
Atom Filtering Algorithm and GPU-Accelerated Calculation of Simulation Atomic Force Microscopy Images


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
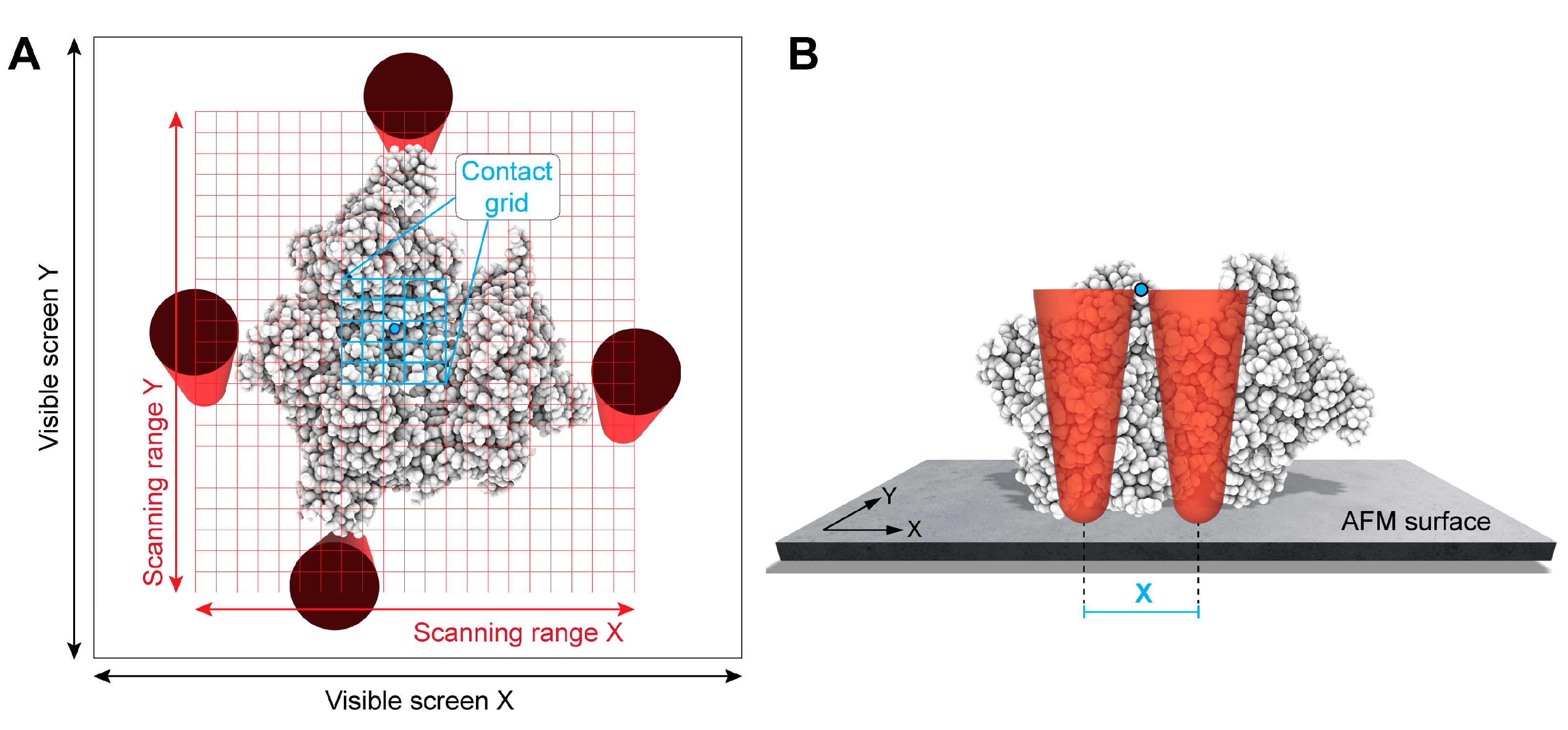
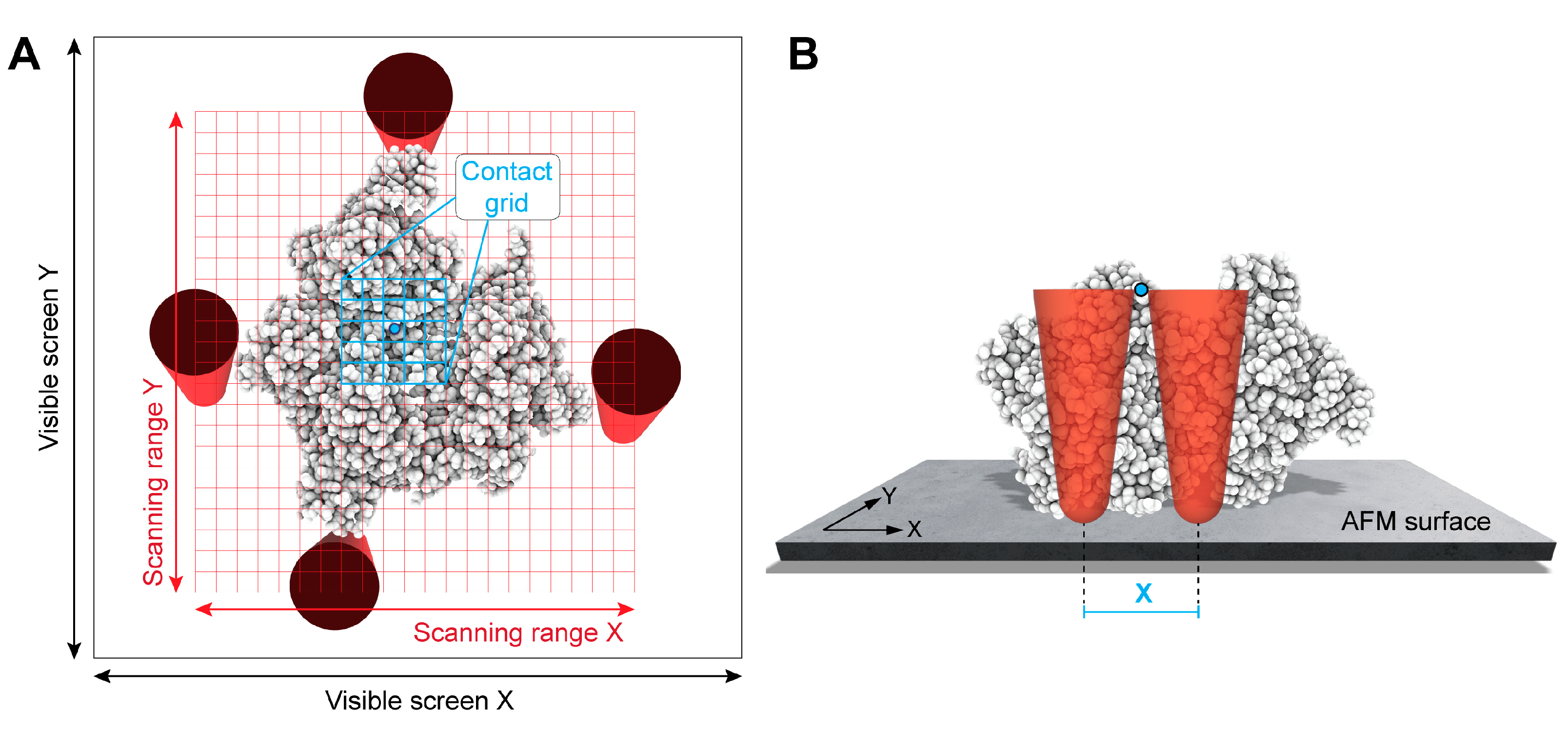
2.1. Primitive X-Y Filtering
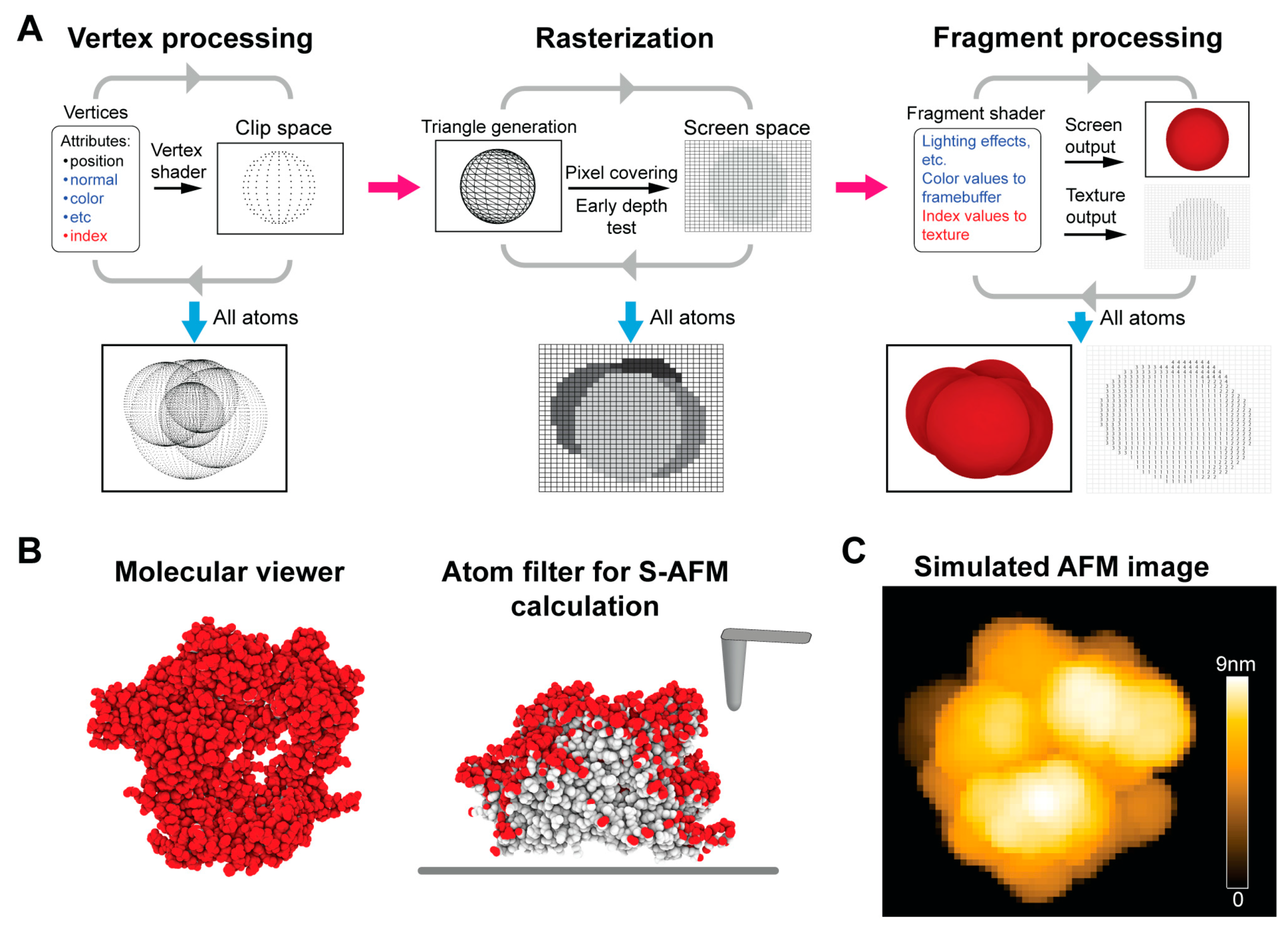
2.2. GPU-Based Lateral Z Direction Filtering of Surface Atoms
2.3. GPU-Based Computation of Simulated AFM Images
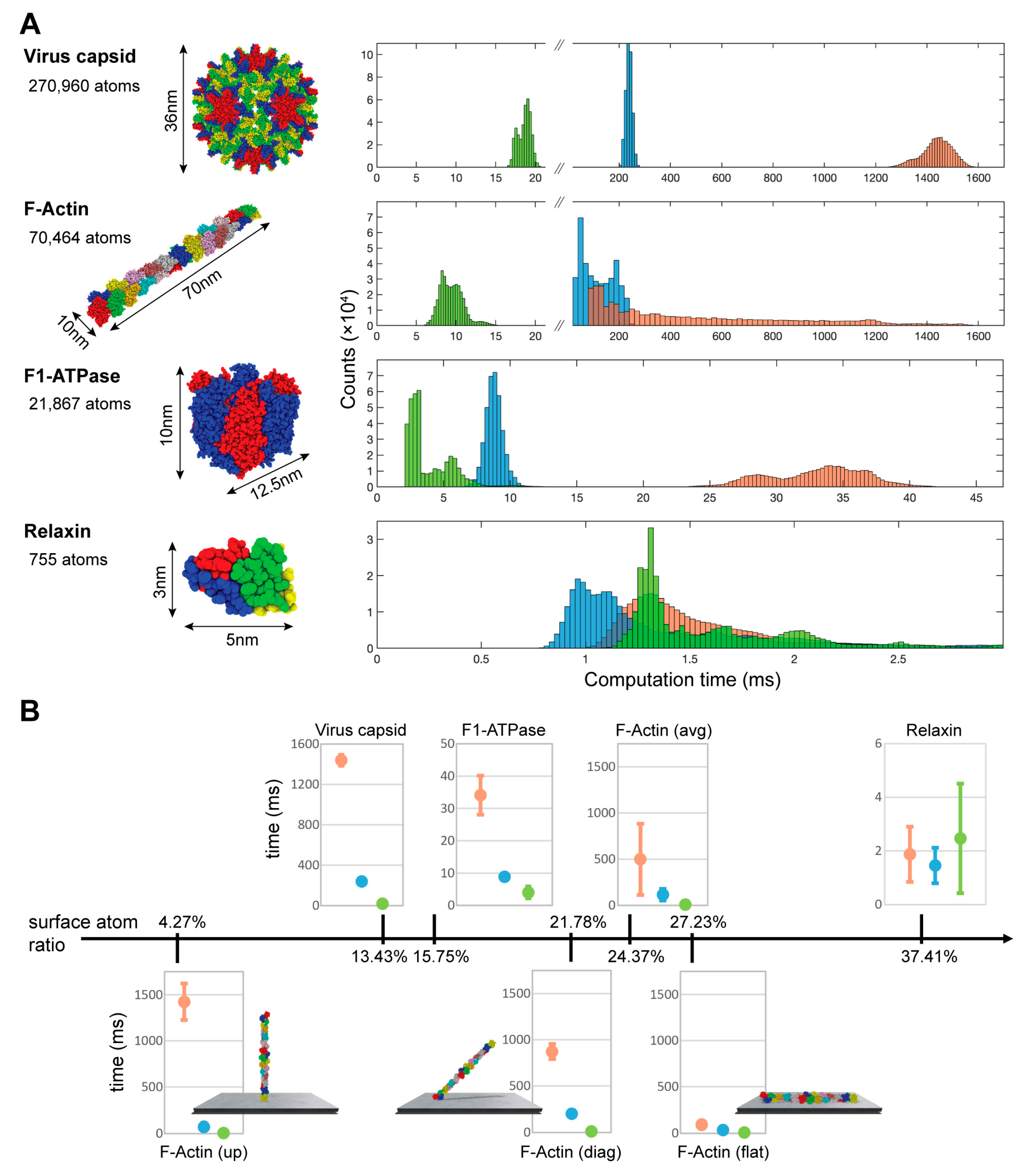
3. Results
Quantification of the Computational Efficiency Gain
- The hepatitis B virus capsid cryo-EM ball-shaped structure with a diameter of ~36 nm and a total of 270,960 atoms (constructed from PDB 6BVF);
- A model of the actin filament structure containing 24 subunits and having a rod-shape with ~10 nm diameter, ~70 nm length, and a total of 70,464 atoms (from Ref. [25]);
- The rotor-less F1-ATPase motor cube-like structure with lengths of ~10 and ~12.5 nm and a total of 21,867 atoms (PDB 1SKY);
- The small global-shaped relaxin protein with lengths of ~3 nm and ~5 nm with just 755 atoms (PDB 6RLX).
4. Discussion
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Müller, D.J.; Dufrêne, Y.F. Atomic force microscopy as a multifunctional molecular toolbox in nanobiotechnology. Nat. Nanotech. 2008, 3, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Ando, T.; Uchihashi, T.; Scheuring, S. Filming biomolecular processes by high-speed atomic force microscopy. Chem. Rev. 2014, 114, 3120–3188. [Google Scholar] [CrossRef] [PubMed]
- Ando, T. Directly watching biomolecules in action by high-speed atomic force microscopy. Biophys. Rev. 2017, 9, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Uchihashi, T.; Ganser, C. Recent advances in bioimaging with high-speed atomic force microscopy. Biophys. Rev. 2020, 12, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Casuso, I.; Redondo-Morata, L.; Rico, F. Biological physics by high-speed atomic force microscopy. Philos. Trans. R. Soc. A 2020, 378, 20190604. [Google Scholar] [CrossRef] [PubMed]
- Ando, T. High-Speed Atomic Force Microscopy in Biology, 1st ed.; Springer: Berlin/Heidelberg, Germany, 2022; pp. 1–319. [Google Scholar] [CrossRef]
- wwPDBconsortium. Protein data bank: The single global archive for 3D macromolecular structure data. Nucleic Acids Res. 2019, 47, D520–D528. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Kenzaki, H.; Koga, N.; Hori, N.; Kanada, R.; Li, W.; Okazaki, K.; Yao, X.-Q.; Takada, S. CafeMol: A coarse-grained biomolecular simulator for simulating proteins at work. J. Chem. Theory Comput. 2011, 7, 1979–1989. [Google Scholar] [CrossRef]
- Takada, S.; Kanada, R.; Tan, C.; Terakawa, T.; Li, W.; Kenzaki, H. Modeling structural dynamics of biomolecular complexes by coarse-grained molecular simulations. Acc. Chem. Res. 2015, 48, 3026–3035. [Google Scholar] [CrossRef]
- Pak, A.J.; Voth, G.A. Advances in coarse-grained modeling of macromolecular complexes. Curr. Opin. Struct. Biol. 2018, 52, 119–126. [Google Scholar] [CrossRef]
- Togashi, Y.; Flechsig, H. Coarse-grained protein dynamics studies using elastic network models. Int. J. Mol. Sci. 2018, 19, 3899. [Google Scholar] [CrossRef] [PubMed]
- Flechsig, H.; Mikhailov, A.S. Simple mechanics of protein machines. J. R. Soc. Interface 2019, 16, 20190244. [Google Scholar] [CrossRef] [PubMed]
- Niina, T.; Fuchigami, S.; Takada, S. Flexible fitting of biomolecular structures to atomic force microscopy images via biased molecular simulations. J. Chem. Theory Comput. 2020, 16, 1349–1358. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, B.; Miyashita, O.; Tama, F. Reconstruction of low-resolution molecular structures from simulated AFM force microscopy images. Biochim. Biophys. Acta—Gen. Subj. 2020, 1864, 129420. [Google Scholar] [CrossRef]
- Niina, T.; Matsunaga, Y.; Takada, S. Rigid-body fitting to atomic force microscopy images for inferring probe shape and biomolecular structure. PLoS Comput. Biol. 2021, 17, e1009215. [Google Scholar] [CrossRef]
- Dasgupta, B.; Miyashita, O.; Uchihashi, T.; Tama, F. Reconstruction of three-dimensional conformations of bacterial ClpB from high-speed atomic-force-microscopy images. Front. Mol. Biosci. 2021, 8, 704274. [Google Scholar] [CrossRef]
- Amyot, R.; Marchesi, A.; Franz, C.M.; Casuso, I.; Flechsig, H. Simulation atomic force microscopy for atomic reconstruction of biomolecular structures from resolution-limited experimental images. PLoS Comput. Biol. 2022, 18, e1009970. [Google Scholar] [CrossRef]
- Ogane, T.; Noshiro, D.; Ando, T.; Yamashita, A.; Sugita, Y.; Matsunaga, Y. Development of hidden Markov modeling method for molecular orientations and structure estimation from high-speed atomic force microscopy time-series images. PLoS Comput. Biol. 2022, 18, e1010384. [Google Scholar] [CrossRef]
- Flechsig, H.; Ando, T. Protein dynamics by the combination of high-speed AFM and computational modeling. Curr. Opin. Struct. Biol. 2023, 80, 102591. [Google Scholar] [CrossRef]
- Owens, J.D.; Houston, M.; Luebke, D.; Green, S.; Stone, J.E.; Phillips, J.C. GPU computing. Proc. IEEE 2008, 96, 879–899. [Google Scholar] [CrossRef]
- Amyot, R.; Flechsig, H. BioAFMviewer: An interactive interface for simulated AFM scanning of biomolecular structures and dynamics. PLoS Comput. Biol. 2020, 16, e1008444. [Google Scholar] [CrossRef] [PubMed]
- Maughan, C.; Wloka, M. Vertex Shader Introduction; Technical report; NVIDIA Corporation: Santa Clara, CA, USA, 2001. [Google Scholar]
- Ebner, M.; Reinhardt, M.; Albert, J. Evolution of vertex and pixel shaders. In Genetic Programming. EuroGP 2005. Lecture Notes in Computer Science; Keijzer, M., Tettamanzi, A., Collet, P., van Hemert, J., Tomassini, M., Eds.; Springer: Berlin/Heidelberg, Germany, 2005; Volume 3447. [Google Scholar] [CrossRef]
- Pirani, A.; Vinogradova, M.V.; Curmi, P.M.G.; King, W.A.; Fletterick, R.J.; Craig, R.; Tobacman, L.S.; Xu, C.; Hatch, V.; Lehman, W. An atomic model of the thin filament in the relaxed and Ca2+-activated states. J. Mol. Biol. 2006, 357, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Fuchigami, S.; Niina, T.; Takada, S. Particle filter method to integrate high-speed atomic force microscopy measurements with biomolecular simulations. J. Chem. Theor. Comput. 2020, 16, 6609–6619. [Google Scholar] [CrossRef] [PubMed]
- Fuchigami, S.; Niina, T.; Takada, S. Case report: Bayesian statistical interference of experimental parameters via biomolecular simulations: Atomic force microscopy. Front. Mol. Biosci. 2021, 8, 636940. [Google Scholar] [CrossRef]
- Matsunaga, Y.; Fuchigami, S.; Ogane, T.; Takada, S. End-to-end differentiable blind tip reconstruction for noisy atomic force microscopy images. Sci. Rep. 2023, 13, 129. [Google Scholar] [CrossRef]
- Amyot, R.; Kodera, N.; Flechsig, H. BioAFMviewer software for simulation atomic force microscopy of molecular structures and conformational dynamics. J. Struct. Biol. X 2023, 7, 100086. [Google Scholar] [CrossRef]
- Kessenich, J.; Sellers, G.; Shreiner, D. OpenGL Programming Guide: The Official Guide to Learning OpenGL, Version 4.5 with SPIR-V; Addison-Wesley Professional: Glenview, IL, USA, 2016. [Google Scholar]
- Rost, R.J.; Licea-Kane, B.; Ginsburg, D.; Kessenich, J.; Lichtenbelt, B.; Malan, H.; Weiblen, M. OpenGL Shading Language; Pearson Education: London, UK, 2009. [Google Scholar]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amyot, R.; Kodera, N.; Flechsig, H. Atom Filtering Algorithm and GPU-Accelerated Calculation of Simulation Atomic Force Microscopy Images. Algorithms 2024, 17, 38. https://doi.org/10.3390/a17010038
Amyot R, Kodera N, Flechsig H. Atom Filtering Algorithm and GPU-Accelerated Calculation of Simulation Atomic Force Microscopy Images. Algorithms. 2024; 17(1):38. https://doi.org/10.3390/a17010038
Chicago/Turabian StyleAmyot, Romain, Noriyuki Kodera, and Holger Flechsig. 2024. "Atom Filtering Algorithm and GPU-Accelerated Calculation of Simulation Atomic Force Microscopy Images" Algorithms 17, no. 1: 38. https://doi.org/10.3390/a17010038
APA StyleAmyot, R., Kodera, N., & Flechsig, H. (2024). Atom Filtering Algorithm and GPU-Accelerated Calculation of Simulation Atomic Force Microscopy Images. Algorithms, 17(1), 38. https://doi.org/10.3390/a17010038