Genetic Analyses of the Laurel Wilt Pathogen, Raffaelea lauricola, in Asia Provide Clues on the Source of the Clone that is Responsible for the Current USA Epidemic

Abstract
:1. Introduction
2. Materials and Methods
2.1. Isolates of Raffaelea lauricola
2.2. Mating-Type Assay
2.3. Microsatellite Markers
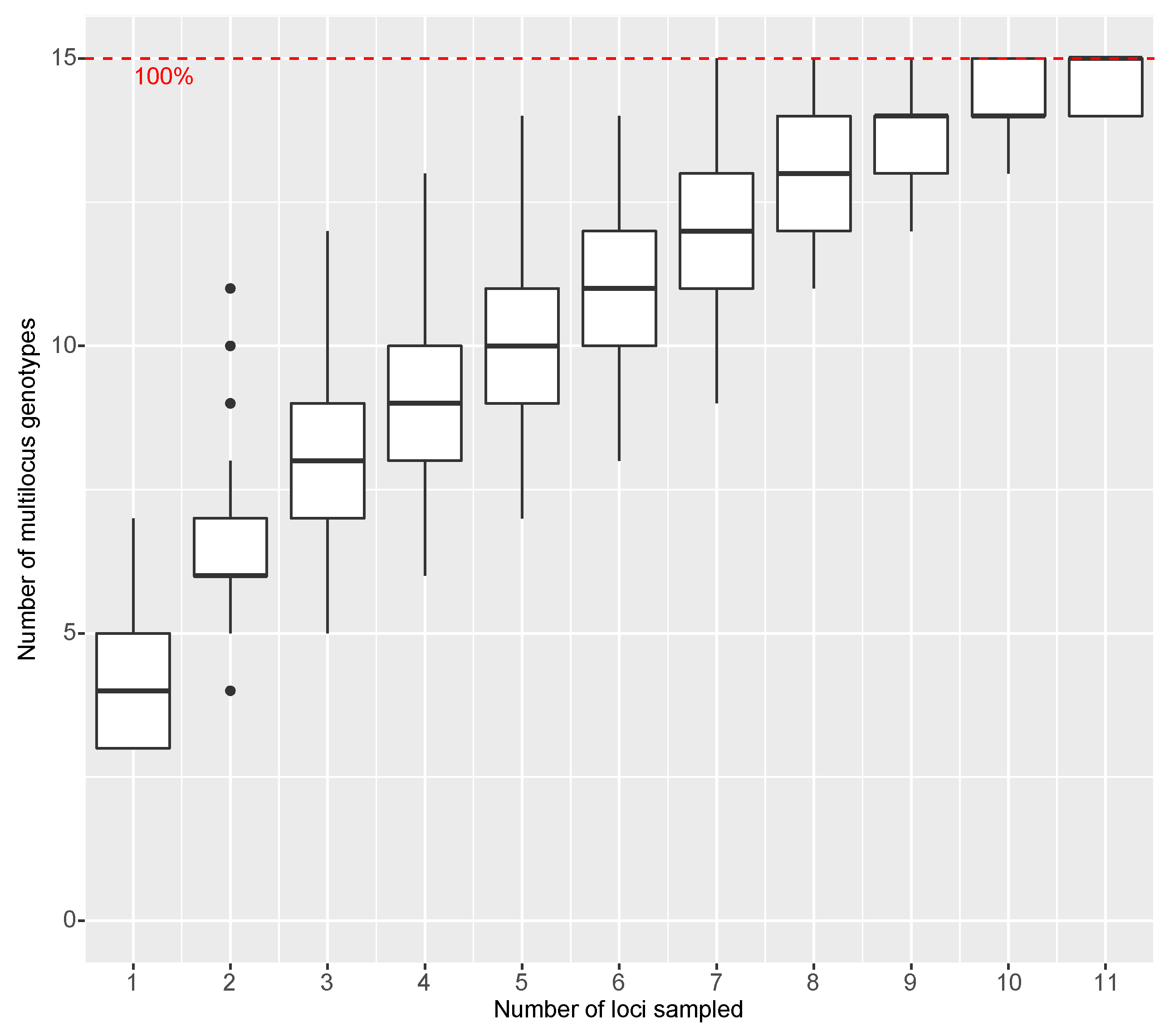
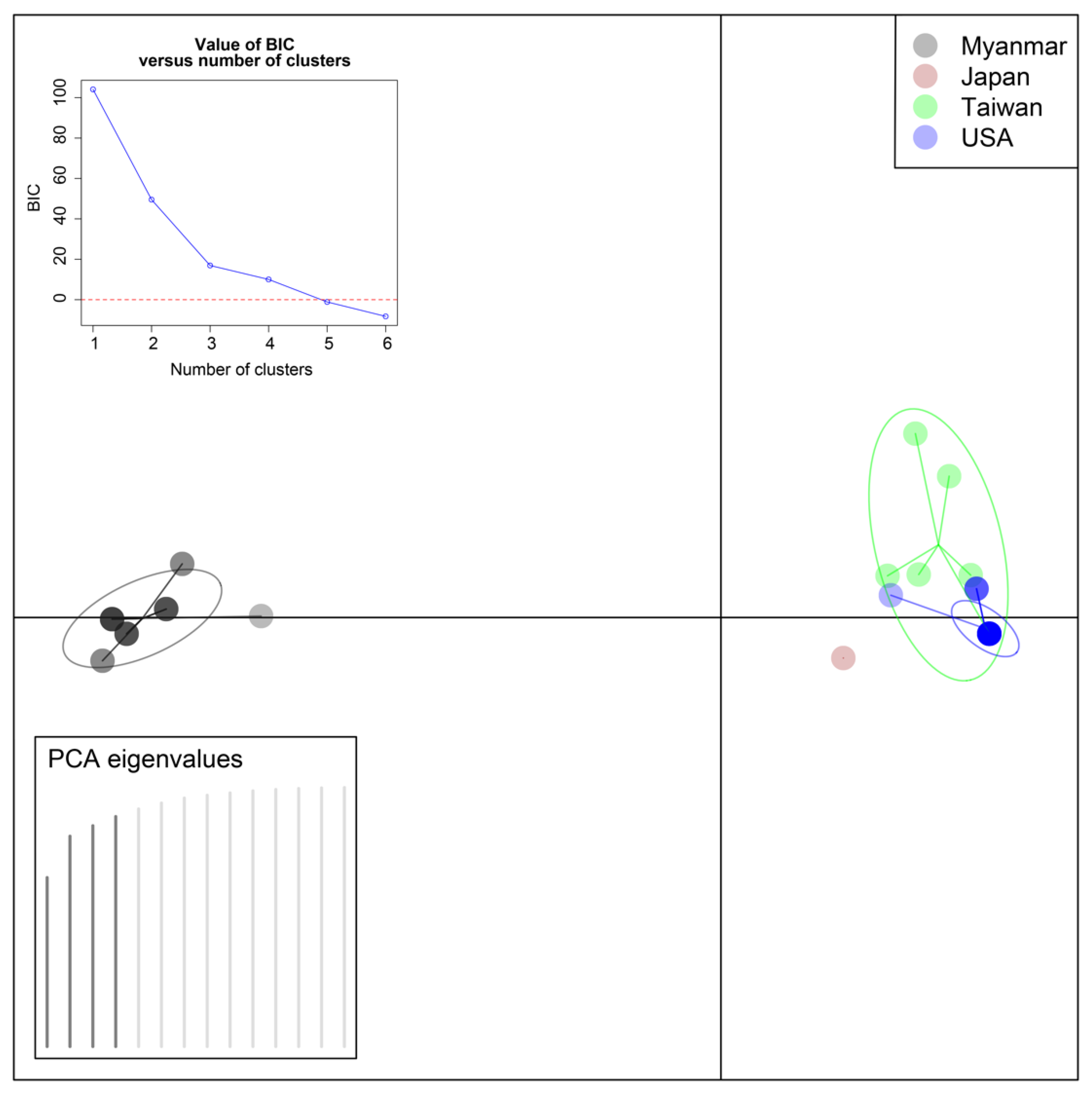
2.4. Statistical Analyses
3. Results
3.1. Mating-Type Assay
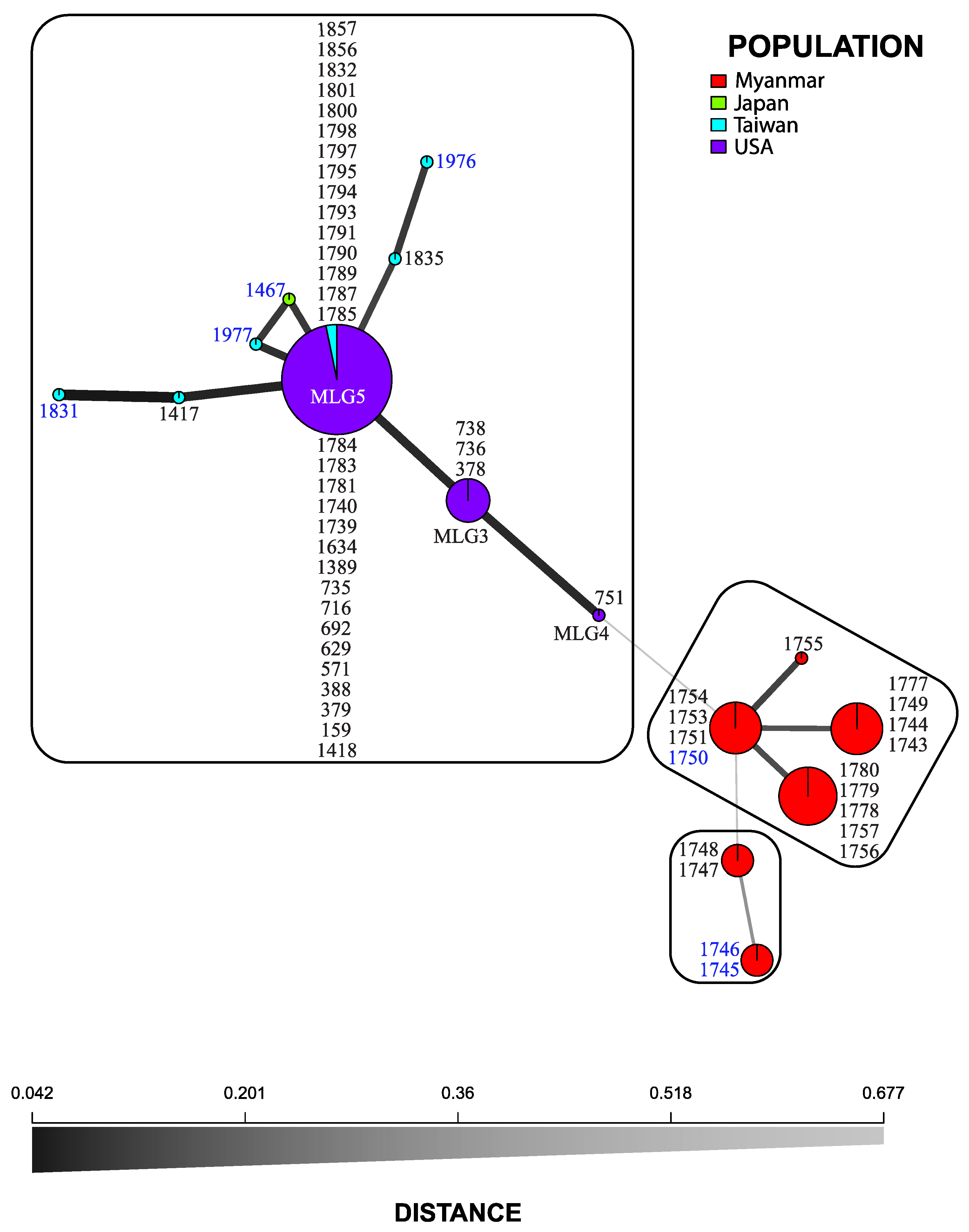
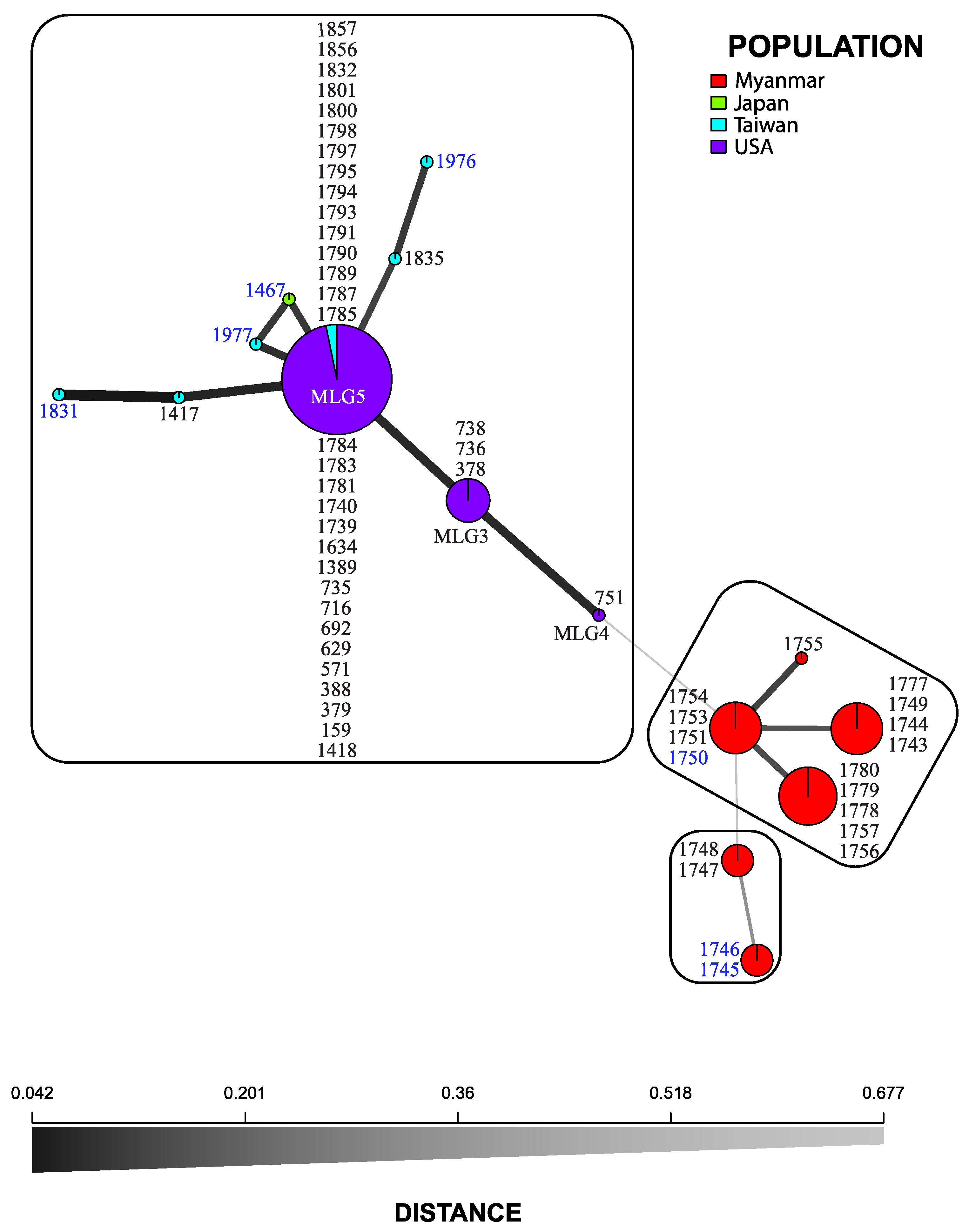
3.2. Microsatellite Markers
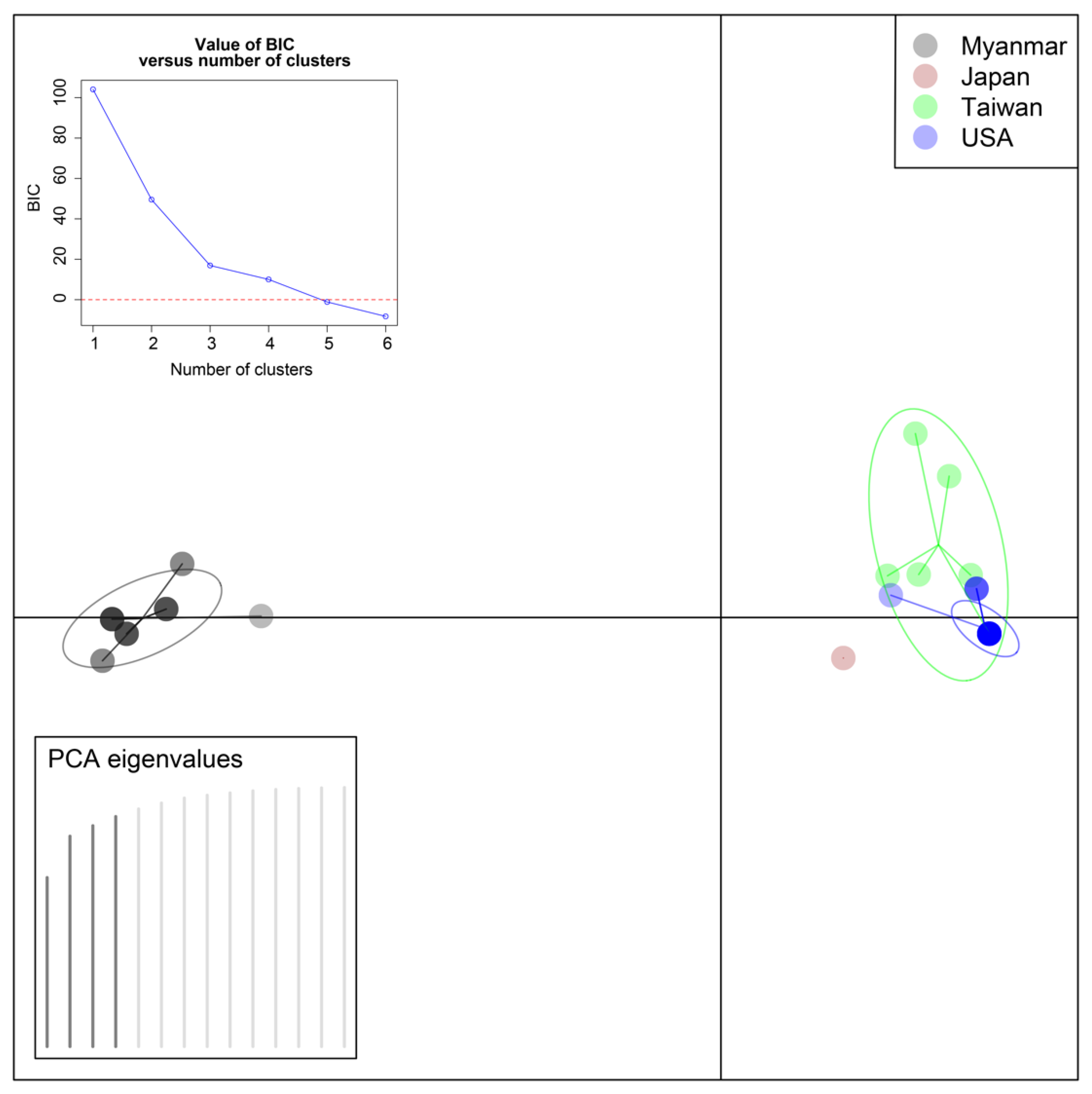
3.3. Statistical Analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fraedrich, S.W.; Harrington, T.C.; Rabaglia, R.J.; Ulyshen, M.D.; Mayfield, A.E., III; Hanula, J.L.; Eickwort, J.M.; Miller, D.R. A fungal symbiont of the redbay ambrosia beetle causes a lethal wilt in redbay and other Lauraceae in the southeastern USA. Plant Dis. 2008, 92, 215–224. [Google Scholar] [CrossRef]
- Harrington, T.C.; Fraedrich, S.W.; Aghayeva, D.N. Raffaelea lauricola, a new ambrosia beetle symbiont and pathogen on the Lauraceae. Mycotaxon 2008, 104, 399–404. [Google Scholar]
- Rabaglia, R.J.; Dole, S.A.; Cognato, A.I. Review of American Xyleborina (Coleoptera: Curculionidae: Scolytinae) occurring North of Mexico, with an illustrated key. Ann. Entomol. Soc. Am. 2006, 99, 1034–1056. [Google Scholar] [CrossRef]
- Hughes, M.A.; Smith, J.A.; Ploetz, R.C.; Kendra, P.E.; Mayfield, A.E., III; Hanula, J.L.; Hulcr, J.; Stelinski, L.L.; Cameron, S.; Riggins, J.J.; et al. Recovery plan for laurel wilt on redbay and other forest species caused by Raffaelea lauricola and disseminated by Xyleborus glabratus. Plant Health Prog. 2015. [Google Scholar] [CrossRef]
- Hughes, M.A.; Riggins, J.J.; Koch, F.H.; Cognato, A.I.; Anderson, C.; Formby, J.P.; Dreaden, T.J.; Ploetz, R.C.; Smith, J.A. No rest for the laurels: Symbiotic invaders cause unprecedented damage to southern USA forests. Biol. Invasions 2017, 79, 22–36. [Google Scholar] [CrossRef]
- Fraedrich, S.W.; Johnson, C.W.; Menard, R.D.; Harrington, T.C.; Olatinwo, R.; Best, G.S. First report of Xyleborus glabratus (Coleoptera: Curculionidae: Scolytinae) and laurel wilt in Louisiana, USA: The disease continues westward on sassafras. Fla. Entomol. 2015, 98, 1266–1268. [Google Scholar] [CrossRef]
- Olatinwo, R.; Barton, C.; Fraedrich, S.W.; Johnson, W.; Hwang, J. First report of laurel wilt, caused by Raffaelea lauricola, on sassafras (Sassafras albidum) in Arkansas. Plant Dis. 2016, 100, 2331. [Google Scholar] [CrossRef]
- Formby, J.P.; Rodgers, J.C.; Koch, F.H.; Krishnan, N.; Duerr, D.D.; Riggins, J.J. Cold tolerance and invasive potential of the redbay ambrosia beetle (Xyleborus glabratus) in the eastern United States. Biol. Invasions 2018, 20, 995–1007. [Google Scholar] [CrossRef]
- Ploetz, R.C.; Hughes, M.A.; Kendra, P.E.; Fraedrich, S.W.; Carrillo, D.; Stelinski, L.L.; Hulcr, J.; Mayfield, A.E., III; Dreaden, T.J.; Crane, J.H.; et al. Recovery plan for laurel wilt of avocado, caused by Raffaelea lauricola. Plant Health Prog. 2017, 18, 51–77. [Google Scholar] [CrossRef]
- Ploetz, R.C.; Kendra, P.; Choudhury, R.A.; Rollins, J.; Campbell, A.; Garrett, K.; Hughes, M.; Dreaden, T. Laurel wilt in native and agricultural ecosystems: Understanding the drivers and scales of complex pathosystems. Forests 2017, 8, 48. [Google Scholar] [CrossRef]
- Hulcr, J.; Lou, Q.-Z. The redbay ambrosia beetle (Coleoptera: Curculionidae) prefers Lauraceae in its native range: Records from the Chinese national insect collection. Fla. Entomol. 2013, 96, 1595–1596. [Google Scholar] [CrossRef]
- Harrington, T.C.; Yun, H.Y.; Lu, S.S.; Goto, H.; Fraedrich, S.W. Isolations from the redbay ambrosia beetle, Xyleborus glabratus, confirm that the laurel wilt pathogen, Raffalea lauricola, originated in Asia. Mycologia 2011, 103, 1028–1036. [Google Scholar] [CrossRef] [PubMed]
- Hulcr, J.; Black, A.; Prior, K.P.; Chen, C.-Y.; Li, H.-F. Studies of ambrosia beetles in their native ranges help predict invasion impact. Fla. Entomol. 2017, 100, 257–261. [Google Scholar] [CrossRef]
- Ploetz, R.C.; Hulcr, J.; Wingfield, M.; de Beer, Z.W. Ambrosia and bark beetle-associated tree diseases: Black Swan events in tree pathology? Plant Dis. 2013, 95, 856–872. [Google Scholar] [CrossRef]
- Fraedrich, S.W.; Harrington, T.C.; Best, G.S. Xyleborus glabratus attacks and systemic colonization by Raffaelea lauricola associated with dieback of Cinnamomum camphora in the southeastern United States. For. Pathol. 2015, 45, 60–70. [Google Scholar] [CrossRef]
- Ploetz, R.C.; Thant, Y.Y.; Hughes, M.A.; Dreaden, T.J.; Konkol, J.L.; Kyaw, A.T.; Smith, J.A.; Harmon, C.L. Laurel wilt, caused by Raffaelea lauricola, is detected for the first time outside the southeastern USA. Plant Dis. 2016, 100, 2166. [Google Scholar] [CrossRef]
- Wuest, C.E.; Harrington, T.C.; Fraedrich, S.W.; Yun, H.-Y.; Lu, S.-S. Genetic variation in native populations of the laurel wilt pathogen, Raffaelea lauricola, in Taiwan and Japan and the introduced population in the United States. Plant Dis. 2017, 101, 619–628. [Google Scholar] [CrossRef]
- Dreaden, T.J.; Davis, J.M.; Harmon, C.L.; Ploetz, R.C.; Palmateer, A.J.; Soltis, P.S.; Smith, J.A. Development of multilocus PCR assays for Raffaelea lauricola, causal agent of laurel wilt disease. Plant Dis. 2014, 98, 379–383. [Google Scholar] [CrossRef]
- Ploetz, R.C.; Konkol, J.L.; Narvaez, T.; Duncan, R.E.; Saucedo, R.J.; Campbell, A.; Mantilla, J.; Carrillo, D.; Kendra, P.E. Presence and prevalence of Raffaelea lauricola, cause of laurel wilt, in different species of ambrosia beetles in Florida, USA. J. Econ. Entomol. 2017, 110, 347–354. [Google Scholar]
- Duong, T.A.; de Beer, Z.W.; Wingfield, B.D.; Wingfield, M.J. Mating type markers reveal high levels of heterothallism in Leptographium sensu lato. Fungal Biol. 2016, 120, 538–546. [Google Scholar] [CrossRef]
- Holleley, C.E.; Geerts, P.G. Multiplex Manager 1.0: A crossplatform computer program that plans and optimizes multiplex PCR. BioTechniques 2009, 46, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Drost, D.R.; Novaes, E.; Boaventura-Novaes, C.; Benedic, C.I.; Brown, R.S.; Yin, T.; Tuskan, G.A.; Kirst, M. A microarray-based genotyping and genetic mapping approach for highly heterozygous outcrossing species enables localization of a large fraction of the unassembled Populus trichocarpa genome sequence. Plant J. 2009, 58, 1054–1067. [Google Scholar] [CrossRef] [PubMed]
- Kamvar, Z.N.; Tabima, J.F.; Grünwald, N.J. Poppr: An R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2014, 2, e281. [Google Scholar] [CrossRef] [PubMed]
- Kamvar, Z.N.; Brooks, J.C.; Grünwald, N.J. Novel R tools for analysis of genome-wide population genetic data with emphasis on clonality. Front. Genet. 2015, 6, 208. [Google Scholar] [CrossRef]
- Excoffier, L.; Smouse, P.E.; Quattro, J.M. Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics 1992, 131, 479–491. [Google Scholar]
- Jombart, T.; Collins, C. A Tutorial for Discriminant Analysis of Principal Components (dapc) Using Adegenet 2.0.0. Available online: http://adegenet.r-forge.r-project.org/files/tutorial-dapc.pdf (accessed on 2 June 2018).
- Bruvo, R.; Michiels, N.K.; D’Souza, T.G.; Schulenburg, H. A simple method for the calculation of microsatellite genotype distances irrespective of ploidy level. Mol. Ecol. 2004, 13, 2101–2106. [Google Scholar] [CrossRef] [PubMed]
- Grünwald, N.J.; Kamvar, Z.N.; Everhart, S.E.; Tabima, J.F.; Knaus, B.J. Population Genetics and Genomics in R. Available online: http://grunwaldlab.github.io/Population_Genetics_in_R/ (accessed on 2 June 2018).
- Brown, A.H.D.; Feldman, M.W.; Nevo, E. Multilocus structure of natural populations of Hordeum spontaneum. Genetics 1980, 96, 523–536. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
| Multiplex PCR | SSR Loci x | Error Rate (Incorrect Calls/Total Calls) | Alleles |
|---|---|---|---|
| 1 | NB6 | 0/15 | 3 |
| 0DS | 0/15 | 4 | |
| ZWC | 1 */15 | 2 | |
| 2 | OCT | 4/13 | |
| F8I | 0/13 | 3 | |
| 3 | ZBI | 0/15 | 5 |
| KTR | 0/15 | 6 | |
| 4 | P86 | 0/14 | 3 |
| 46Z | 2/14 | ||
| 5 | QI5 | 1/17 | |
| V5I | 0/17 | 5 | |
| 6 | EC6 | 0/13 | 4 |
| 03B | 0/13 | 4 | |
| IFW | 1 */13 | 6 | |
| 7 | X21 | - | |
| 9V8 | - | ||
| 8 | MNT | 1 */15 | 3 |
| REH | 2/14 |
| No Clone Correction | With Clone Correction | |||||||
|---|---|---|---|---|---|---|---|---|
| df | Sigma | % Variance | p | df | Sigma | % Variance | p | |
| Between Populations | 3 | 2.98 | 77 | 0.001 | 3 | 1.66 | 54 | 0.001 |
| Within Population | 55 | 0.86 | 22 | 12 | 1.42 | 46 | ||
| Total | 58 | 3.85 | 15 | 3.07 | ||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dreaden, T.J.; Hughes, M.A.; Ploetz, R.C.; Black, A.; Smith, J.A. Genetic Analyses of the Laurel Wilt Pathogen, Raffaelea lauricola, in Asia Provide Clues on the Source of the Clone that is Responsible for the Current USA Epidemic. Forests 2019, 10, 37. https://doi.org/10.3390/f10010037
Dreaden TJ, Hughes MA, Ploetz RC, Black A, Smith JA. Genetic Analyses of the Laurel Wilt Pathogen, Raffaelea lauricola, in Asia Provide Clues on the Source of the Clone that is Responsible for the Current USA Epidemic. Forests. 2019; 10(1):37. https://doi.org/10.3390/f10010037
Chicago/Turabian StyleDreaden, Tyler J., Marc A. Hughes, Randy C. Ploetz, Adam Black, and Jason A. Smith. 2019. "Genetic Analyses of the Laurel Wilt Pathogen, Raffaelea lauricola, in Asia Provide Clues on the Source of the Clone that is Responsible for the Current USA Epidemic" Forests 10, no. 1: 37. https://doi.org/10.3390/f10010037
APA StyleDreaden, T. J., Hughes, M. A., Ploetz, R. C., Black, A., & Smith, J. A. (2019). Genetic Analyses of the Laurel Wilt Pathogen, Raffaelea lauricola, in Asia Provide Clues on the Source of the Clone that is Responsible for the Current USA Epidemic. Forests, 10(1), 37. https://doi.org/10.3390/f10010037