1. Introduction
Phylogenetic tree estimation is a basic part of many biological research studies, due to the centrality of the evolutionary perspective in biology. These analyses are typically based on probabilistic models of evolution (e.g., the Generalized Time Reversible Model [
1]), and maximum likelihood (ML) tree estimation under these models is a standard approach. Yet, ML tree estimation is NP-hard [
2], so exact solutions are infeasible; hence, local search heuristics are used that seek good (hopefully close to globally optimal) solutions.
Over the last several decades, many different software packages have been developed for ML tree estimation, with several of these (e.g., RAxML [
3,
4], IQ-TREE [
5], PAUP* [
6], PhyML [
7], and FastTree 2 [
8]) in frequent use. Studies comparing the ML methods have generally been limited to relatively small trees (i.e., up to 100 or so leaves), but they have found that, in general, RAxML and IQ-TREE produce better ML scores than other heuristics, and that FastTree 2 has been the fastest of these heuristics [
9,
10,
11]. In general, FastTree 2 (more commonly referred to as “FastTree”) is the only ML heuristic that scales well to very large numbers of sequences (e.g., more than 10,000), but it is not as commonly used because of its relatively poor ML scores as compared to RAxML and other heuristics.
At the same time, the sizes of estimated phylogenies are increasing, and the interest in constructing very large phylogenetic trees has also increased. One reason for this is that advances in sequencing technologies have made it relatively inexpensive to produce large sequence datasets. A recent example is the fast-increasing number of SARS-CoV-2 sequences, for which “keeping the phylogenetic trees up to date is becoming increasingly difficult” [
12]. Another reason for the increased size in phylogenies is the realization that phylogenetic accuracy is improved through dense taxonomic sampling, so that adding sequences to a dataset is expected to help provide improved resolution around challenging internal nodes in the phylogeny [
13]. A third reason is that many genes evolve with duplications and losses, so that gene families have multiple copies of each gene within a given individual and the true “gene family tree” will have multiple leaves for each species. In consequence, the size of some gene family trees can be in the thousands or more [
14]. Thus, for multiple reasons, large-scale gene trees are increasingly of interest to biologists.
The question of interest in this study is how well the best ML codes perform with respect to accuracy and computational effort on large datasets, and whether better methods can be designed. Liu et al. [
9] compared RAxML and FastTree 2 on simulated datasets for single genes with 1000 or more sequences using many different estimated alignments, and found that the two methods had essentially the same topological accuracy. Lees et al. [
15] used simulations to explore ML heuristics and found RAxML and IQ-TREE to be close in accuracy and both better than FastTree 2. However, their study examined genome-scale data, rather than single gene data, which makes the study not quite relevant to the question that we ask here. Two studies [
16,
17] evaluated FastTree 2 and RAxML on datasets, where some of the sequences are fragmentary, and both found that FastTree 2 was less accurate than RAxML when the input multiple sequence alignment had a high proportion of fragmentary sequences.
Overall, these studies have shown that IQ-TREE and RAxML are both very good ML heuristics with respect to ML scores, but which of these two methods is better in terms of tree topology is not clear. Furthermore, while all of the studies have shown that FastTree 2 is indeed very fast, it is not very good at ML score optimization, and it is less topologically accurate than RAxML when given datasets with a high proportion of fragmentary sequences. However, whether there are other conditions where FastTree 2 also degrades in topological accuracy when compared to RAxML is not known, and it is also not known how IQ-TREE handles fragmentary datasets.
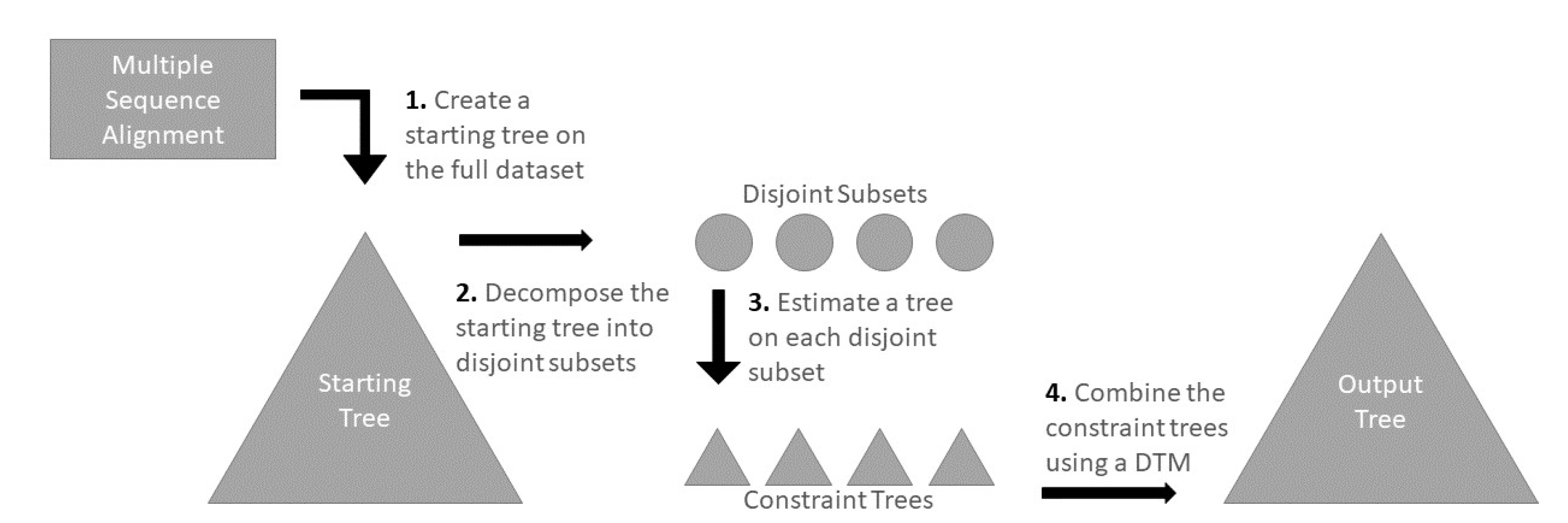
Divide-and-conquer techniques to scale phylogeny estimation methods to large datasets have also been developed, with the recent "Disjoint Tree Mergers” (DTMs) among the most promising. The DTM pipelines operate by dividing the set of taxa (e.g., species or individuals that will label the leaves of the final tree) into disjoint sets, computing trees on the subsets using a selected phylogeny estimation method, and then using the selected DTM to combine the subset trees. Because the subset trees are disjoint, this requires providing the DTM with some auxiliary information (e.g., a distance matrix or estimated guide tree) that spans all of the subsets.
Note that, in a DTM pipeline, the subset trees are treated as absolute constraints on the output tree, so that the final tree is required to induce the subset trees. The design of DTMs is motivated by the observation that the most accurate methods are often the most computationally intensive, so that, while they can be used easily on small datasets, it becomes infeasible to use the most accurate methods on even just moderately large datasets. Hence, the final tree can have high accuracy while being computationally feasible if the combination step (how the subset trees are combined together) can be performed in polynomial time without (a considerable) loss of accuracy. Thus, DTMs are designed to be used in divide-and-conquer pipelines and they have the potential to provide advantages for phylogeny estimation when the datasets are too large for the most accurate methods to be used on the entire dataset.
The first of these DTMs was Constrained-INC [
18], which was followed by NJMerge [
19], TreeMerge [
20], and the Guide Tree Merger (GTM) [
21]. TreeMerge is a direct improvement on NJMerge (that can fail on some inputs with three or more constraint trees due to its algorithmic design). However, of these methods, only Constrained-INC allows full “blending” (which means that, after merging, the subset trees can be intermingled). TreeMerge allows for partial blending, but GTM does not allow any blending at all (in GTM, the subset constraint trees are combined by adding edges between the subset trees). Thus, the different DTMs have different algorithmic designs and constraints.
The importance of blending can be seen in a simple example. Suppose that the true tree is a caterpillar tree, with leaves labelled , and we are given the true tree on the even-numbered leaves and the the true tree on the odd numbered leaves. These two smaller subset trees can be combined together into the original tree, but only if full blending is allowed. For example, if the two trees can only be combined by adding an edge between them, then the best that can be done will still have a large topological distance to the original true tree.
Constrained-INC, TreeMerge, and GTM have all been shown to provide benefits for species tree estimation from multi-locus datasets, where gene trees can differ from the species tree due to incomplete lineage sorting (ILS) [
22]. For example, [
21] showed that using GTM with ASTRAL [
23] or concatenation using RAxML maintained or improved the accuracy and reduced the running time on large datasets.
However, less is known regarding using DTM methods for gene tree estimation. A limited study conducted by Le et al. [
24] explored the use of a pipeline using Constrained-INC for gene tree estimation. Their pipeline divides the input sequence dataset into subsets using an estimated tree and then computes subset constraint trees using either RAxML or FastTree 2. This divide-and-conquer approach is referred to as “INC-ML” in [
24], where “INC” refers to the incremental technique that it uses of adding the species to the growing tree, one-by-one, while obeying the input constraint trees that it computes.
Le et al. [
24] showed that the INC-ML tree was much less accurate than RAxML and often not as accurate as FastTree 2. Overall, their results of using Constrained-INC for gene tree estimation were disappointing. However, their study had several important limitations: (1) the study did not evaluate any other DTM method for gene tree estimation beyond Constrained-INC, (2) only a few variations to the parameters of the pipeline were considered, and (3) only one model condition had more than 1000 sequences. These limitations are significant, since other approaches for designing divide-and-conquer pipelines, along with the use of other DTM methods, might provide better accuracy, and it is also possible that the most important usage of DTM pipelines might be found on the largest datasets.
Being motivated by the new DTM methods that have been developed in the last years, we aim to revisit the question of whether DTM pipelines can be useful in large-scale maximum likelihood gene tree estimation. We include three ML heuristics (RAxML-NG, IQ-TREE 2, and FastTree 2) and compare them to divide-and-conquer pipelines using the three current leading DTMs (TreeMerge, GTM, and Constrained-INC). We explore topological accuracy and runtime on simulated datasets with 1000 to 50K sequences. Our study shows that the improved DTM pipelines we developed for this study are more accurate than FastTree 2 and IQ-TREE 2 under the model conditions we explored (which constrain the running time and available memory to 64 GB). Furthermore, our new DTM pipelines are faster than RAxML-NG and they can analyze ultra-large datasets that IQ-TREE 2 or RAxML-NG either fail on or return a poor tree, potentially as a result of the limitation on computational resources. However, for those datasets small enough for RAxML-NG, the DTM pipelines that we developed do not reliably match or improve the accuracy of RAxML-NG, which suggests that further development is needed to achieve the goal of enabling fast, scalable, and highly accurate ML gene tree estimation. Therefore, our study also provides insight into design strategies for use in divide-and-conquer approaches to large scale phylogeny estimation, and it provides directions for future research. Finally, our study, although limited to a small number of model conditions, enables us to make some preliminary recommendations about method choice using existing methods and DTM pipelines.
2. Materials and Methods
2.1. Overview
We compared three well known maximum likelihood codes (RAxML-NG v.1.0.1, IQ-TREE v.2.0.6, and FastTree 2 v. 2.1.10) to divide-and-conquer pipelines using three Disjoint Tree Merger (DTM) methods (Constrained-INC, TreeMerge, and GTM, commit IDs for github repositories that are provided in the
Appendix C).
Table 1 provides an overview of the DTM methods that we compared and
Figure 1 describes the DTM pipeline.
We used simulated datasets to evaluate phylogeny estimation methods, so that the true alignment and true tree are known. In each case, we provided the true alignment to the different methods and recorded the tree error as well as the running time. Our simulated datasets come from five model conditions and they range in terms of size and difficulty. One of these model conditions evolved under the standard i.i.d. Generalized Time Reversible (GTR) model equipped with insertions and deletions, but others evolved under models with additional complexity (e.g., heterotachy across the gene and across the tree, or under a model that includes selection). We also specifically explored a model condition that contains fragmentary sequences (e.g., a case that arises in real biological analyses when combining full-length sequences with reads or partially assembled sequences as a result of using high-throughput sequencing technologies).
We performed three experiments. The initial experiment was performed on just one model condition to determine (a) how we would define distances for use within two of the DTM pipelines that require guide distance matrices and (b) the maximum subset size for the decomposition. The second experiment was performed on three model conditions with 1000 to 2341 sequences to evaluate the impact of the starting tree on DTM pipelines and compare them to the three ML heuristics that we selected. Our third experiment evaluated the two faster pipelines with respect to the speed and accuracy on datasets with 10K and 50K sequences, and then compared them to the three ML heuristics that we selected.
2.2. Methods
2.2.1. Maximum Likelihood Codes
We use three existing codes for maximum likelihood (ML): FastTree 2, RAxML-NG, and IQ-TREE 2; for the sake of simplicity, we will subsequently refer to these methods as FastTree, RAxML, and IQ-TREE. All of these methods are run under the GTR+G (i.e., GTR with gamma-distributed rates across sites) model of evolution.
FastTree does not make a strong attempt to find the best solution to ML and, instead, stops the search (using nearest neighbor interchange, or NNI, moves) after a polynomial number of moves. This approach makes it faster than the other methods, which continue searching until there is evidence of having found a good local optimum. Moreover, in our study, FastTree completed on all datasets. The other methods did not complete on some of the datasets within the allowed run-time limit (24 h for the datasets with at most 2341 sequences, and one week for the datasets with 10K or 50K sequences), using the available memory (generally limited to 64 Gb). In such cases, we used the best tree (i.e., the tree with the best ML score, according to the code) found for that method on that dataset.
2.2.2. Disjoint Tree Mergers Pipelines
Given an input dataset (here, a multiple sequence alignment), a DTM pipeline has four steps:
Step 1: compute a starting tree
Step 2: use the starting tree to decompose the set of sequences into disjoint sets
Step 3: construct the trees on the different sets using a selected phylogeny estimation method, and
Step 4: merge the trees using a DTM method along with some auxiliary information (e.g., guide tree or distance matrix)
Thus, Step 4 is where the DTM method is used. We begin by describing how TreeMerge, Constrained-INC, and GTM operate at a very high level, and then describe how they are used in divide-and-conquer pipelines.
The input to each DTM is a set of disjoint subset trees, which are treated as constraint trees, as well as some auxiliary information. For the purpose of this study, each DTM is able to use the starting tree for its auxiliary information, and so we describe these methods with that modification.
2.2.3. TreeMerge
TreeMerge takes as input the set of
k disjoint trees and a guide tree, and the guide tree is used to compute a matrix of leaf-to-leaf distances. TreeMerge then operates in two stages, where the first stage produces a set of
pairwise merged constraint trees, and the second stage merges these
constraint trees into a tree on the full dataset. The first stage is operated, as follows. First, a spanning tree on the input constraint trees is computed using the distance matrix, and the edges of the spanning tree then define which pairs of constraint trees will merged into larger (and now overlapping) trees. These overlapping pairs are then merged together using NJMerge, while using the provided or computed distance matrix. Note that NJMerge allows for the full blending of two trees. In Stage 2, the larger constraint trees are combined, two at a time, until all of the larger constraint trees are merged together, which results in a tree on the full dataset that is compatible with all of the original input constraint trees. To merge two trees that overlap, TreeMerge computes the branch lengths on the trees and then uses these branch lengths to determine how to merge the trees on the region in which they overlap. This step allows for a partial, but not full, blending (see [
20] for full details). We also made a small modification to the TreeMerge approach. Specifically, we discovered that the technique that was used by TreeMerge (i.e., PAUP*) to compute branch lengths in order to merge overlapping trees was a computationally expensive step. We evaluated the use of RAxML instead of PAUP* for this branch length estimation and found it reduced the running time without changing accuracy (see
Table A2). Hence, we made this modification in our TreeMerge analyses.
2.2.4. Constrained-INC
Constrained-INC takes as input the set of disjoint constraint trees and a starting tree, and it incrementally builds a tree that obeys the constraint trees by (a) computing a distance matrix from the starting tree and (b) computing quartet trees that will vote on where to place each species into the growing tree. Although the set of quartet trees Constrained-INC uses can be estimated from a distance matrix or by recomputing quartet trees using an ML method, better accuracy is obtained using induced quartet trees from a starting tree estimated using an ML heuristic [
24], and so we describe the algorithm in this context. Constrained-INC operates by building a tree that obeys the constraint trees by incrementally adding the species, one-by-one, using a distance matrix (which, in our study, is computed from the starting tree). To place a new species into the tree, it uses selected quartet trees that are defined by the starting tree, with a voting scheme that weights the quartet trees using the distance matrix. The final tree is returned once all of the species are included. Each placement is guaranteed to obey the constraint trees, so that the final tree is guaranteed to obey the constraint trees. By design, Constrained-INC (where “INC” refers to incremental) allows full blending.
2.2.5. Guide Tree Merger (GTM)
The input to GTM is a set of constraint trees and a guide tree (in this study, we use the starting tree as the guide tree to GTM). GTM constructs a tree on the full set of species by adding edges between the constraint trees; hence, GTM does not allow any blending. Furthermore, GTM returns a tree T that is guaranteed to minimize the total Robinson–Foulds distance to the guide tree, and it does so in polynomial time.
2.2.6. Steps 1–3 Pipeline Details
To complete the description of the existing DTM pipelines we explore, we now describe Steps 1–3. In Step 1, an initial tree is computed on the alignment using an existing maximum likelihood (ML) tree estimation method. In Step 2, the starting tree is decomposed into subtrees by repeatedly removing centroid edges (i.e., edges that decompose the tree roughly equally into two parts) until the size of each subtree is below a user-specified maximum subset size (this is the same decomposition as used in several multiple sequence alignment methods, including SATé-II [
27], PASTA [
28], and MAGUS [
29]). The leaves in each subtree define a subset of the sequences in the input sequence alignment, and a ML tree is computed on each subset of sequences using the ML method (Step 3). Finally, in Step 4, these subtrees are merged together using the selected DTM method (i.e., TreeMerge, GTM, or Constrained-INC). These DTM methods require auxiliary information, with TreeMerge requiring a distance matrix, GTM requiring a guide tree, and Constrained-INC requiring either a distance matrix or a guide tree that can be used for defining the distance matrix.
For this study, the starting trees are computed using IQ-TREE or FastTree, we decompose these starting trees using the centroid decomposition with varying maximum subset size, and the subset trees are computed using IQ-TREE. We use the starting tree from Step 1 for the guide trees.
2.3. Datasets
Our datasets range in size from 1000 to 50,000 sequences, and they vary in complexity and difficulty; see
Table 2. The RNASim datasets evolve under a model that incorporates fitness and positive selection to maintain the RNA structure [
28], and so evolve under a non-
i.i.d. model. The Cox1-HET dataset evolves with heterotachy across the tree, and it was created for this study. The 1000M1-HF datasets evolve under a standard GTR substitution model with indels, and have a high rate of evolution, and some of the sequences are fragmentary; this makes the inference of the tree challenging for these datasets. In each case, we use the true alignment as input to the tree estimation method. Note that only one of the model conditions that we explore (1000M1-HF) evolves under a standard GTR+indel model. By including models that incorporate selection or variability of the model process across the tree as well as across the sites, we have expanded the model space to incorporate more biological realism.
1000M1-HF. These 1000-sequence datasets are from a prior study [
16], where half of the sequences have been fragmentary. We picked the first five replicates only.
RNASim1000. We sampled five 1000-sequence subsets of the single million-sequence replicate from the RNASim million-sequence dataset studied in [
28].
Cox1-HET. This is a dataset developed explicitly for this study containing 2341 sequences, and described in detail below. We created 10 replicates.
RNASim10K. We sampled 10,000-sequence subsets of the RNASim million-sequence datasets (ten replicates).
RNASim50K. We used the same strategy as for the RNASim10K analysis but sampled 50,000-sequence subsets (ten replicates).
2.3.1. 1000M1-HF
This dataset was used in [
16], and it was created by fragmenting half of the sequences in each replicate of the 1000M1 model condition from [
30], making each fragmentary sequence 25% of the original median sequence length. The reference trees provided with this dataset are the “potentially inferrable model trees”, which means that the zero-event branches (i.e., branches on which no substitution or indel occurs) are collapsed, hence creating a non-binary reference tree. This dataset is publicly available, and we use the first five replicates from this collection.
2.3.2. RNASim1000
RNASim1000 was created for this study by randomly sampling five subsets with 1000 sequences each from the single million-sequence replicate of the RNASim dataset used in [
28] to evaluate multiple sequence alignment methods. Once the sequences were sampled, the reference trees were induced from the original RNASim million-sequence reference tree on the same set of taxa that the sequences represent.
Appendix C provides the code used to generate these sequences.
2.3.3. Cox1-HET
We created a dataset for which the sequences would evolve under different model parameters across the tree, in order to evaluate tree estimation methods in the presence of heterotachy [
31]. We began by estimating a species-level biological
cox1 barcode gene tree (see
Appendix B). The tree of 2341 species was decomposed into 324 subtrees using centroid edge decomposition, with maximum subset size 10, using the decompose.py script (see
Appendix C). For each of the 324 subsets, we estimated the numeric GTR+G parameters using IQ-TREE v1.6.12. Each set of numeric model parameters (i.e., substitution rate matrix, stationary frequencies, and discrete gamma model) was then assigned to the corresponding subset in the model tree following the INDELible format [
32]. Subsequently, we used INDELible V1.03 [
33] to simulate new sequences down the model tree (with 324 different model parameters). Because the proportion of invariant sites cannot be changed across the tree in INDELible V1.03, we fixed this proportion at 15.78%, the overall proportion of invariant sites in the original sequence dataset. We repeated the sequence evolution for 10 replicates, and name this dataset “Cox1-HET”.
2.3.4. RNASim10K
We took the first ten trials of the million-sequence RNASim dataset from [
34] and randomly extracted a sub-sample of 10,000 for each trial. The result is a dataset of 10 replicates of 10,000 sequences. Once the sequences were sampled, the reference trees were induced from the original RNASim million-sequence reference tree on the same set of taxa that the sequences represent.
2.3.5. RNASim50K
We used the same procedure as for the RNASim10K dataset, but sampled subsets with 50,000 sequences, producing 10 replicate datasets.
2.4. Computational Platform
Most of the analyses were run on the Illinois Campus Cluster, with the exception of the TreeMerge analyses on the Cox1-HET dataset as well as the RNASim10k and RNASim50k datasets, which were run on NCSA’s Blue Waters. All of the analyses were allowed between 54 Gb and 64 Gb of memory, an amount that is sufficient for all alignments of the sizes that we study, with the exception of one model conditions which involves 50,000 sequences. Unless otherwise specified, no method came close to being bottlenecked by the limitations on allocated memory.
Most of the multi-threaded methods were allowed between 10 and 16 cores, with few exceptions. Each constraint tree estimation for the 1000M1-HF and RNASim1000 datasets was allowed to run with up to four cores, with IQ-TREE deciding the appropriate number of cores. For the Cox1-HET dataset, constraint trees estimated with IQ-TREE were allowed to run up to 16 cores, and we let IQ-TREE select the appropriate number of cores; most of the constraint tree calculations only used 1–3 cores.
Constrained-INC and GTM were used in their currently available implementations, taken from the developers’ Github repositories, and are single-threaded. TreeMerge was modified to take in pre-estimated RAxML branch lengths (that were computed with two cores), and is otherwise run single-threaded.
2.5. Evaluation Criteria
Our focus is on the topological error in the estimated tree in comparison to the reference tree for the simulation study. Every tree t can be represented by its set of edge-induced bipartitions; hence, the set , where t is the estimated tree and T is the true tree, is the set of “missing branches” or “false negatives”. Conversely, the set of false positives is . The false negative (FN) rate is the number of false negatives divided by the number of internal branches in the true tree, and the false positive (FP) rate is the number of false positives divided by the number of internal edges in the estimated tree. These rates are identical when the estimated and true tree are both fully resolved (i.e., binary), in which case they are also equal to the commonly used Robinson–Foulds (RF) error rate.
1000M1-HF is the only model condition we explore that has non-binary reference trees, where the reference trees are about 0.4% unresolved (
Table A1); all other model conditions have binary reference trees. In addition, all of the estimated trees are also binary. Hence, for the model conditions other than the 1000M1-HF condition, the RF error rate is exactly the same as the FN error rate. We report FN rates for all methods here, and note that the difference between FP and FN rates on the 1000M1-HF datasets is approximately 0.3%. To compute these error rates, we use a script that was coded by Erin K. Molloy (see
Appendix C).
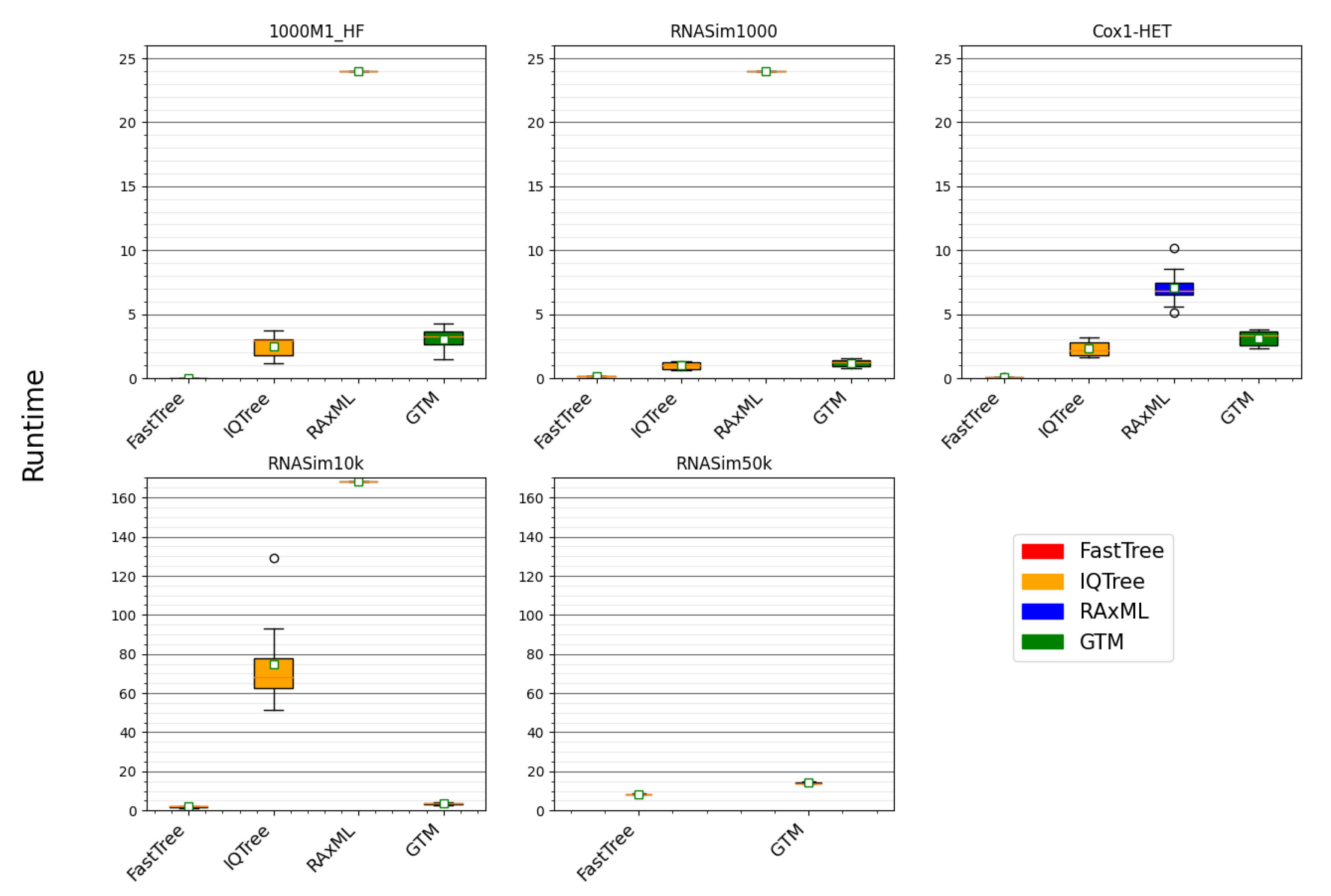
We also evaluate methods with respect to running time. In all of our analyses, we assume a maximum of 16 core parallelism, i.e., when running constraint tree calculations in parallel, only those analyses that will fit under 16 cores are considered to be parallel, assuming that a typical computational platform would have access to 16 cores. In cases where multiple methods are used in stages, such as the DTM pipelines, the same assumption of 16 core parallelism applies. In a DTM pipeline, the run-time calculation is the sum of running times of computing the starting tree, constraint trees, and the running time of disjoint tree merger. No given method or a set of methods running in parallel was allowed to use more than 16 cores.
4. Discussion
This study provides some insights into the conditions that impact the relative accuracy and running times of three leading maximum likelihood (ML) codes, and into the potential for Disjoint Tree Merger (DTM) pipelines to provide improved accuracy and/or scalability when compared to these ML codes. Our simulation included some model conditions that were relatively “easy” (i.e., the RNASim1000 condition) and then model conditions that were more challenging due to the inclusion of fragmentary sequences (i.e., the 1000M1-HF condition), heterotachous sequence evolution model (i.e., Cox1-HET), or very large or ultra-large datasets (i.e., RNASim10K and RNASim50K, respectively).
Our study showed differences between the basic ML heuristics—RAxML, IQ-TREE, and FastTree—that depended, in part, on the dataset size and properties, as well as between the different DTM pipelines. However, the main focus of this study was to determine whether any of the current DTM pipelines provide improved accuracy over the better ML codes in the context of gene tree estimation. Hence, we begin by discussing what we learned about the differences between the three ML codes.
4.1. Comparison of the Three ML Codes
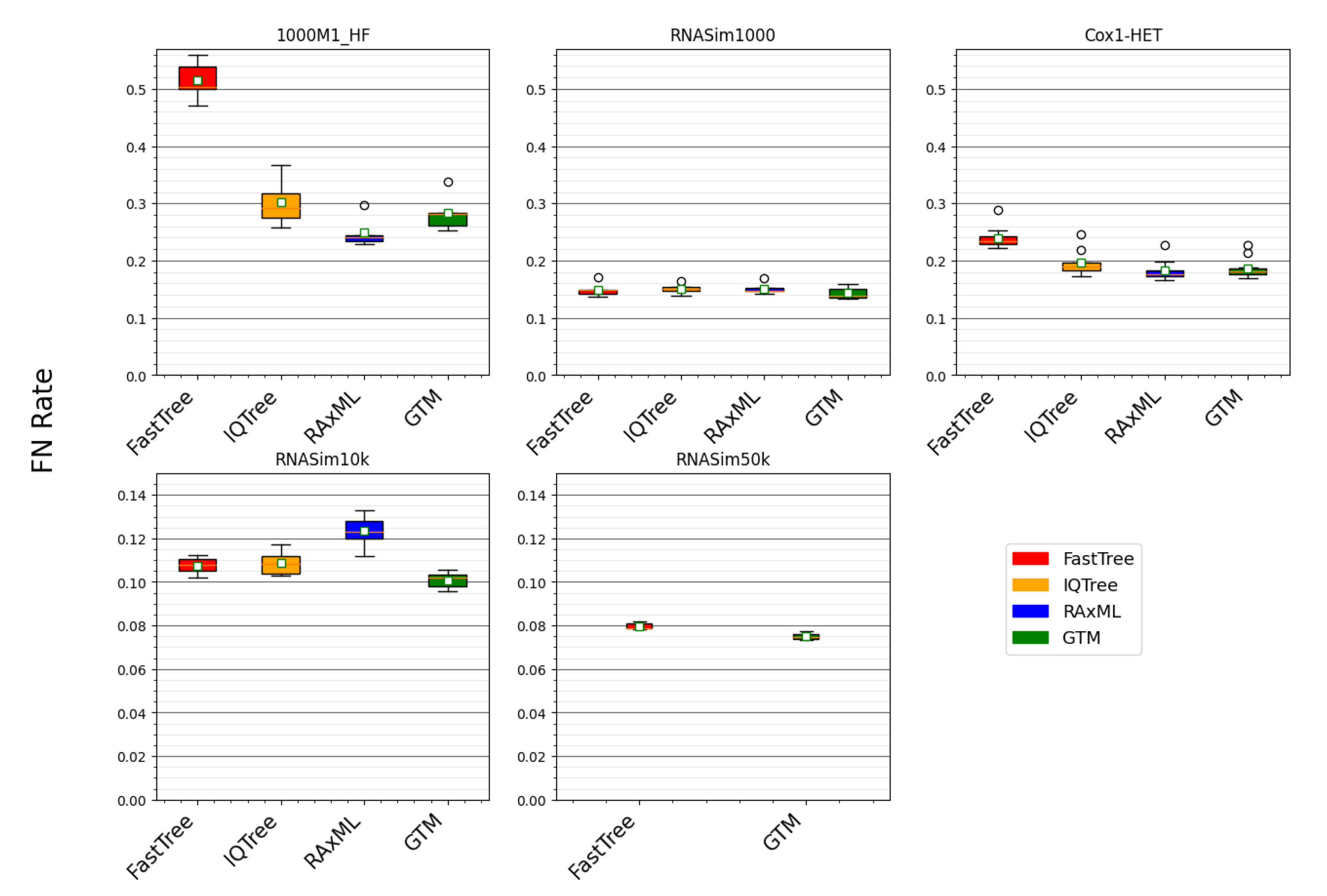
The running time comparison shows RAxML to be clearly the most computationally intensive, followed by IQ-TREE, and then by FastTree, which is, by far, the fastest method. Therefore, we begin by comparing FastTree to the other methods (
Figure 3). On two conditions (i.e., RNASim1000 and RNASim10k datasets), FastTree ties for best among these methods (
Table 3 and
Table 6). However, on the datasets with fragmentary sequences (1000M1-HF) or with heterotachy (Cox1-HET), FastTree is clearly less accurate than both RAxML and IQ-TREE (
Table 4 and
Table 5). Hence, FastTree, although, by far, the fastest of these methods, is less robust to these challenging conditions.
A comparison between RAxML and IQ-TREE is also interesting. RAxML and IQ-TREE have nearly identical accuracy on RNASim1000, and there is only a small advantage to RAxML on the Cox1-HET datasets (
Table 3 and
Table 4). However, RAxML is much more accurate than IQ-TREE on the 1000M1-HF dataset, and it is much less accurate on the RNASim10k dataset (
Table 5 and
Table 6). Hence, the relative performance of RAxML and IQ-TREE depends on the dataset properties, with size and sequence length heterogeneity distinguishing the two methods.
This study shows that very large datasets present specific challenges to ML heuristics. On the RNASim10K dataset, all of the tested methods were able to return reasonably accurate trees within the limitations on running time and available memory, but the same did not hold for the RNASim50K dataset (
Table 6 and
Table 7). This trend suggests that algorithmic strategies for ML heuristics that have been successful on smaller datasets (with at most 1000 or so sequences) may be counterproductive on ultra-large datasets. Specifically, when the sequence dataset is ultra-large, with many thousands (or tens of thousands) of sequences, methods, such as RAxML and IQ-TREE, which make a significant effort to produce good likelihood scores through numeric parameter optimization may not be competitive (in terms of topological accuracy) with methods (such as FastTree) that do not make as serious an effort to optimize the numeric parameters and, instead, focus on exploring more model tree topologies.
4.2. Comparison between DTM Pipelines
Our study showed that the algorithmic choices within the DTM pipelines (starting tree, maximum subset size, and how the distance matrix is computed for use with Constrained-INC and TreeMerge) impact accuracy. In general, we found that using a highly accurate starting tree and larger maximum subset sizes, as well as computing the distance matrix using topological distances, produces better final topological accuracy when compared to the alternative variants of DTM pipelines that we tested. The first two observations (impact of starting tree and subset size) were also noted in [
24], but using a different set of options for each algorithmic step. Our third observation, that using topological distances in the guide tree instead of other ways of defining pairwise distances of sequences (for use within Constrained-INC or TreeMerge) is beneficial, has not been communicated in any prior publication in the context of gene tree estimation; however, similar findings were reported in the context of species tree estimation in [
25].
One of the surprising findings in this study is that we did not see substantial differences in accuracy in the trees that were computed using our better pipelines once everything other than the DTM method (Constrained-INC, TreeMerge, or GTM) is fixed. This is interesting since the DTM methods use very different strategies for merging disjoint subtrees, especially with respect to the degree of blending that is permitted. Specifically, GTM does not allow any blending, TreeMerge allows partial blending, and only Constrained-INC allows full blending. Given the close accuracy between the DTM pipelines, computational issues become important and, thus, pipelines using GTM are superior to the other DTM pipelines, which are slower.
Another trend observed in this study is that DTM pipelines are impacted (even if only by a small amount) by the choice of the starting tree. The impact of a poor decomposition on GTM makes sense, since, if the subsets are not convex in the true tree (i.e., separable from each other by edge deletions), then the requirement to not blend subset trees means that the true tree cannot be constructed using GTM. Therefore, it is noteworthy that the other DTM methods, which do allow blending, are also negatively impacted by poor starting trees.
4.3. Comparing DTM Pipelines to ML Heuristics
Our study showed that the best DTM pipelines we explored (which use IQ-TREE starting and constraint trees) provide improved accuracy as compared to both FastTree and IQ-TREE, but they are not as reliably accurate as RAxML, a finding that should be contrasted with the trends (and interpretation) reported in the prior study evaluating Constrained-INC pipelines for gene tree estimation [
24]. It is worth noting that [
24] never found their best pipeline using Constrained-INC to be competitive with RAxML and, indeed, their pipelines were not reliably more accurate than FastTree. This is very different from what we observe, where the best GTM pipeline is reliably more accurate than FastTree and IQ-TREE and, in some cases, as accurate as RAxML (or even more accurate than RAxML).
The difference in trends can be explained by the choices made in [
24] for the Constrained-INC pipeline, which—in many cases—reduced accuracy when compared to the choices that we made. Of the many differences, the three most likely to have negatively impacted the Constrained-INC pipeline in [
24] as compared to the better pipelines that we studied are (1) the use of Constrained-INC instead of GTM, (2) the smaller maximum subset size (they used 200) compared to ours (we used 500), and (3) the use of FastTree instead of IQ-TREE as the starting tree in all analyses. Thus, the choice of algorithmic parameters has a substantial impact on pipeline accuracy, and our choices improved accuracy when compared to theirs.
Thus, unlike [
24], we find that the better DTM pipelines can provide competitive accuracy with leading ML heuristics. Moreover, the best DTM pipeline that we developed—GTM using IQ-TREE for starting tree and constraint trees–was competitive with RAxML, and only clearly less accurate than RAxML on the high fragmentary 1000M1-HF condition. Furthermore, for that high fragmentary condition, the problem was that the starting tree (that was produced using IQ-TREE) had high error—30.2%. The GTM pipeline reduced this error by 1.8%, but this was not enough to come close to RAxML, which had error of only 24.9%. In other words, the GTM pipeline that we presented was only able to reduce the error over the starting tree by a small amount, and when the gap in accuracy between the starting tree and RAxML is large; this means that the pipeline will not be competitive with RAxML.
This examination reveals that DTM pipelines can be competitive with RAxML in accuracy (and much faster) when IQ-TREE or FastTree can produce a tree that is close to the accuracy of the RAxML tree. However, as the high fragmentary condition (1000M1-HF) shows that there are conditions where neither FastTree nor IQ-TREE are nearly as accurate as RAxML. For such cases, DTM pipelines will fail to be competitive in terms of accuracy with RAxML.
4.4. Limitations of This Study
This study was limited to only a few model conditions (and, further, three of the five model conditions are based on subsets of the RNASim million-sequence simulation), and so trends observed here may not hold under very different conditions. In particular, the relative performance between methods may be very different for much smaller datasets (where optimizing the ML may be achieved well by some methods) or on much larger datasets. In particular, if the running time and available memory are limited, then it may be that only FastTree will be able to analyze very large datasets, and both IQ-TREE and RAxML will be infeasible. Another trend we saw that may not hold under other conditions is that pipelines using GTM were as accurate as the pipelines using TreeMerge or Constrained-INC; it is possible that other conditions will be found where GTM is not as accurate as TreeMerge or Constrained-INC, due to GTM’s inability to blend its constraint trees.
4.5. Future Work
This study suggests several directions for future work. First, to design a DTM pipeline that has the potential to consistently and reliably match or improve on the accuracy of RAxML, it will be necessary to change the DTM pipeline. One possibility is to change how we obtain the initial decomposition. Using a good starting tree is obviously desirable, but, as RAxML is very expensive, this is not computationally feasible on large datasets. Instead, other techniques for decomposing the input dataset should be considered, including fast clustering methods or sequence similarity based decompositions. A second change that might improve accuracy is to use RAxML instead of IQ-TREE to compute the constraint trees. However, that substitution in the pipeline design would increase the running time, as RAxML is significantly more expensive than IQ-TREE.
This study is also suggestive of other directions for improving large-scale phylogeny estimation. For example, the results that are shown here suggest that iteration might result in improved accuracy, since the DTM pipelines that we presented reliably improved on their starting trees. Divide-and-conquer pipelines are embarrassingly parallel due to the fact that the constraint trees on the disjoint subsets can be computed independently. Hence, a natural direction for future work in terms of runtime gain is a careful parallel implementation of these pipelines, in order to achieve improved wall-clock running times when compared to existing approaches.
Finally, evaluating ML methods for gene tree estimation presents specific challenges, especially when topological accuracy is the main objective. Benchmark biological datasets where the true gene tree is known (or where at least most of the branches are reliably known) would be helpful, but, since gene tree discord with the species tree is a common and well established phenomenon [
22], relying on comparisons to the species tree (which itself may not be known with high reliability) is not an acceptable substitute. Alternatively, developing additional large-scale single gene simulations that are biologically realistic and, hence, can be used to characterize method performance would be useful.
5. Conclusions
This study provides new insight into maximum likelihood (ML) tree estimation on large gene sequence datasets. We compared three leading ML heuristics—RAxML-NG, IQ-TREE 2, and FastTree 2—on five different model conditions with 1000 or more sequences with respect to the topological accuracy and running time. We found that RAxML-NG and IQ-TREE 2 were more robust to challenging conditions that were characterized by sequence length heterogeneity and substantial heterotachy than FastTree 2. We also observed that RAxML-NG was the most computationally intensive of the three ML methods and FastTree 2 was the fastest. The comparison between RAxML-NG and IQ-TREE 2 showed that they had similar accuracy under some conditions, but RAxML-NG was more accurate on the datasets with fragmentary conditions (thus displaying greater robustness to this challenging condition than IQ-TREE 2), but less accurate on the RNASim10K datasets. Thus, the choice between RAxML-NG and IQ-TREE 2 may depend on the dataset size (with IQ-TREE 2 being preferred for the largest datasets), as well as the available computational resources. Moreover, our study suggests that gene tree maximum likelihood estimation using RAxML-NG is not likely to be feasible for the very large datasets.
Our study then explored the design space for divide-and-conquer pipelines using three DTM methods. In contrast to an earlier study [
24], which found that a divide-and-conquer pipeline using Constrained-INC did not provide an advantage over its starting tree for gene tree estimation, our new pipeline reliably produces trees more accurately than both IQ-TREE 2 and FastTree 2 starting trees. However, we also saw that, to consistently match or improve on of RAxML-NG while being substantially faster, will require changes to the current divide-and-conquer strategy and most likely will require a new approach to decomposing the input set of sequences into disjoint sets that is fast and suitable for subsequent use in a DTM pipeline.
Although this study was only based on a few model conditions and much work is still needed to understand the relative performance of different methods and the conditions under which each method is generally reliable, this study suggests the following guidelines for the selection of ML heuristic:
When the dataset sizes and computational resources make it feasible to use RAxML-NG or IQ-TREE 2, these methods should be used rather than FastTree 2, due to their (generally) higher level of topological accuracy when analyzing datasets that evolve under non-i.i.d. models (which may be typical of biological datasets). The choice between IQ-TREE 2 and RAxML-NG is less clear, as the relative accuracy of these two methods depended on the model condition. However, when datasets have substantial numbers of fragmentary sequences, RAxML-NG may be preferable to IQ-TREE 2.
For very large datasets where RAxML-NG and IQ-TREE 2 are not feasible to run, then FastTree 2 or one of the DTM pipelines that we presented could provide reasonable accuracy.
In summary, this study is very positive with respect to the potential for DTM pipelines to improve the scalability of gene tree estimation. First, the current best DTM pipeline improves on IQ-TREE 2 and FastTree 2, which had not been seen before. Second, we have identified an algorithmic challenge—the fast and accurate clustering of sequence datasets—that may provide the key to improving the DTM pipeline accuracy and scalability to large and more challenging datasets. Advances on this basic challenge could lead to substantial breakthroughs for large-scale phylogeny estimation. This study also showed that the phylogenetic estimation error rates can decrease with increased taxonomic sampling (a trend that has been observed in other studies [
13]), provided that good tree estimation methods are used, showing the value of developing methods that can scale well to large datasets. Overall, therefore, this study suggests new designs for large-scale phylogeny estimation that have both high accuracy and speed.




{kind=link}
{kind=link}
{kind=link}


