
An Investigation of Alternatives to Transform Protein Sequence Databases to a Columnar Index Schema

 ,
,  , , and
, , and
Abstract
1. Introduction
- Database schema: We propose a suitable schema for protein sequence data in a real-time cloud system as an extension to our prior paper [13].
- Protein data transformation: We review the process of protein data transformation into the proposed database schema and detail suitable techniques for the transformation steps.
- Indexing: We propose a radix tree for efficient deduplication (including an extended description compared to our prior paper [13]).
- Performance comparison: We evaluate the best method to transform the protein sequence data using the four proposed approaches.
2. Background
2.1. Protein Data

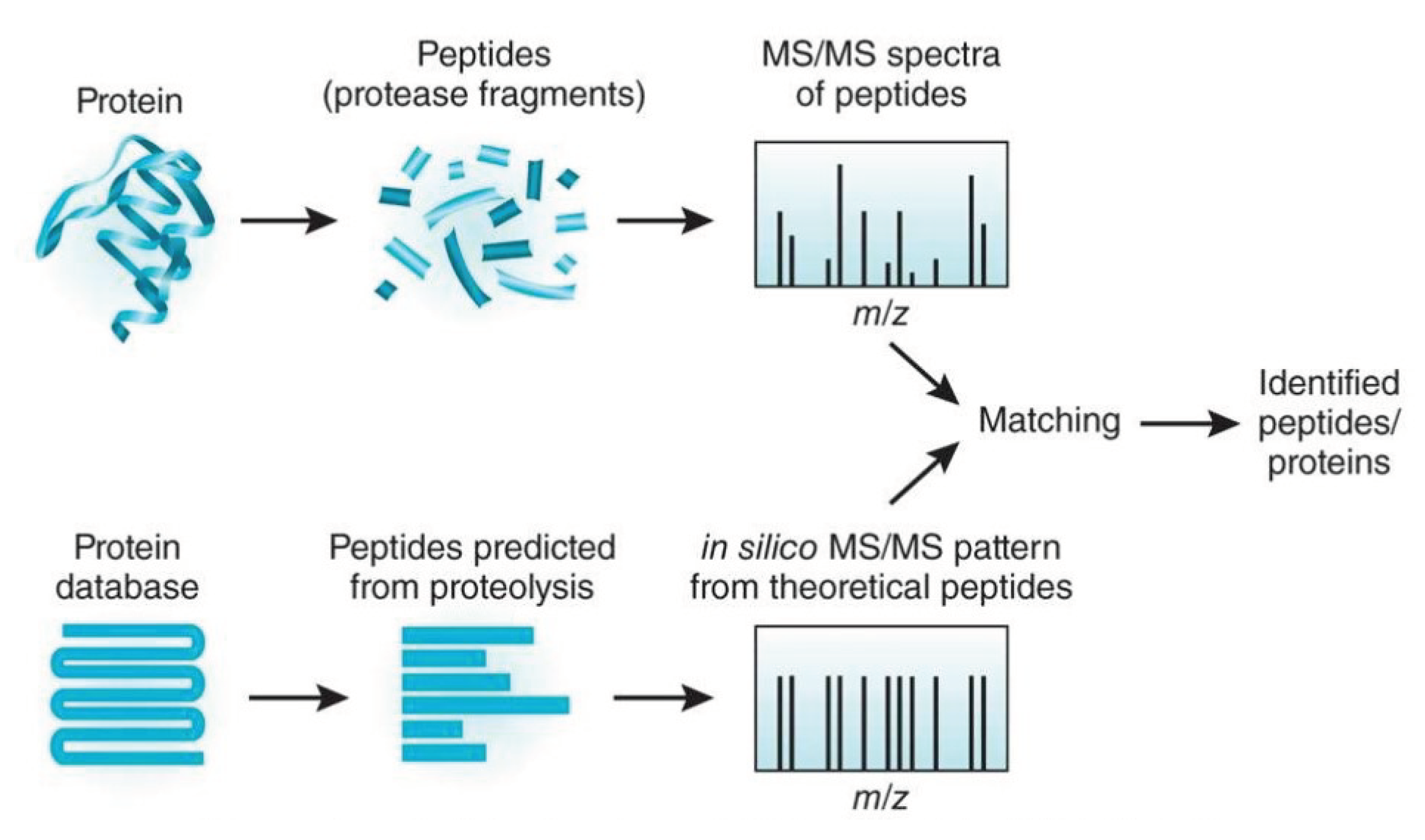
2.2. Protein Identification
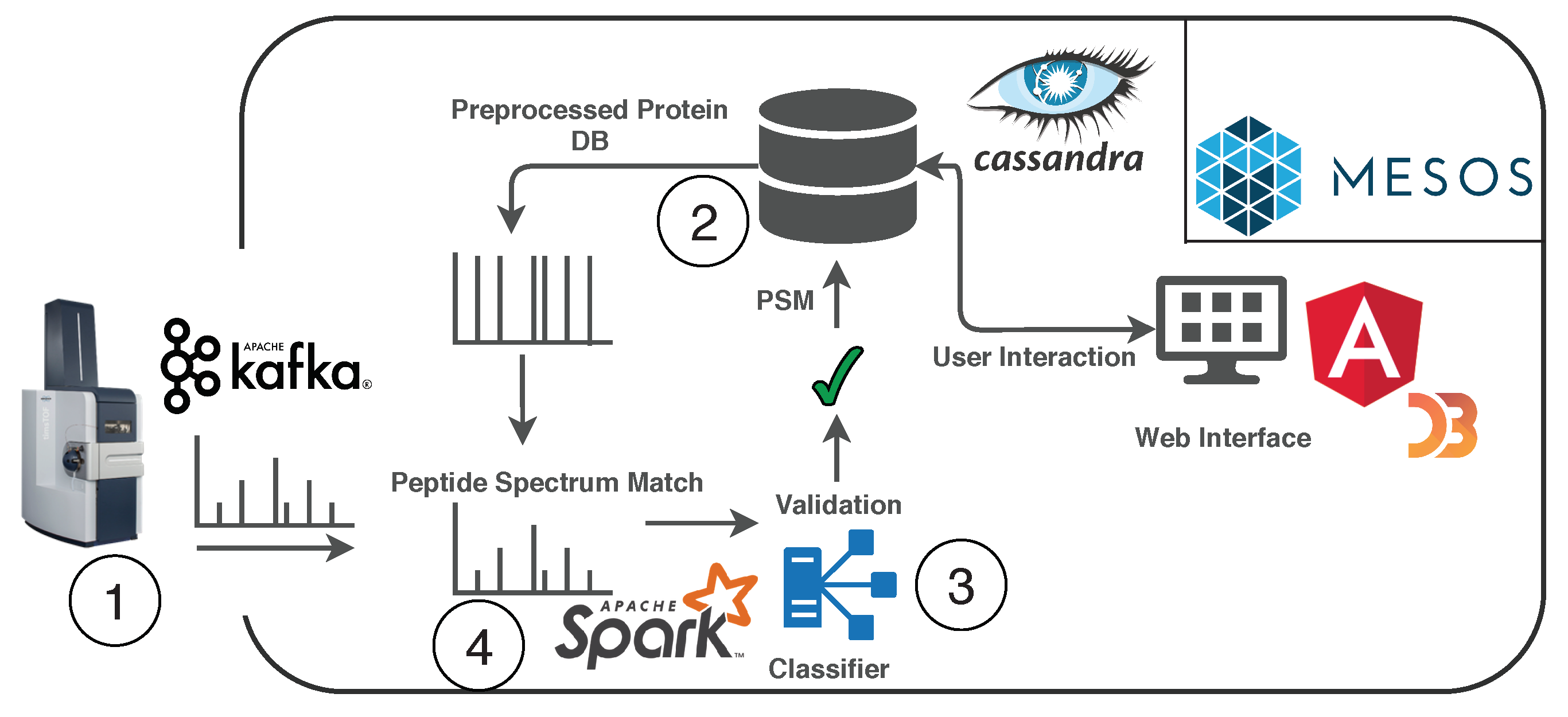
2.3. Real-Time Analysis of Mass Spectrometry Data
2.4. Radix Tree Data Structure
3. Data Preparation for Real-Time Protein Identification
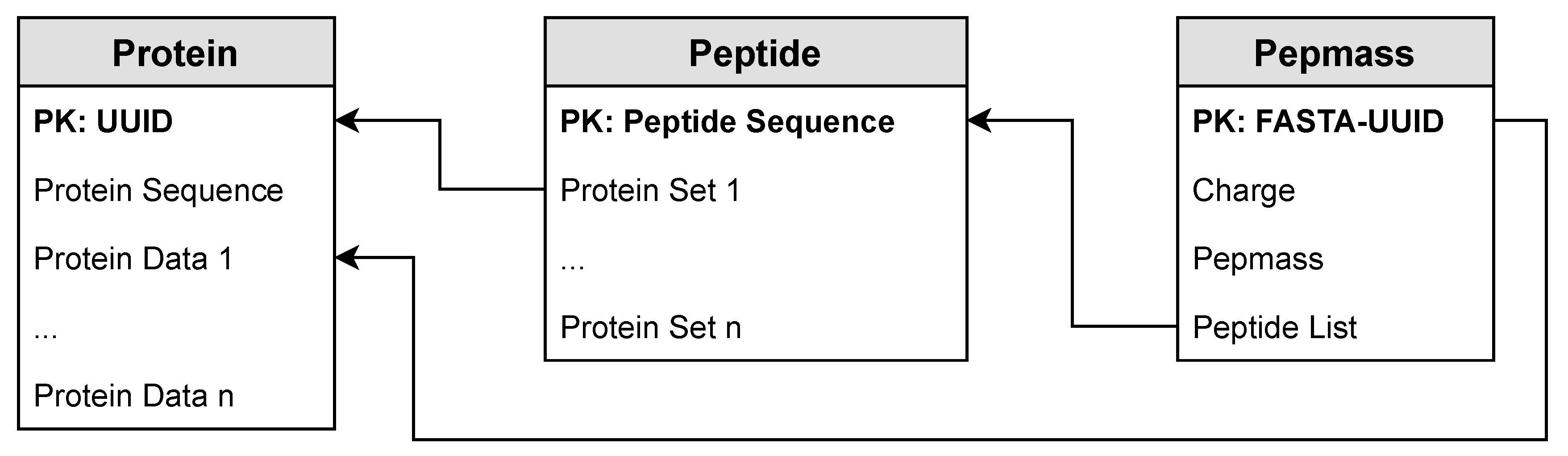
3.1. Indexed Masses of Peptides
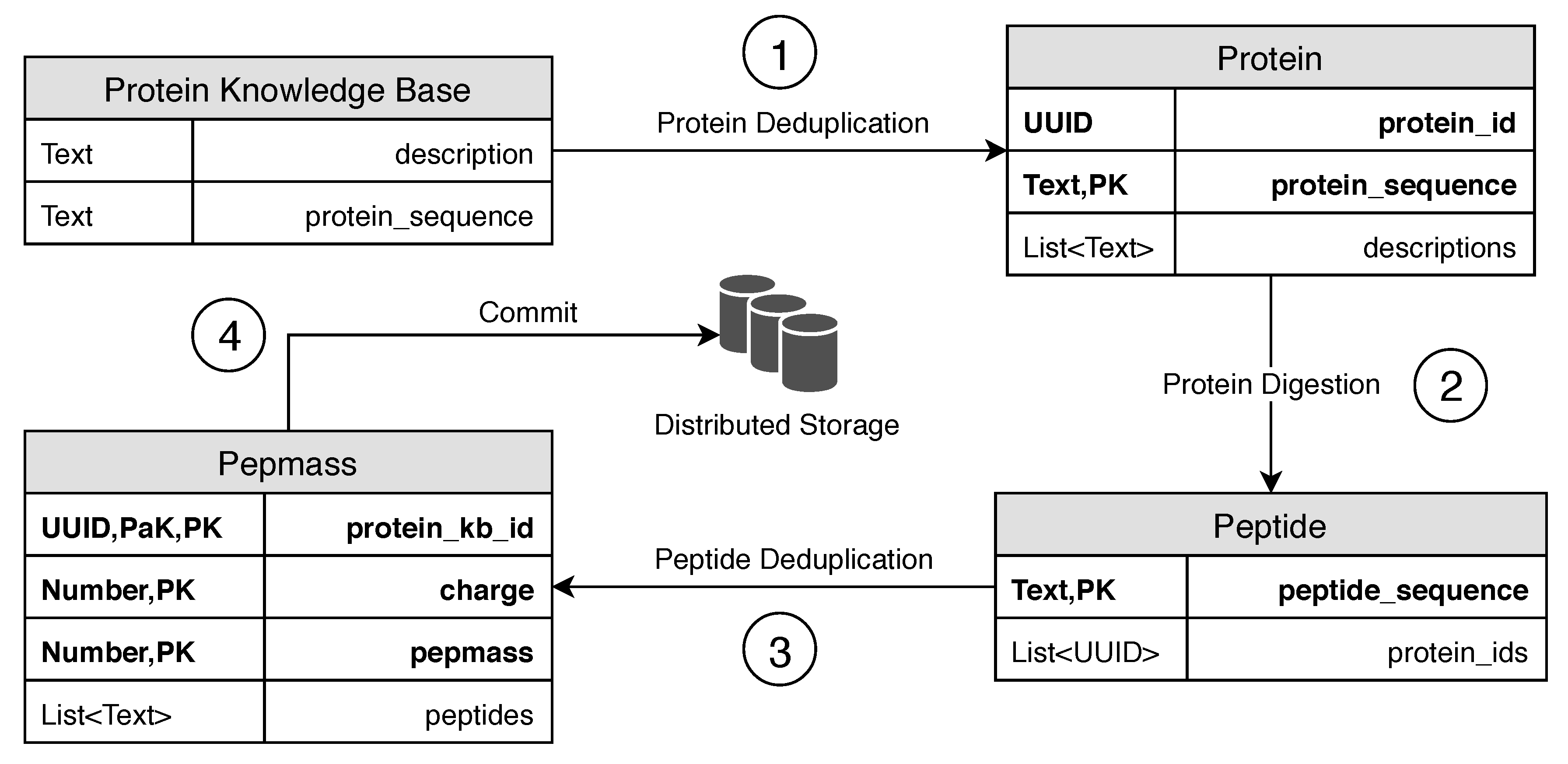
3.1.1. Protein Table
3.1.2. Peptide Table
3.1.3. Pepmass Table
3.2. Data Transformation
3.2.1. Protein Deduplication

3.2.2. Protein Digestion
3.2.3. Peptide Deduplication and Mass Calculation
3.2.4. Approaches for Data Transformation
4. Implementation
4.1. Transformation Using a Map Structure
4.2. Transformation Using DBMS Queries

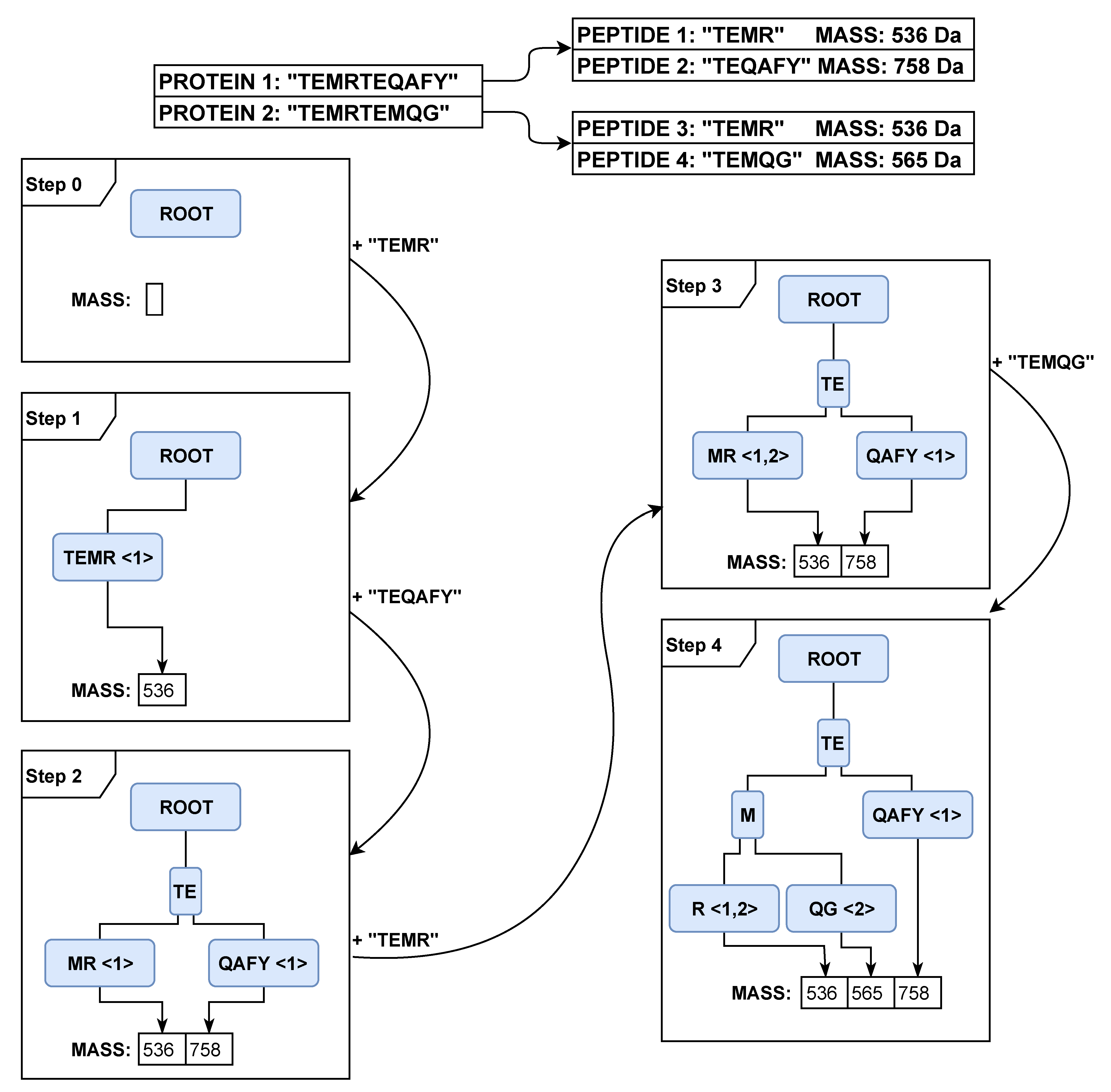
4.3. Transformation Using the Extended Radix Tree Structure

4.4. Our Radix Tree Adaptation
5. Evaluation
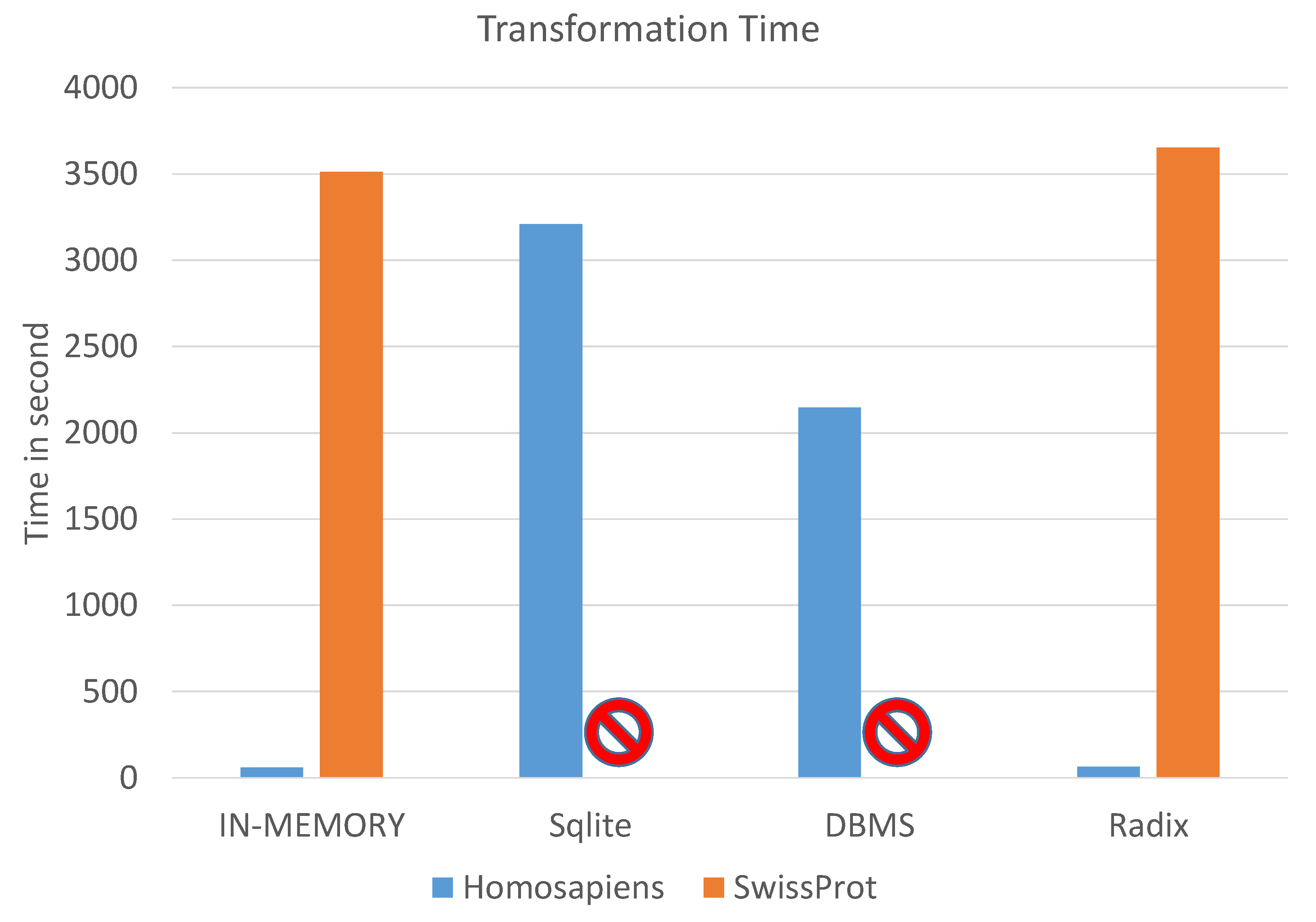
5.1. Time Evaluation
5.1.1. Homo sapiens Data Set
5.1.2. UniProtKB/SwissProt Data Set
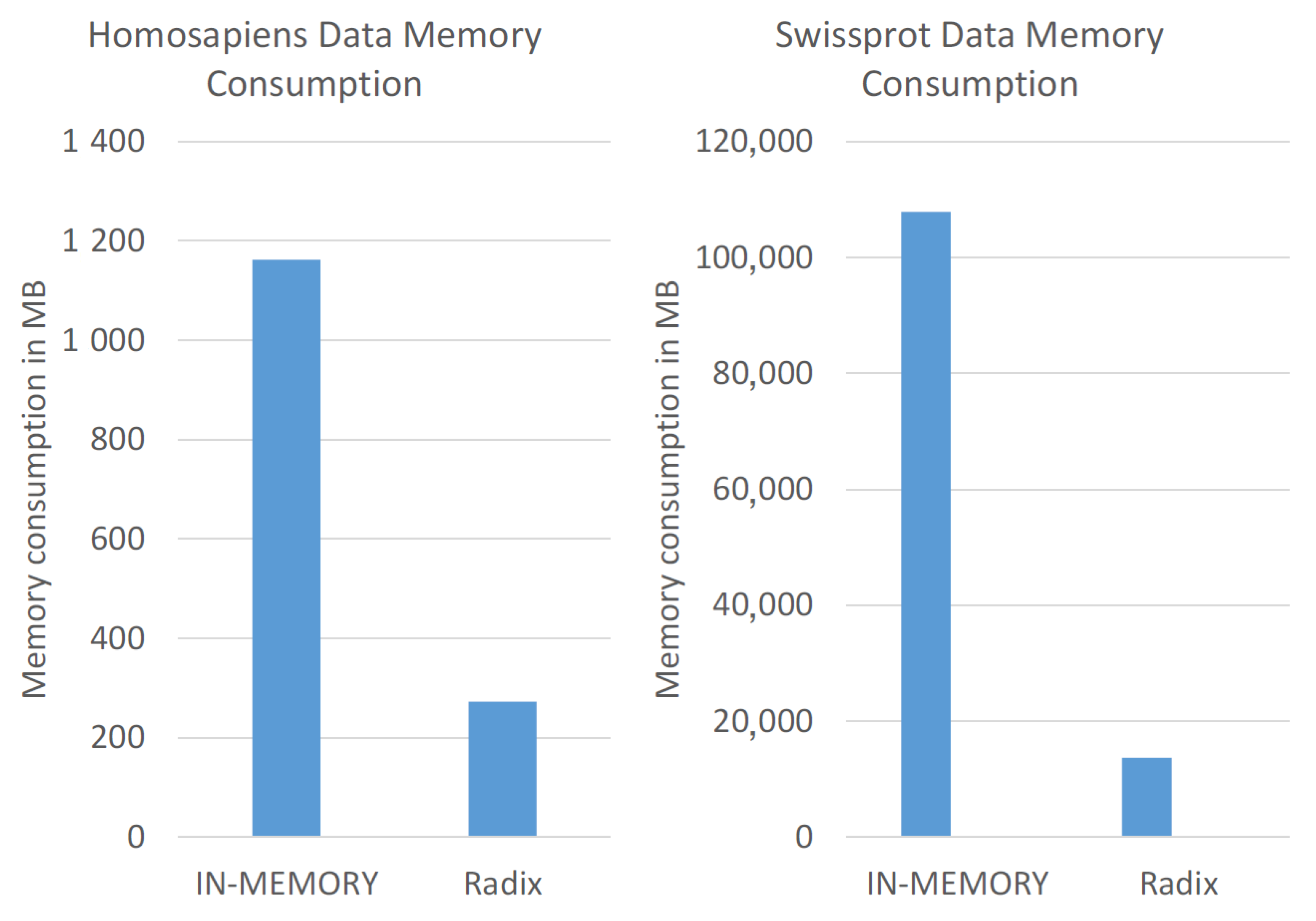
5.2. Memory Consumption
5.3. Result Summary
6. Related Work
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Global Mass Spectrometry Market Size, Market Share, Application Analysis, Regional Outlook, Growth Trends, Key Players, Competitive Strategies and Forecasts, 2015 to 2025. 2017. Available online: https://www.researchandmarkets.com/reports/4313373/global-mass-spectrometry-market-size-market (accessed on 10 February 2021).
- Aebersold, R.; Mann, M. Mass spectrometry-based proteomics. Nature 2003, 422, 198. [Google Scholar] [CrossRef]
- Ashcroft, A.E. An Introduction to Mass Spectrometry; The University of Leeds: Leeds, UK, 2011. [Google Scholar]
- Heyer, R.; Kohrs, F.; Reichl, U.; Benndorf, D. Metaproteomics of complex microbial communities in biogas plants. Microb. Technol. 2015, 8, 749–763. [Google Scholar] [CrossRef] [PubMed]
- Heyer, R.; Schallert, K.; Siewert, C.; Kohrs, F.; Greve, J.; Maus, I.; Klang, J.; Klocke, M.; Heiermann, M.; Hoffmann, M.; et al. Metaproteome analysis reveals that syntrophy, competition, and phage-host interaction shape microbial communities in biogas plants. Microbiome 2019, 7, 69. [Google Scholar] [CrossRef]
- Petriz, B.A.; Franco, O.L. Metaproteomics as a Complementary Approach to Gut Microbiota in Health and Disease. Front. Chem. 2017, 5, 4. [Google Scholar] [CrossRef]
- Lehmann, T.; Schallert, K.; Vilchez-Vargas, R.; Benndorf, D.; Püttker, S.; Sydor, S.; Schulz, C.; Bechmann, L.; Canbay, A.; Heidrich, B.; et al. Metaproteomics of fecal samples of Crohn’s disease and Ulcerative Colitis. J. Proteom. 2019, 201, 93–103. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, G.; Palmieri, F. Discovering genomic patterns in SARS-CoV-2 variants. Int. J. Intell. Syst. 2020, 35, 1680–1698. [Google Scholar] [CrossRef]
- Millioni, R.; Franchin, C.; Tessari, P.; Polati, R.; Cecconi, D.; Arrigoni, G. Pros and cons of peptide isolectric focusing in shotgun proteomics. J. Chromatogr. A 2013, 1293, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Zoun, R.; Schallert, K.; Janki, A.; Ravindran, R.; Durand, G.C.; Fenske, W.; Broneske, D.; Heyer, R.; Benndorf, D.; Saake, G. Streaming FDR Calculation for Protein Identication. In Advances in Databases and Information Systems; Springer: Budapest, Hungary, 2018; pp. 80–87. [Google Scholar]
- Zoun, R.; Durand, G.C.; Schallert, K.; Patrikar, A.; Broneske, D.; Fenske, W.; Heyer, R.; Benndorf, D.; Saake, G. Protein Identification as a Suitable Application for Fast Data Architecture. In Proceedings of the DEXA 2018 International Workshops, BDMICS, BIOKDD, and TIR, Regensburg, Germany, 3–6 September 2018; pp. 168–178. [Google Scholar]
- Zoun, R.; Schallert, K.; Broneske, D.; Fenske, W.; Pinnecke, M.; Heyer, R.; Brehmer, S.; Benndorf, D.; Saake, G. MSDataStream-Connecting a Bruker Mass Spectrometer to the Internet. Available online: https://new-dl.gi.de/handle/20.500.12116/21719 (accessed on 10 February 2021).
- Zoun, R.; Schallert, K.; Broneske, D.; Trifonova, I.; Chen, X.; Heyer, R.; Benndorf, D.; Saake, G. Efficient Transformation of Protein Sequence Databases to Columnar Index Schema; Database and Expert Systems Applications; Springer International Publishing: Cham, Switzerland, 2019; pp. 67–72. [Google Scholar]
- Banerjee, S.; Mazumdar, S. Electrospray Ionization Mass Spectrometry: A Technique to Access the Information beyond the Molecular Weight of the Analyte. Int. J. Anal. Chem. 2012, 2012, 282574. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, E.W. File formats commonly used in mass spectrometry proteomics. Mol. Cell. Proteom. 2012, 11, 1612–1621. [Google Scholar] [CrossRef] [PubMed]
- McDonald, W.H.; Tabb, D.L.; Sadygov, R.G.; MacCoss, M.J.; Venable, J.; Graumann, J.; Johnson, J.R.; Cociorva, D.; Yates, J.R., III. MS1, MS2, and SQT—three unified, compact, and easily parsed file formats for the storage of shotgun proteomic spectra and identifications. Rapid Commun. Mass Spectrom. 2004, 18, 2162–2168. [Google Scholar] [CrossRef]
- Matrix Science. Data File Format. 2016. Available online: http://www.matrixscience.com/help/data_\file_help.html (accessed on 10 February 2021).
- FASTA Format. 2002. Available online: https://blast.ncbi.nlm.nih.gov/Blast.cgi?CMD=Web&PAGE_TYPE=BlastDocs&\DOC_TYPE=BlastHelp (accessed on 10 February 2021).
- Wampler, D. Fast Data Architectures for Streaming Applications, 1st ed.; O’Reilly Media: Sebastopol, CA, USA, 2016. [Google Scholar]
- Zoun, R. Analytic Cloud Platform for Near Real-Time Mass Spectrometry Processing on the Fast Data Architecture. Ph.D. Thesis, University of Magdeburg, Magdeburg, Germany, 2020. [Google Scholar]
- De La Briandais, R. File Searching Using Variable Length Keys. In Proceedings of the Western Joint Computer Conference, San Francisco, CA, USA, 3–5 March 1959. [Google Scholar]
- Leis, V.; Kemper, A.; Neumann, T. The Adaptive Radix Tree: ARTful Indexing for Main-memory Databases. In Proceedings of the 2013 IEEE International Conference on Data Engineering (ICDE 2013), Brisbane, Australia, 8–11 April 2013; pp. 38–49. [Google Scholar]
- Shishibori, M.; Okuno, M.; Ando, K.; Aoe, J.I. An efficient compression method for Patricia tries. In Proceedings of the 1997 IEEE International Conference on Systems, Man, and Cybernetics, Computational Cybernetics and Simulation, Orlando, FL, USA, 12–15 October 1997; Volume 1, pp. 415–420. [Google Scholar] [CrossRef]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A Peptide Search Engine Integrated into the MaxQuant Environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef] [PubMed]
- Muth, T.; Behne, A.; Heyer, R.; Kohrs, F.; Benndorf, D.; Hoffmann, M.; Lehtevä, M.; Reichl, U.; Martens, L.; Rapp, E. The MetaProteomeAnalyzer: A Powerful Open-Source Software Suite for Metaproteomics Data Analysis and Interpretation. J. Proteome Res. 2015, 14, 1557–1565. [Google Scholar] [CrossRef] [PubMed]
- Siragusa, E. Approximate String Matching for High-Throughput Sequencing. Ph.D. Thesis, Freie Universität Berlin, Berlin, Germany, 2015. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
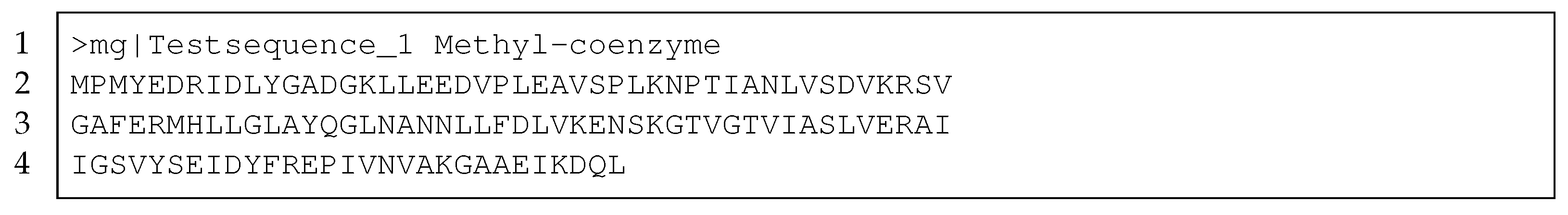{kind=link}
{kind=link}
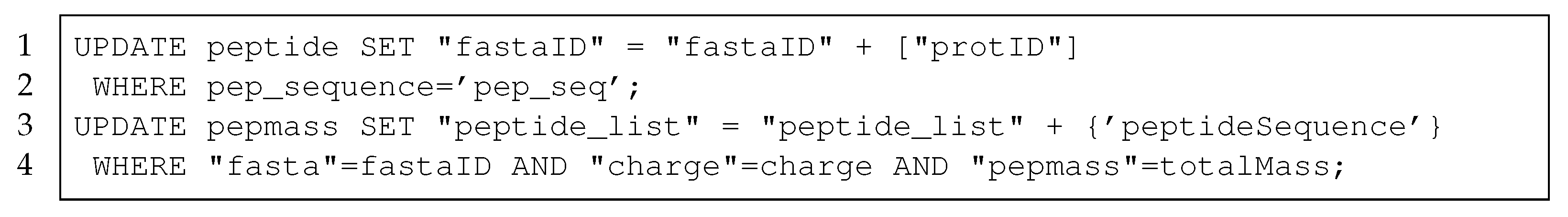{kind=link}
{kind=link}
| prot_id | FILE_1 | FILE_2 | prot_seq |
|---|---|---|---|
| PROTEIN_1 | null | [’json-data’] | TEMRTEQAFY |
| PROTEIN_2 | [’json-data’] | [’json-data’] | TEMRTEMQG |
| pep_seq | FILE_1 | FILE_2 |
|---|---|---|
| TEMR | [PROTEIN_2] | [PROTEIN_1, PROTEIN_2] |
| TEQAFY | null | [PROTEIN_1] |
| TEMQG | [PROTEIN_2] | [PROTEIN_2] |
| Fasta | Charge | Pepmass | Peptide_List |
|---|---|---|---|
| FILE_1 | 1 | 536 | [’TEMR’] |
| FILE_1 | 1 | 565 | [’TEMQG’] |
| FILE_2 | 1 | 536 | [’TEMR’] |
| FILE_2 | 1 | 768 | [’TEQAFY’] |
| FILE_2 | 1 | 565 | [’TEMQG’] |
| Method | Protein Deduplication | Protein Digestion | Peptide Deduplication | Commit |
|---|---|---|---|---|
| In-Memory Map | No | Yes | Yes | No |
| HDD-resident Map | No | Yes | Yes | No |
| Database Engine | Yes | Yes | Yes | Yes |
| Radix Tree | No | Yes | Yes | No |
| Homo Sapiens | SwissProt | |
|---|---|---|
| Size in MB | 2.88 | 255 |
| Number of proteins | 4794 | 556,196 |
| Number of peptides | 484,479 | 37,403,696 |
| Number of unique peptides | 248,996 | 23,254,068 |
| Number of radix nodes | 295,602 | 28,481,207 |
| Method | Protein Deduplication | Protein Digestion | Peptide Deduplication | Commit |
|---|---|---|---|---|
| In-Memory Map | No | Yes | Yes | No |
| HDD Map | No | Yes | Yes | No |
| Database Engine | Yes | Yes | Yes | Yes |
| Radix Tree | No | Yes | Yes | No |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zoun, R.; Schallert, K.; Broneske, D.; Trifonova, I.; Chen, X.; Heyer, R.; Benndorf, D.; Saake, G. An Investigation of Alternatives to Transform Protein Sequence Databases to a Columnar Index Schema. Algorithms 2021, 14, 59. https://doi.org/10.3390/a14020059
Zoun R, Schallert K, Broneske D, Trifonova I, Chen X, Heyer R, Benndorf D, Saake G. An Investigation of Alternatives to Transform Protein Sequence Databases to a Columnar Index Schema. Algorithms. 2021; 14(2):59. https://doi.org/10.3390/a14020059
Chicago/Turabian StyleZoun, Roman, Kay Schallert, David Broneske, Ivayla Trifonova, Xiao Chen, Robert Heyer, Dirk Benndorf, and Gunter Saake. 2021. "An Investigation of Alternatives to Transform Protein Sequence Databases to a Columnar Index Schema" Algorithms 14, no. 2: 59. https://doi.org/10.3390/a14020059
APA StyleZoun, R., Schallert, K., Broneske, D., Trifonova, I., Chen, X., Heyer, R., Benndorf, D., & Saake, G. (2021). An Investigation of Alternatives to Transform Protein Sequence Databases to a Columnar Index Schema. Algorithms, 14(2), 59. https://doi.org/10.3390/a14020059