.J.; Chung, K.M.; Woo, H.R. Arabidopsis VIM proteins regulate epigenetic silencing by modulating DNA methylation and histone modification in cooperation with MET1. Mol. Plant 2014, 7, 1470–1485. [Google Scholar] [CrossRef]
- Kuhlmann, M.; Finke, A.; Mascher, M.; Mette, M.F. DNA methylation maintenance consolidates RNA-directed DNA methylation and transcriptional gene silencing over generations in Arabidopsis thaliana. Plant J. 2014, 80, 269–281. [Google Scholar] [CrossRef]
- Ngezahayo, F.; Wang, X.L.; Yu, X.M.; Jiang, L.L.; Chu, Y.J.; Shen, B.H.; Yan, Z.K.; Liu, B. Habitat-induced reciprocal transformation in the root phenotype of Oriental ginseng is associated with alteration in DNA methylation. Chin. Sci. Bull. 2011, 56, 1685–1690. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Sequence (5′–3′) | Use | Expected Amplification Length |
|---|---|---|---|
| CFPSJqs5 | AAAACTCAGATCTGGTCAAAAAGGG | Primers for PCR amplification of FPS in unsulfite treated samples | 277 bp |
| CFPSJqx5 | CCTGGAAACCAAGCACCAAATGCA | ||
| CFPSJhs5 | AAAATTTAGATTTGGTTAAAAAGGG | Primers for PCR amplification of FPS in sulfite treated samples | |
| CFPSJhx5 | CCTAAAAACCAAACACCAAATACA | ||
| CSSJqs5 | AAAAAACTTAAAAATGTGAGCCAACAG | Primers for PCR amplification of unsulfite treated sample SS | 309 bp |
| CSSJqx5 | GAATGAAAATGGAGCAGCAATTTGT | ||
| CSSJhs5 | AAAAAATTTAAAAATGTGAGTTAATAG | Primers for PCR amplification of bisulfite treated sample SS | |
| CSSJhx5 | AAATAAAAATAAAACAACAATTTAT | ||
| CSEJqs | GGGGTTGTTCCAGATCTGGAATACTA | Primers for PCR amplification of unsulfite treated sample SE | 547 bp |
| CSEJqx | CAGCCACCAGAGCCACAGAATTCCG | ||
| CSEJhs | GGGGTTGTTTTAGATTTGGAATATTA | Primers for PCR amplification of sulfite treated sample SE | |
| CSEJhx | AACCACCAAAACCACAAAATTCCG | ||
| ActrtF | GCAAGAGCTTGAAACAGCAAAG | Primers for amplification of actin | 128 bp |
| AcrrtR | TCAAAGATGGCTGGAAAAGGA | ||
| FPSrtF | TCTCTCCCTCTTTCCCTCCTCT | Primers for amplifying FPS Gene | 132 bp |
| FPSrtR | ATCGCTCATTCTGGCTGGTT | ||
| SSrtF | TCACGACTAGATGGACAAGCAAA | Primers for amplifying SS | 143 bp |
| SSrtR | GCCAATAACAAGAGCAGGAATG | ||
| SErtF | TAAAGTCGGAAGGAGTTCG | Primers for amplification of SE | 82 bp |
| SErtR | CTTAGTGAATGAATGGGAGG |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Z.; Guo, H.; Zhang, Y.; Lin, L.; Cui, M.; Long, Y.; Xing, Z. DNA Methylation of Farnesyl Pyrophosphate Synthase, Squalene Synthase, and Squalene Epoxidase Gene Promoters and Effect on the Saponin Content of Eleutherococcus Senticosus. Forests 2019, 10, 1053. https://doi.org/10.3390/f10121053
Wang Z, Guo H, Zhang Y, Lin L, Cui M, Long Y, Xing Z. DNA Methylation of Farnesyl Pyrophosphate Synthase, Squalene Synthase, and Squalene Epoxidase Gene Promoters and Effect on the Saponin Content of Eleutherococcus Senticosus. Forests. 2019; 10(12):1053. https://doi.org/10.3390/f10121053
Chicago/Turabian StyleWang, Zhuo, Hongyu Guo, Yantong Zhang, Limei Lin, Minghui Cui, Yuehong Long, and Zhaobin Xing. 2019. "DNA Methylation of Farnesyl Pyrophosphate Synthase, Squalene Synthase, and Squalene Epoxidase Gene Promoters and Effect on the Saponin Content of Eleutherococcus Senticosus" Forests 10, no. 12: 1053. https://doi.org/10.3390/f10121053