The Prediction of Intrinsically Disordered Proteins Based on Feature Selection


Abstract
1. Introduction
2. Materials and Methods
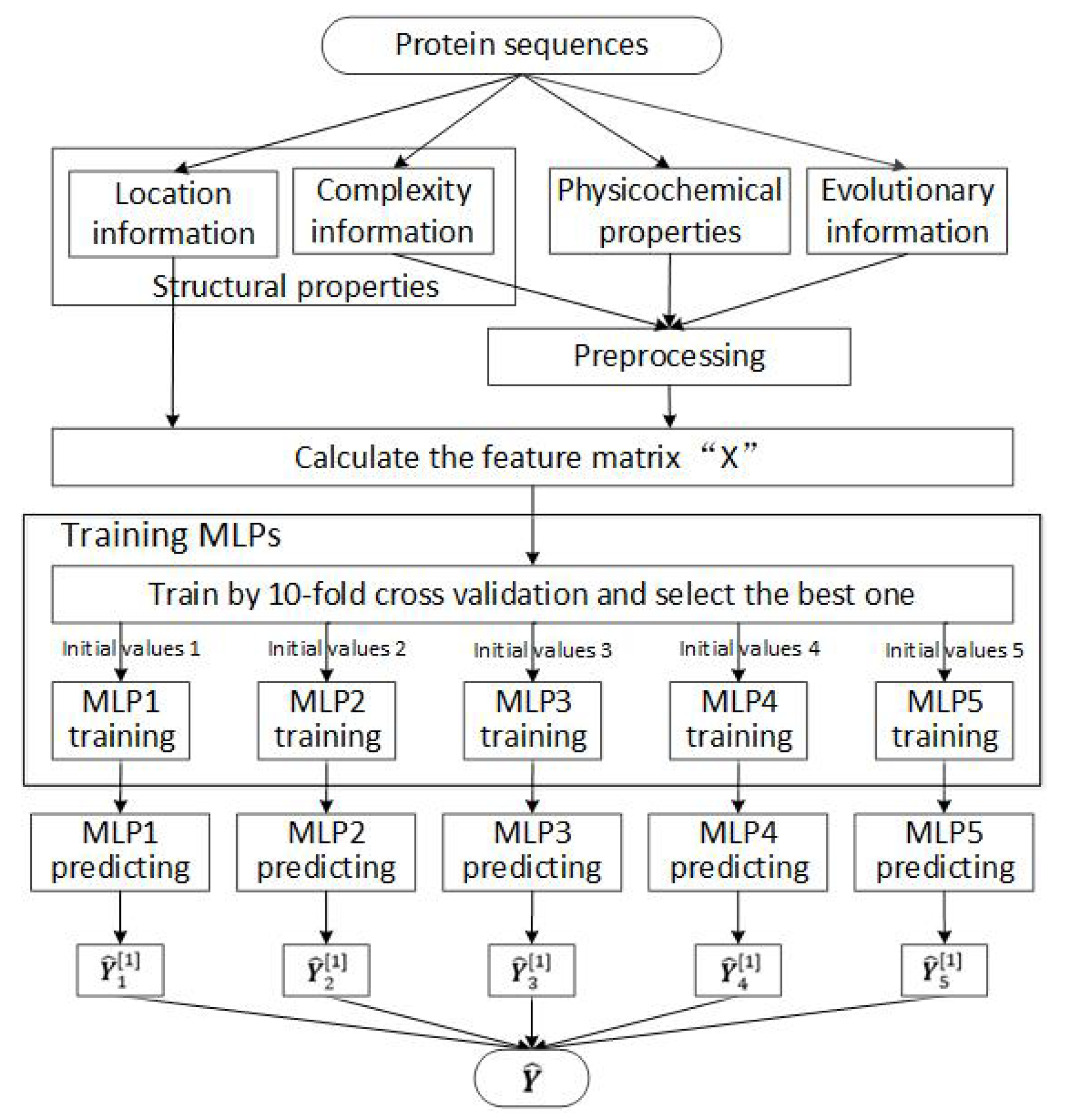
2.1. Feature Selection
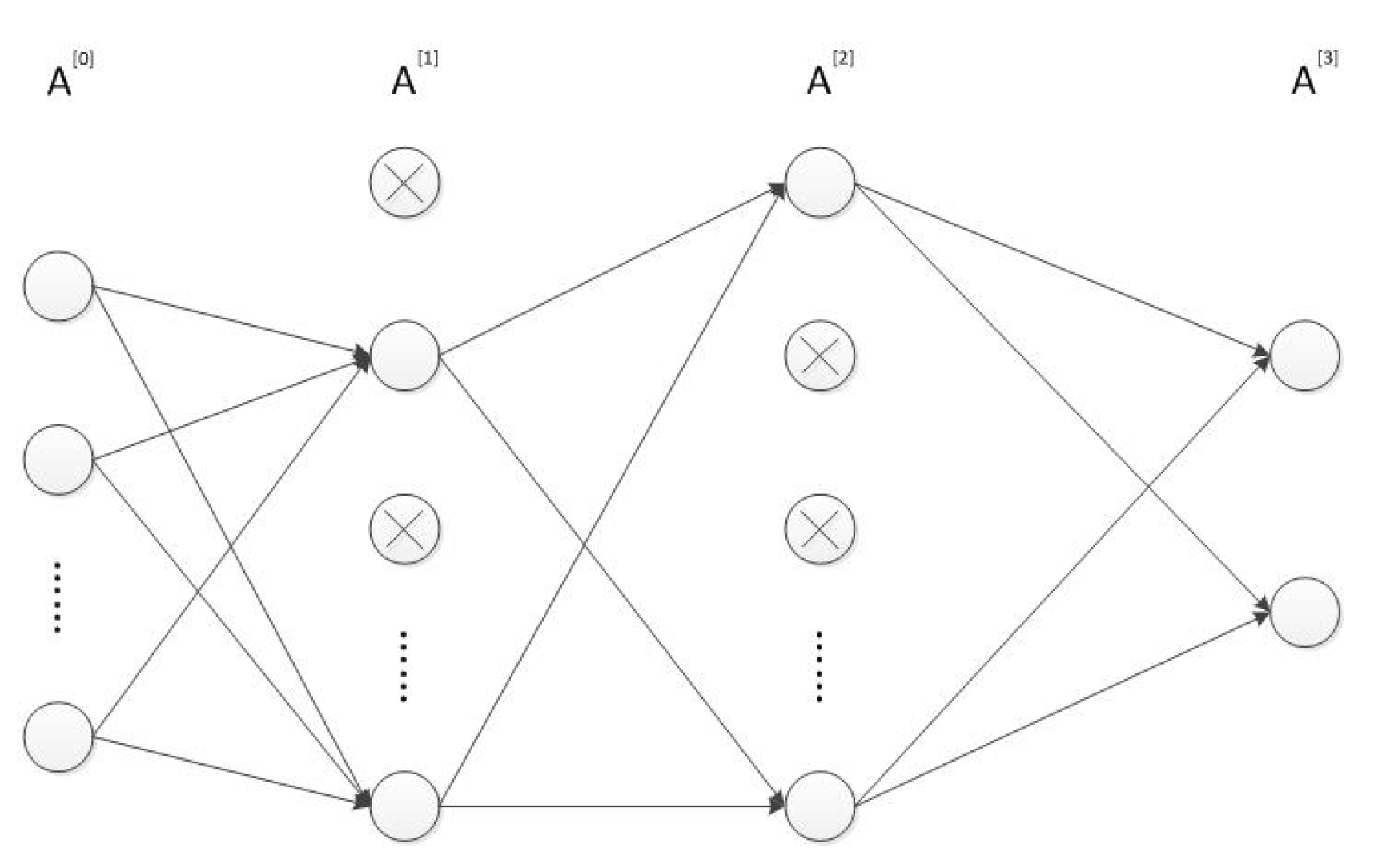
2.2. Designing and Training the MLP Neural Network
- Input and compute the forward propagation throughwhere is the activation function in the l-th layer . In our MLP neural network, both and are ReLU functions and is softmax function. represents the dropout vector which obeys the Bernoulli distribution with probability . The symbols ∗ and · respectively represent the scalar and matrix multiplication. Furthermore, is equal to while is the prediction result . The structure of the MLP network is shown in Figure 1.
- After obtaining , compute the cost function which is the cross entropy. If the cost function is converged or the max iteration number is achieved, then stop and output .
- Employ Adam algorithm [22] to perform the back propagation. Update and and repeat 1.
2.3. Datasets for Training and Testing
2.4. Performance Evaluation
3. Results and Discussion
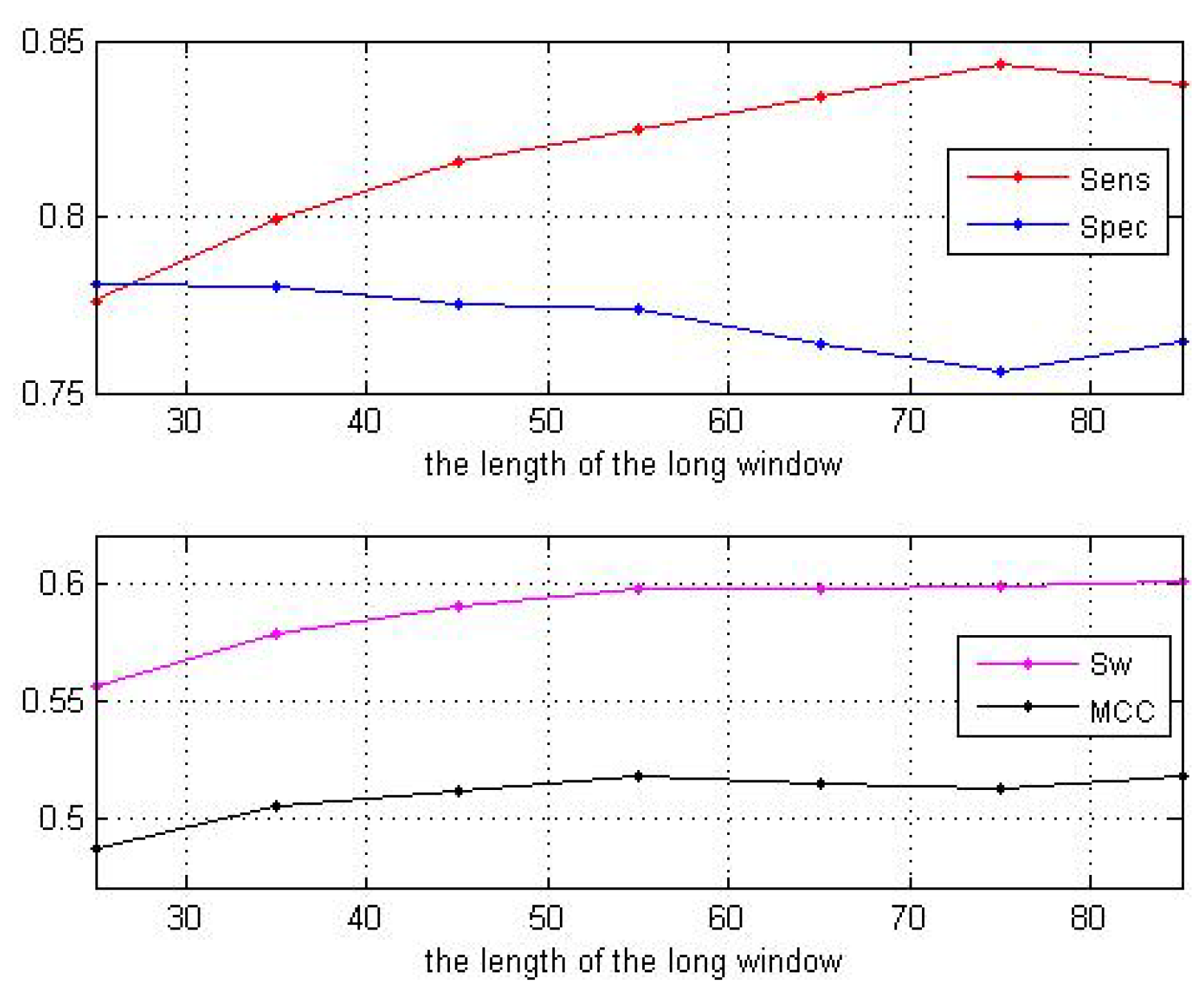
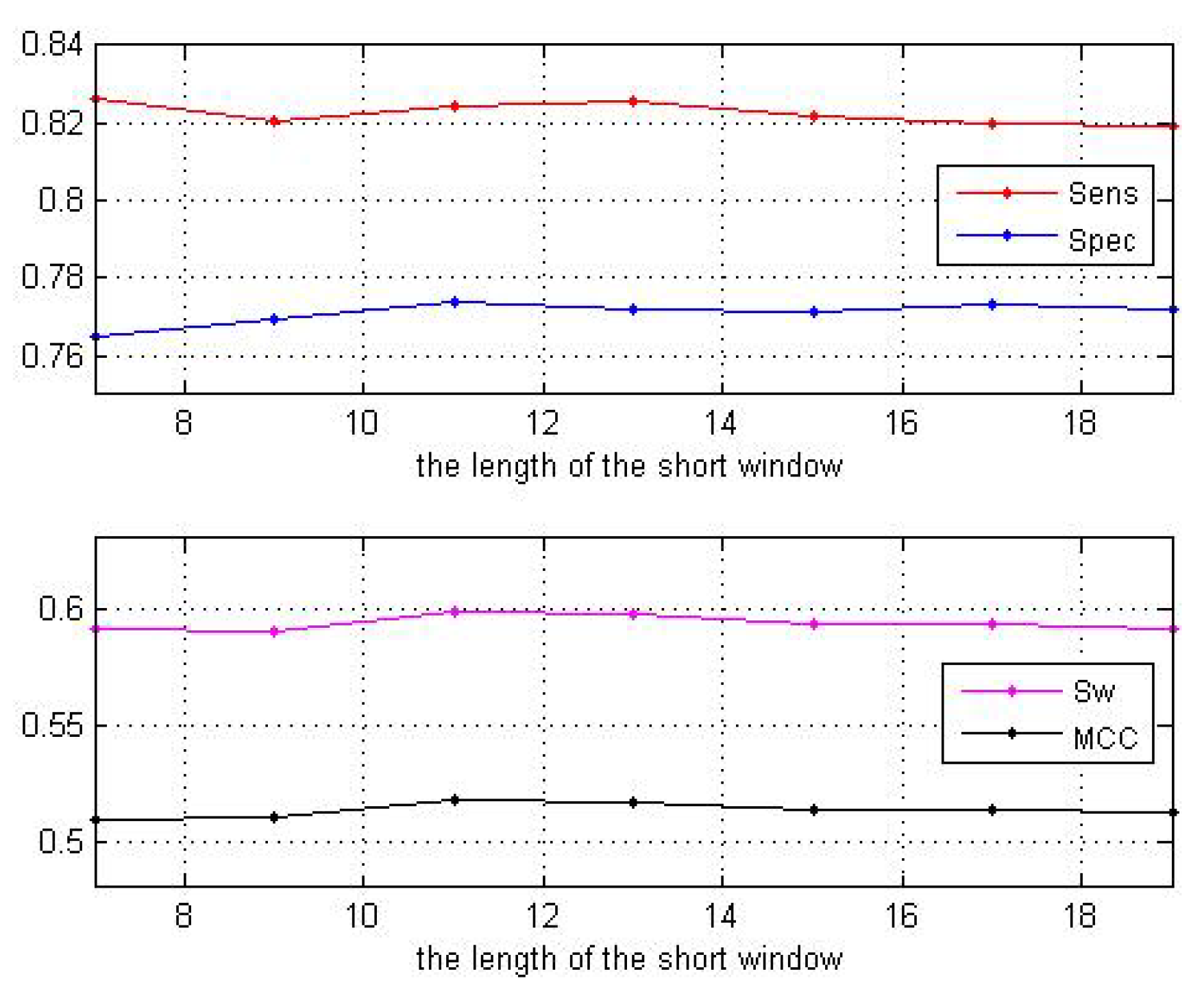
3.1. Impact of the Sliding Window Sizes
3.2. Impact of the MLP Parameters
3.3. Impact of the Preprocessing
3.4. Impact of the Evolutionary Information
3.5. Comparing with Other Classification Algorithms
3.6. Testing Performances
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Uversky, V.N. The mysterious unfoldome: structureless, underappreciated, yet vital part of any given proteome. J. Biomed. Biotechnol. 2010, 2010, 568068. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Kriwacki, R.W. The orderly chaos of proteins. Sci. Am. 2011, 304, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Oldfield, C.J.; Dunker, A.K. Intrinsically Disordered Proteins and Intrinsically Disordered Protein Regions. Annu. Rev. Biochem. 2014, 83, 553–584. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Functional roles of transiently and intrinsically disordered regions within proteins. FEBS J. 2015, 282, 1182–1189. [Google Scholar] [CrossRef] [PubMed]
- Wright, P.E.; Dyson, H.J. Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J. Mol. Biol. 1999, 293, 321–331. [Google Scholar] [CrossRef]
- Kaya, I.E.; Ibrikci, T.; Ersoy, O.K. Prediction of disorder with new computational tool: BVDEA. Expert Syst. Appl. 2011, 38, 14451–14459. [Google Scholar] [CrossRef]
- Oldfield, C.J.; Ulrich, E.L.; Cheng, Y.G.; Dunker, A.K.; Markley, J.L. Addressing the intrinsic disorder bottleneck in structural proteomics. Proteins 2005, 59, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Prilusky, J.; Felder, C.E.; Zeev-Ben-Mordehai, T.; Rydberg, E.H.; Man, O.; Beckmann, J.S.; Silman, I.; Sussman, J.L. FoldIndex: A simple tool to predict whether a given protein sequence is intrinsically unfolded. Bioinformatics 2005, 21, 3435–3438. [Google Scholar] [CrossRef]
- Linding, R.; Russell, R.B.; Neduva, V.; Gibson, T.J. Globplot: Exploring Protein Sequences for Globularity and Disorder. Nucleic Acids Res. 2003, 31, 3701–3708. [Google Scholar] [CrossRef]
- Dosztanyi, Z.; Csizmok, V.; Tompa, P.; Simon, I. IUPred: Web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics 2005, 21, 3433–3434. [Google Scholar] [CrossRef]
- Galzitskaya, O.V.; Garbuzynskiy, S.O.; Lobanov, M.Y. FoldUnfold: web server for the prediction of disordered regions in protein chain. Bioinformatics 2006, 22, 2948–2949. [Google Scholar] [CrossRef]
- Lobanov, M.Y.; Galzitskaya, O.V. The Ising model for prediction of disordered residues from protein sequence alone. Phys. Biol. 2011, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- PONDR: Predictors of Natural Disordered Regions. Available online: http://www.pondr.com/ (accessed on 20 February 2019).
- Yang, Z.R.; Thomson, R.; McNeil, P.; Esnouf, R.M. RONN: The bio-basis function neural network technique applied to the detection of natively disordered regions in proteins. Bioinformatics 2005, 21, 3369–3376. [Google Scholar] [CrossRef]
- Ward, J.J.; Sodhi, J.S.; Mcguffin, L.J.; Buxton, B.F.; Jones, D.T. Prediction and Functional Analysis of Native Disorder in Proteins from the Three Kingdoms of Life. J. Mol. Biol. 2004, 337, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Su, C.T.; Chen, C.Y.; Ou, Y.Y. Protein disorder prediction by condensed pssm considering propensity for order or disorder. BMC Bioinform. 2006, 7, 319. [Google Scholar] [CrossRef]
- Zhang, T.; Faraggi, E.; Xue, B.; Dunker, A.K.; Uversky, V.N.; Zhou, Y. SPINE-D: Accurate prediction of short and long disordered regions by a single neural-network based method. J. Biomol. Struct. Dyn. 2012, 29, 799–813. [Google Scholar] [CrossRef]
- Walsh, I.; Martin, A.J.; Di Domenico, T.; Tosatto, S.C. ESpritz: Accurate and fast prediction of protein disorder. Bioinformatics 2012, 28, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Mizianty, M.J.; Stach, W.; Chen, K.; Kedarisetti, K.D.; Disfani, F.M.; Kurgan, L. Improved sequence-based prediction of disordered regions with multilayer fusion of multiple information sources. Bioinformatics 2010, 26, i489–i496. [Google Scholar] [CrossRef]
- Ishida, T.; Kinoshita, K. Prediction of disordered regions in proteins based on the meta approach. Bioinformatics 2008, 24, 1344–1348. [Google Scholar] [CrossRef]
- Schlessinger, A.; Punta, M.; Yachdav, G.; Kajan, L.; Rost, B. Improved disorder prediction by combination of orthogonal approaches. PLoS ONE 2009, 4, 4433. [Google Scholar] [CrossRef]
- Kingma, D.P.; Ba, J.L. Adam: A method for stochastic optimization. In Proceedings of the 3rd International Conference on Learning Representations (ICLR), San Diego, CA, USA, 7 May 2015; pp. 1–15. [Google Scholar]
- Srivastava, N.; Hinton, G.; Krizhevsky, A.; Sutskever, I.; Salakhutdinov, R. Dropout: A Simple Way to Prevent Neural Networks from Overfitting. J. Mach. Learn. Res. 2014, 15, 1929–1958. [Google Scholar]
- Sickmeier, M.; Hamilton, J.A.; LeGall, T.; Vacic, V.; Cortese, M.S.; Tantos, A.; Szabo, B.; Tompa, P.; Chen, J.; Uversky, V.N.; et al. DisProt: the database of disordered proteins. Nucleic Acids Res. 2007, 35, 786–793. [Google Scholar] [CrossRef] [PubMed]
- Burges, C.J.C. A tutorial on support vector machines for pattern recognition. Data Min. Knowl. Discov. 1998, 2, 121–167. [Google Scholar] [CrossRef]
- Mika, S.; Ratsch, G.; Weston, J.; Scholkopf, B.; Mullers, K.R. Fisher discriminant analysis with kernels. In Proceedings of the Neural Networks for Signal Processing IX: Proceedings of the 1999 IEEE Signal Processing Society Workshop, Madison, WI, USA, 25 August 1999; pp. 41–48. [Google Scholar]
- He, H.; Zhao, J.X. A Low Computational Complexity Scheme for the Prediction of Intrinsically Disordered Protein Regions. Math. Probl. Eng. 2018, 2018, 8087391. [Google Scholar] [CrossRef]
- Shimizu, K.; Muraoka, Y.; Hirose, S.; Noguchi, T. Feature selection based on physicochemical properties of redefined n-term region and c-term regions for predicting disorder. In Proceedings of the 2005 IEEE Symposium on Computational Intelligence in Bioinformatics and Computational Biology, La Jolla, CA, USA, 15 November 2005; pp. 262–267. [Google Scholar]
- Meiler, J.; Muller, M.; Zeidler, A.; Schmaschke, F. Generation and evaluation of dimension-reduced amino acid parameter representations by artificial neural networks. J. Mol. Model. 2001, 7, 360–369. [Google Scholar] [CrossRef]
- Jones, D.T.; Ward, J.J. Prediction of Disordered Regions in Proteins from Position Specific Score Matrices. Proteins 2003, 3, 573–578. [Google Scholar] [CrossRef] [PubMed]
- Pruitt, K.D.; Tatusova, T.; Klimke, W.; Maglott, D.R. NCBI Reference Sequences: current status, policy and new initiatives. Nucleic Acids Res. 2009, 37, 32–35. [Google Scholar] [CrossRef] [PubMed]
- Monastyrskyy, B.; Fidelis, K.; Moult, J.; Tramontano, A.; Kryshtafovych, A. Evaluation of disorder predictions in CASP9. Proteins 2011, 79, 107–118. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Window Sizes | 25 | 35 | 45 | 55 | 65 | 75 | 85 |
|---|---|---|---|---|---|---|---|
| 0.7762 | 0.7991 | 0.8156 | 0.8245 | 0.8344 | 0.8430 | 0.8373 | |
| 0.7806 | 0.7805 | 0.7752 | 0.7739 | 0.7637 | 0.7556 | 0.7645 | |
| 0.5568 | 0.5796 | 0.5908 | 0.5984 | 0.5980 | 0.5990 | 0.6018 | |
| 0.4872 | 0.5053 | 0.5124 | 0.5181 | 0.5147 | 0.5133 | 0.5180 |
| Window Sizes | 7 | 9 | 11 | 13 | 15 | 17 | 19 |
|---|---|---|---|---|---|---|---|
| 0.8264 | 0.8204 | 0.8245 | 0.8254 | 0.8221 | 0.8199 | 0.8191 | |
| 0.7649 | 0.7694 | 0.7738 | 0.7718 | 0.7714 | 0.7734 | 0.7722 | |
| 0.5913 | 0.5898 | 0.5984 | 0.5972 | 0.5933 | 0.5933 | 0.5913 | |
| 0.5096 | 0.5098 | 0.5181 | 0.5165 | 0.5131 | 0.5139 | 0.5119 |
| Nneur1 | ||||
|---|---|---|---|---|
| 0.8291 | 0.7679 | 0.5967 | 0.5148 | |
| 0.8245 | 0.7739 | 0.5984 | 0.5181 | |
| 0.8237 | 0.7749 | 0.5986 | 0.5186 |
| 0.8245 | 0.7739 | 0.5984 | 0.5181 | |
| 0.8231 | 0.7676 | 0.5907 | 0.5100 | |
| 1 | 0.7633 | 0.8106 | 0.5738 | 0.5123 |
| Schemes | ||||
|---|---|---|---|---|
| With preprocessing | 0.8245 | 0.7739 | 0.5984 | 0.5181 |
| Without preprocessing | 0.7291 | 0.7723 | 0.5014 | 0.4399 |
| Schemes | Speed(s) | ||||
|---|---|---|---|---|---|
| Including PSSM | 0.8245 | 0.7739 | 0.5984 | 0.5181 | 1340.0 |
| Excluding PSSM | 0.7508 | 0.7809 | 0.5317 | 0.4671 | 2.9160 |
| Schemes | ||||
|---|---|---|---|---|
| MLP | 0.8245 | 0.7739 | 0.5984 | 0.5181 |
| SVM | 0.7643 | 0.7765 | 0.5408 | 0.4729 |
| Fisher | 0.8127 | 0.7660 | 0.5787 | 0.5000 |
| Schemes | ||||
|---|---|---|---|---|
| DISpre | 0.7910 | 0.7836 | 0.5746 | 0.5006 |
| ESpritz | 0.7255 | 0.8135 | 0.5389 | 0.4840 |
| IsUnstruct | 0.7513 | 0.7855 | 0.5368 | 0.4711 |
| Schemes | ||||
|---|---|---|---|---|
| DISpre | 0.7482 | 0.8618 | 0.6100 | 0.4707 |
| ESpritz | 0.6884 | 0.9115 | 0.6000 | 0.5178 |
| IsUnstruct | 0.6882 | 0.8845 | 0.5727 | 0.4663 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, H.; Zhao, J.; Sun, G. The Prediction of Intrinsically Disordered Proteins Based on Feature Selection. Algorithms 2019, 12, 46. https://doi.org/10.3390/a12020046
He H, Zhao J, Sun G. The Prediction of Intrinsically Disordered Proteins Based on Feature Selection. Algorithms. 2019; 12(2):46. https://doi.org/10.3390/a12020046
Chicago/Turabian StyleHe, Hao, Jiaxiang Zhao, and Guiling Sun. 2019. "The Prediction of Intrinsically Disordered Proteins Based on Feature Selection" Algorithms 12, no. 2: 46. https://doi.org/10.3390/a12020046
APA StyleHe, H., Zhao, J., & Sun, G. (2019). The Prediction of Intrinsically Disordered Proteins Based on Feature Selection. Algorithms, 12(2), 46. https://doi.org/10.3390/a12020046