Abstract
In this paper, we present our generalized kinetic Monte Carlo (kMC) framework for the simulation of organic semiconductors and electronic devices such as solar cells (OSCs) and light-emitting diodes (OLEDs). Our model generalizes the geometrical representation of the multifaceted properties of the organic material by the use of a non-cubic, generalized Voronoi tessellation and a model that connects sites to polymer chains. Herewith, we obtain a realistic model for both amorphous and crystalline domains of small molecules and polymers. Furthermore, we generalize the excitonic processes and include triplet exciton dynamics, which allows an enhanced investigation of OSCs and OLEDs. We outline the developed methods of our generalized kMC framework and give two exemplary studies of electrical and optical properties inside an organic semiconductor.
1. Introduction
Organic semiconductors represent a promising candidate for cost-efficient, environmentally-friendly materials and renewable energy conversion devices. The advantage compared to currently widely-used inorganic semiconductor-based devices is captured in their mechanical flexibility, light weight and electrical and optical controllability at the molecular level. Organic semiconductors have exhibited a considerable potential for application in organic thin-film transistors (OTFT) [1], light-emitting diodes (OLEDs) [2] and solar cells (OSCs) [3,4]. Experimental research on organic-based devices is accompanied by computational methods providing a better understanding of the underlying processes in energy conversion devices made from nanostructured organic materials.
In the simulation of organic materials and devices, several methods on different length scales are of interest. While drift-diffusion (DD) simulations yield a good understanding at a macroscopic continuum level of several , they can neither provide insight into the microscopic, internal properties, nor track the particle dynamics. Tools solving molecular dynamics give detailed insight into the processes occurring on an atomistic length scale (), but the computational demand is not feasible for the simulation of bulk materials or thin films of a thickness. Kinetic Monte Carlo (kMC) simulations have gained more and more attention in the field of materials science in recent years. Due to strong localization of charge carriers on molecules or molecular subunits, kMC is highly feasible for the modeling of organic disordered semiconductors. The aim of understanding the internal processes in organic devices and the rising available computational power has driven the development of the above microscopic numerical methods. Existing kMC models can reproduce the main macroscopic characteristics of organic optoelectronic devices and provide insight into fundamental physical properties considering charge transport and optical processes. Thus, kMC provides a bridge between different length scales from the atomistic level up to a macroscopic device level.
Ries and Bässler have presented one of the first applications of kMC in the study of charge transport in organic materials [5]. From that point on, kMC has been employed for many theoretical studies covering charge and exciton transport [6,7,8], recombination of charge carriers [9,10] and the impact of all processes on the photocurrent generation [11,12,13,14,15]. kMC simulations have been frequently utilized to study full devices such as bulk-heterojunction (BHJ) organic solar cells [10,11,12,13,15,16]. There are several groups working on kMC models describing all properties mentioned above to a certain extent. Many of them consist of a cubic lattice model, which cannot represent all geometrical features and the impact of the structure of organic materials on the physical processes. Furthermore, many do not consider the directionality of charge transport along, e.g., polymer chains. Another limitation of existing models is the simplified treatment of excitonic processes, which are of high importance regarding organic devices. Most kMC models either account for singlet [10,17,18,19] or triplet exciton dynamics [20,21,22,23] for the use in OSCs or OLEDs, respectively. However, the interplay between singlets and triplets can be used to modify exciton lifetimes and diffusion lengths, which is a promising way to tailor efficiencies in organic devices and therefore requires a better understanding [24,25,26].
In this work, we present our in-house-developed generalized kMC framework for the simulation of organic semiconductors and devices. Our model overcomes the mentioned limitations by the inclusion of a non-cubic lattice model, based on the Voronoi tessellation, and a model of polymer chains. Both yield a better geometrical description of organic materials and herewith provide new insight into the electrical properties. Charge transport is a strongly percolative process [27,28]. Thus, a generalized lattice model is of high importance in the kMC method. Considering the excitonic properties, we present a detailed model of exciton dynamics including both singlet and triplet excitons and their interaction. We present two exemplary studies on electrical and optical properties of organic materials using results obtained with our generalized kMC model.
In Section 2, we give an overview of the kMC method and show its applicability for the simulation of organic materials. A detailed description of the non-cubic Voronoi set, polymer chains, the model of the energetic landscape and excitonic processes is presented in Section 3. Section 4 discusses two exemplary studies. Firstly, the charge transport properties of organic materials with the inclusion of polymer chains is presented. We investigate the impact of the directionality of hopping processes on the mobility. Secondly, the effective lifetime of excitons inside an organic material is studied. We focus here on the impact of the dopant volume concentration on the effective exciton lifetime.
2. Kinetic Monte Carlo Method
Monte Carlo (MC) simulations refer to a broad class of numerical algorithms utilizing random numbers to solve real-world problems. The first Monte Carlo methods were developed during the 1940s within the Manhattan project to study neutron diffusion in fissionable materials. During that time, the Metropolis algorithm [29] originated, which is until today a convenient technique to study equilibrium properties of physical systems. However, the Metropolis algorithm is not capable of modeling the time-dependent evolution of a system. The kinetic Monte Carlo method is a specific MC method that allows propagating a physical system from state to state including the corresponding time information of the propagation steps. Therefore, kMC simulations are often termed simulated experiments because they essentially mimic the explicit evolution of a real-world system. The step by step evolution, where each time-step depends only on the previous one, fulfills the properties of a Markov chain and makes the kMC algorithm a sequential algorithm. kMC methods have gained large popularity in a variety of fields due to their ease of implementation and the increasing computational power of modern computers. The modern version of the kMC method is based on the work of Gillespie [30] and Bortz et al. [31]. For further information for the reader, an illustrative description of the method was provided by Voter [32].
To model a system, one has to identify the spatial dimensions and the time scales on which the processes determining the properties of the system occur. The kMC method commonly is referred to as a simulation technique at the mesoscale, in between molecular dynamics (MD) on the microscopic and hydrodynamic models on the macroscopic scale. In comparison to MD, which typically resolves systems on the timescale on which molecular vibrations and rotations occur (of the order of femtoseconds), the kMC method is based on the concept of coarse-graining the system dynamics. A system is characterized by a set of long-term states with discrete transitions between them (so-called infrequent events); all transitions on smaller timescales are neglected as long as they do not change a long-term state (illustrated in Figure 1a). Coarse-graining allows one to neglect a sufficient amount of (fast) processes and, thus, reach much longer overall simulation times up to seconds. Only transitions between long-term states determine the dynamic evolution of the system. It needs to be carefully considered which processes are implemented in the model and which ones are combined in long-term states. This way, the kMC can accomplish a tradeoff between accuracy and simulation time.
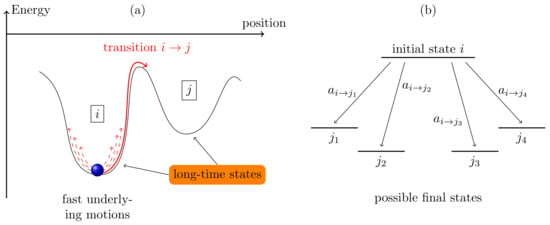
Figure 1.
(a) Coarse-graining of the system dynamics and illustration of long-term system states; (b) transition from initial state i to final states j as employed in the kinetic Monte Carlo (kMC) algorithm.
For the application of the kMC method, the system to be modeled needs to be characterized by a set of long-time system states . These can be, for example, the entirety of positions and energies of all particles currently in the system. For a system being in a certain state i, a set of possible states j exists to which a system may evolve (illustrated in Figure 1b). Every transition is accompanied by a transition rate (given in units of ), describing the physical process leading to the transition . The magnitude of the rates is proportional to their probability of execution, i.e., larger rates (faster processes) are more likely to occur than smaller rates (slower processes).
The temporal evolution of discretized transitions between states is phenomenologically described by the master equation [33]:
where gives the probability to find the system in state at time t. An analytic or numerical solution of the master equation is often not feasible. The kMC method provides a numerical approach to essentially execute the master equation to stochastically propagate the system between its long-term states. If the coarse-graining is chosen accurately such that the correlation time of the environment in the continuous-time dynamics is much shorter than the typical rate constants, the discretized system evolution can be regarded as Markovian. A single trajectory of state transitions does not yield the same system behavior as described by the master equation. Averaging over several trajectories provides an equivalent system behavior one would obtain by solving the master equation.
The dynamical evolution of the system is governed by the transitions rates. The rates are inputs to the kMC model and can either come from experimental measurements or are based on underlying theoretical models. Given rates, the kMC algorithm is straightforward and can be reduced to the following scheme. For a detailed description of the derivation, we refer to [30,34].
- Initialize the system, and define all parameters that are required for the transition rates of all processes in the kMC. Define termination conditions such as the maximum simulation time .
- Identify the activated transitions for the current system state i and calculate the transition rates of all physical processes that can occur. The rates mirror the probability of the occurrence of the transition to state j. The sum of all rates is calculated.
- Two uniformly-distributed random numbers and are chosen. The first number chooses the transition from the set of events , which fulfills the condition:The second number gives the time passed before the transition is performed, given by:
- The simulation time t is advanced by , and the system configuration is updated following the transition .
- If the simulation time exceeds , the simulation stops, and all particle trajectories and the counters tracking the events are evaluated. If , the kMC loop (Step 2–4) is repeated.
For simple systems such as unipolar single layer devices, the master equation has been applied successfully [35,36,37]. However, critical properties such as the electrostatic Coulomb interaction between charge carriers can only be captured by an effective potential in the framework of the master equation. With the particular application to OSCs in mind, particles like excitons, electrons and holes reside in localized states of either monomers of a polymer or small organic molecules. Charge motion can occur by processes on shorter timescales, but as long as they do not perform a hopping process to another localized state, the particles are spatially assigned to that state. The overall dynamical behavior is determined by hopping events between localized states. This concept has been applied to model charge transport in disordered organic semiconductors, as well as organic devices such as OSCs or OLEDs. A significant contribution of describing charge transport has been conducted by Bässler [6]. Several models for OSCs have been set up investigating active layer morphology and exciton separation [11,17], recombination [10,14,38], Coulomb interaction [12] and charge densities [13].
3. kMC Framework for Organic Materials and Devices
In this section, we focus on the new features of our kMC framework extending the previously shown model [10,13]. A generalized representation of the organic material is presented, which covers the model of a non-cubic Voronoi set and polymer chains. Furthermore, dopants are included, which need to be taken into account for the simulation of all excitonic processes in organic devices. Correlations of the energy landscape are of importance for many materials. We present a simple weighted average approach in Section 3.2. The Voronoi tessellation and the correlation procedure have been discussed recently in more detail [39]. As they are part of our generalized kMC model, we provide a concise summary. We conclude this section with a detailed description of the particles and transitions containing the information of the physical properties.
3.1. Modeling of the Organic Material
3.1.1. Voronoi Tessellation
With the rising amount of computational power and powerful clusters, investigations on the detailed structure of organic materials and their influence on the physical properties using molecular dynamics (MD) have gained rising interest in the community [40,41,42]. This raises the demand of a generalized lattice representation for kMC studies [6,43,44,45].
For a representative model of the active organic layer, the use of the Voronoi tessellation generalizes the cubic lattice model [39,46]. Using the Voronoi cell library of Rycroft [47], next-neighbors of each lattice site, as well as the distance and all face-areas are computed. In Figure 2, the procedure is shown for the cubic lattice (left) and the non-cubic, Voronoi set (right) in a simple 2D representation. Starting from an arbitrary grid point, the perpendicular bisectors to all neighboring sites in proximity are computed. The algorithm obtains the volume closest to the considered starting point, named the Voronoi cell, as known from the Wigner–Seitz cell in periodic crystals. All neighboring sites sharing one face with the considered Voronoi cell are taken as next-neighbors. The knowledge of these is fundamental for the computation of all physical rates.
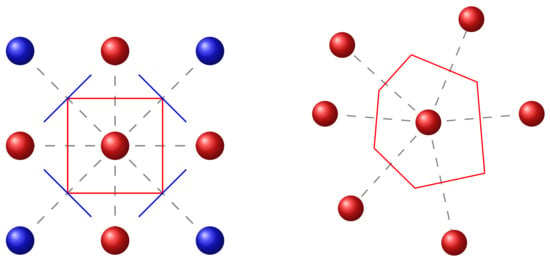
Figure 2.
Visualization of the next neighbor generation in a cubic lattice (left) and a Voronoi set (right). For most physical processes, only the nearest-neighbors, highlighted in red color, are considered. The blue lattice sites do not define the Voronoi cell; thus, they are not labeled as next neighbors.
Comparing the cubic with the off-lattice approach, some obvious differences and their importance are to be highlighted. Due to the spatial disorder of the sites, the number of next-neighbors increases. Moreover, the major drawback of an equidistant spacing between lattice points in the cubic model is overcome with the use of the Voronoi set [48]. The local distribution of lattice sites can be imported from MD simulations using the center of masses of the molecules, providing a sophisticated multi-scale simulation method comparable to [42,43]. In the case of available quantum chemical simulations, the localized quantum state could directly be used as the input of the sites. Herewith, the information depth of the structural alignment of, e.g., polymers or small molecules down to the nanoscale can be represented on a mesoscopic level. Besides, amorphous domains in both polymers and small molecules can be explicitly investigated, which makes the study of the physical properties more realistic. In the case of the exciton diffusion (see Section 3.3.2), non-nearest neighbors are required. The Voronoi tessellation supports a fast iteration method to obtain all neighbors within a certain radius around the centroid.
Existing off-lattice models take into account all adjacent molecules within a certain radius [43,49]. Depending on the density of sites, this may cause a strong increase in simulation time. Using the Voronoi tessellation, only adjacent sites are considered, which keeps the computation time feasibly low. In the case of a strong disorder, a common radius might lead to the case that certain sites are fully disconnected from the surrounding material. This is avoided with the Voronoi tessellation. The tessellation furthermore allows the computation of local current and charge densities as a well-defined area of the faces, and the volume of the Voronoi cells is obtained [39]. Besides all the advantages, it still needs to be considered that the cell boundaries are purely based on the distance between centroids and do not contain any physical information, which, depending on the modeled system, has to be evaluated carefully for the computation of local quantities.
3.1.2. Polymer Chains
Organic materials used in organic devices can be distinguished into two separate categories, small molecules and polymers. Polymers are composed of repeating macromolecules called monomers forming long chains. One prominent example for polymers used in OSCs is poly(3-hexylthiophene) (P3HT) [3]. In this section, we briefly emphasize the importance of the correct model of polymer chains in our kMC model and present the algorithm for the chain morphology generation.
The spatial overlap of molecular orbitals leads to weaker bound and less localized electron states along polymer chains [50]. Therefore, charge transport along polymer chains, called intrachain transport, is faster than charge transport between two separate polymer chains, which we refer to as inter-chain transport [51,52,53]. Vanlaeke et al. showed that crystallization of P3HT increases the mobility [54]. Sirringhaus et al. have demonstrated that the mobility strongly depends on the orientation of the micro-crystalline domains in semi-crystalline P3HT [55,56]. In situ grazing incidence X-ray scattering techniques can be exploited to gain information on the polymer crystallization and ordering [57].
Many present kMC models treat every site as an independent localized state, and charge transport to all of its neighbors is described with the same mechanism [6,58]. Multi-scale simulation models combining molecular dynamics and kMC include the distance dependence and directionality captured in the coupling integrals calculated from MD simulations, but did not investigate the effect of polymer chains on mobility [42,43]. Mollinger et al. model single charge transport between crystalline regions based on kMC along a generated polymer chain and investigate the impact of the electric field on the single charge mobility [44]. However, the probability of leaving the chain is modeled with an effective, field-dependent exit rate and does not account for neighboring chains. To account for both inter-chain and intra-chain transport, it is necessary to generate a morphology in which all the sites belonging to a polymer chain are connected.
The algorithm for the chain morphology generation is based on a self-avoiding random walk [59]. It starts at a random free site, a node that is not yet part of a polymer chain. Then, all free neighbors of the site are determined. Free neighbors are next-neighbors, which are not already part of another polymer chain. One of these free neighbors is randomly chosen and connected to the chain. Every node stores a link to the previous and next chain element. From this newly-added node, again, the free neighbors are determined, and one is added to the chain. This procedure is repeated until the desired chain length is reached or there are no longer any free neighbors. Then, a new free site is searched, and the next polymer chain is generated until the whole simulation box is filled with chains. In our model, we assume that the chain length in organic materials follows a Gaussian distribution. Therefore, the average chain length and its standard deviation are necessary input parameters for the algorithm. The desired chain length for every single chain will then be calculated according to the Gaussian distribution function. Chain length and distribution can be measured experimentally using, e.g., gel permeation chromatography [60] or static light scattering [61].
According to Carbone and Troisi [51], the orientation of the polymer chains is of great importance for the overall transport characteristics of organic materials. Therefore, we account for a preferred direction and the amount of orientation of the polymer chains, including three distinguished chain models. Gaussian chains are flexible and show no preferred orientation. They are obtained by the random walk algorithm described above. Rigid chains are entirely directed polymers pointing in one direction. The crossover between these two modes is described by the worm-like chain model, where a local stiffness is added to the polymer yielding to semi-flexible chains with a preferred direction. The parameter controls the amount of stiffness, where a value of zero results in flexible Gaussian chains and a value of one leads to rigid chains where the polymers can only be generated along the preferred axis. For values in between, the probability for connecting sites to the chain along the preferred axis is increased, while in-plain probability or backward growth is reduced. We use the equations:
to account for these probabilities. Figure 3 shows the morphologies generated with our algorithm. The two single material morphologies show the Gaussian chain model () on the left and the worm-like chain model with high directionality of in the middle. The 2D case with regular grid is chosen for a better visibility, while the algorithm can be used to generate chains in 3D. For an irregular Voronoi set, the distance vector to the neighboring site is decomposed into in-plane and parallel components. The probability for adding a site to the polymer is obtained by averaging its components calculated with Equations (4)–(6).

Figure 3.
Chain morphologies on a two-dimensional grid with an average polymer chain length of six monomers. Single material with Gaussian chains (left), worm-like chains with preferred orientation (middle) and chains in a BHJ where only the blue material is a polymer and the red material is small molecules (right).
It is also possible to generate polymer chains in BHJs. The desired chain length and orientation can be specified for each material individually. In the right part of Figure 3, we visualize a blend of one polymeric material (e.g., P3HT) and one small molecule material (e.g., PCBM). This tool for morphology generation leads to a more realistic representation of a polymeric material and herewith provides better insight into charge transport properties in organic materials and devices on a mesoscopic scale.
3.1.3. Phosphorescent Dopants
OSCs are one representative of the class of excitonic devices. In contrast to their inorganic counterparts, an absorbed photon does not directly excite a free electron-hole pair, but forms an exciton with binding energies exceeding . A morphology that introduces a high exciton splitting efficiency is the bulk-heterojunction (BHJ) [3,62]. The BHJ reduces the distance between the generation position of an exciton and the donor-acceptor interface. This distance should be as short as possible, as singlets have short diffusion lengths of 3–6 [63]. If the distance between origin and interface exceeds the diffusion length, singlet excitons probably decay before reaching an interface and cannot be converted into usable electrical energy.
Another possibility of enhancing the splitting efficiency is to enhance the exciton lifetime and diffusion length. Triplet excitons exhibit an effective lifetime of for P3HT, which corresponds to a diffusion length of [63]. The spin selection rules forbid the radiative decay of triplet excitons. However, due to the spin-orbit coupling triplets may radiatively decay, but this is very improbable for commonly-used organic materials. Triplet excitons cannot be generated by the photon absorption as it is forbidden by spin-selection rules [64]. In the presence of strong spin-orbit coupling, the spin-selection rules are lifted. Thus, singlet excitons can be converted into triplet excitons. This process is known as intersystem crossing (ISC) [64].
Incorporating heavy metal atoms into the organic layer induces a strong spin-orbit coupling. Doping of a BHJ with nanoparticles can increase the effective exciton lifetime and thus improve the power conversion efficiency [24,25,65,66]. Furthermore, in phosphorescent OLEDs, organometallic complexes are used to improve the efficiency. The electrically-generated triplet excitons decay radiatively and help thus to enhance the efficiency of the OLED [67,68].
Dopant sites are implemented to investigate the impact of doping with strong spin-orbit materials in OSCs or OLEDs. Dopant sites are assigned randomly to the lattice sites in the simulation box. The ratio of dopant to organic sites gives the volume concentration of the dopant. The spin-orbit coupling of an atom affects excitons only in its proximity. Thus, a radius is introduced, in which the spin-orbit coupling influences the excitons. As explained in Section 3.3, the intersystem crossing is activated within this radius.
3.2. Energetic Landscape
The energetic landscape of an organic semiconductor plays a significant role in the charge transport and exciton dissociation [10,27,28]. In contrast to inorganic semiconductors, the molecular orbital energies inside an organic material are strongly disordered due to the non-crystalline arrangement of the molecules and polymers [28]. Energetic disorder values of are present in many materials [6]. It is widely accepted that the shape of the density of states (DOS) can be modeled using a Gaussian distribution [15,27,36,58,69,70]. Some experimental observations require a correlation in the site energies, as described in the correlated disorder model (CDM) [15,36,71,72], while many studies are based on the uncorrelated, Gaussian disorder proposed by the Gaussian disorder model (GDM) [6,58,72].
We implement a correlated disorder model based on a simple averaging method of site energies. Figure 4 visualizes the CDM for an organic system consisting of PCBM (black dots) and P3HT (red dots). First, for every site, we choose an energy randomly from a Gaussian distribution:
with the molecular orbital energy and the global energetic disorder . Herewith, the energy distribution in the GDM is obtained. For the CDM, the following procedure is performed.
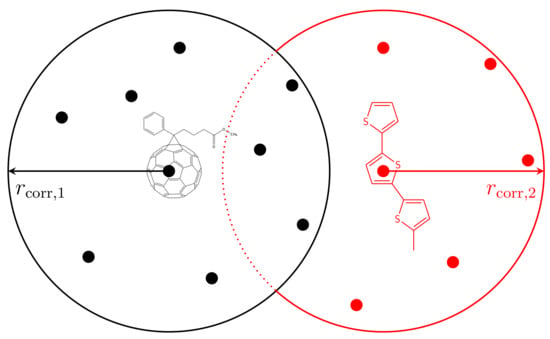
Figure 4.
Correlation of site energies inside a blend. The black and red dots represent lattice sites of the small molecule PCBM and the polymer P3HT, respectively. A weighted average is performed over all sites of the same material type within the radius of .
- Starting from the randomly Gaussian-distributed energetic landscape, we compute the weighted average of the energies of all sites k with for every site i using:Note that the average is only performed over sites k representing the same material as site i. The average energies are stored in an intermediate vector.
- Replace the site energies by the average energies .
- Scaling of the site energies: The averaging procedure reduces the energetic disorder. To keep the initially chosen , we compute the energetic disorder after the averaging process. Then, the site energies are scaled by:
- The level of correlation can be varied by either changing or repeating Steps 1–3.
A measure of the level of correlation is defined by the correlation function . The correlation of sites i and j can be obtained with:
If we take the average of the correlation of the ensemble of pairs () with the same distance , we obtain a measure of the correlation of the whole organic material. Using this correlation mechanism, we have shown that a high correlation in the site energies gives rise to filamentary structures of the local currents [39]. For distances below , the correlation function obtained by atomistic simulations [73] has been reproduced with our mechanism, while for long-range hopping, other procedures, e.g., a dipole-based correlation [36], give better results. While a dipole-based correlation is obtained by summation over all dipoles within the simulation box [36,71], the presented method is computationally favorable as only sites within a certain radius are considered.
3.3. Particles and Transitions
3.3.1. Charge Carriers
In disordered organic materials, charge carriers are spatially localized due to the lack of long-range order. Charge transport is modeled as a hopping process from one localized state to a neighboring localized state. This state can be seen as local energy valleys where charges locate in a long-term, which e.g., represent a small molecule or a few monomers along a polymer chain.
Miller and Abrahams describe hopping as a combination of thermal activation and tunneling [74]. The hopping rate between states i and j reads:
where is the attempt-to-escape frequency, is the inverse localization length, is the Boltzmann constant, T gives the temperature, the distance and the energy difference between the initial configuration i and the final configuration j, before and after a hopping process, respectively. When hopping upwards in energy, the Boltzmann term accounts for thermal activation [74]. This rate model is usually used when the coupling between electrons and phonons is weaker [75,76].
Marcus has introduced a model that accounts for stronger coupling between lattice and charges [77]. Because of the low permittivity of organic materials, charges lead to local polarizations of the lattice, which are described as quasi-particles called polarons. The formula for polaron transitions reads:
with the reduced Planck’s constant ℏ, transfer integral and the reorganization energy describing the reconfiguration of the molecule due to the deformation by the charge. The effect of a spatial disorder in an irregular Voronoi set is included via the site to site distance in Miller–Abrahams rates (Equation (11)) and via the distance-dependent transfer integral in Marcus rates (Equation (12)). The hopping rates are calculated for every particle and all free neighbors. If a particle already occupies a neighboring site, the hopping rate is zero. When a hopping transition occurs, the occupation information of origin and destination sites and the particle position have to be updated.
To calculate hopping rates, it is necessary to compute the electric energy for every site i. The energy at site i is given by:
with being the energy level of the molecular orbital and the energetic disorder . The energy arising from the external applied field is referred to as and represents the Coulomb energy. The Coulomb interaction between all particles is calculated using the Ewald sum [12,78]. The Ewald sum makes use of the principle of image charges. As the potential at the contact layers has to be constant, all the field vectors have to end perpendicular to contacts. This can be achieved by removing the contact and introducing an image charge of opposite sign and the same distance behind the contact. Since an OSC device has two contacts, every image charge behind one contact also induces an image charge behind the other contact, leading to a periodic scheme. The electrostatic potential at node i can be calculated by:
where the outer sum corresponds to the periodic replica in the x-, y- and z-direction and the inner one sums over all charged particles inside the box . The prime in the inner sum notes that self-interactions are excluded. Here, is the vacuum permittivity and is the permittivity of the material.
The Miller–Abrahams rate provides a generic model for the hopping process modeling the contribution of tunneling and thermal activation by the parameters , and the Boltzmann term, respectively. It is used if hopping is mainly caused by single phonon-assisted tunneling between sites of energy and . Using Marcus theory, one implicitly accounts for molecular details including and . It is frequently used in multiscale studies, as for given organic molecules, the molecular positions and transfer integrals need to be calculated by molecular dynamics and quantum chemical calculations, respectively [42,43,79,80]. Marcus theory accounts for multiphonon hopping processes.
Recombination of charge carriers is possible when an electron and a hole are located at neighboring sites. Charge recombination is one of the primary loss processes in OSCs besides exciton decay. When recombination occurs, the two particles are removed, and the sites are free again.
3.3.2. Excitons
An exciton is an intramolecular excited state between a Coulomb-bound electron-hole pair [64]. Compared to the weakly-bound Wannier–Mott excitons present in inorganic semiconductors, strongly-bound Frenkel excitons are formed in organic materials due to the low dielectric constant [3,81]. The excited state is a singlet (triplet) state, if the spins of the electron and the hole are anti-parallel (parallel). We will now describe all physical rates modeled for both singlets and triplets, as well as the conversion rates between these excitons. For a detailed discussion on the photophysical processes, please refer to [64,82]. A schematic overview of the rates and the corresponding energetic levels are depicted in Figure 5. The singlet (blue) and triplet excitons (red), residing at the energetic levels and , respectively, as well as the separated electron-hole pair forming a charge-transfer (CT) state and the ground state are visualized.
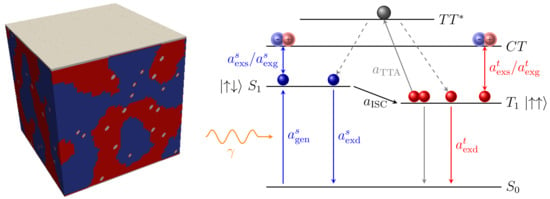
Figure 5.
(Left) Bulk-heterojunction including dopant sites with a volume concentration of . (Right) Jablonski diagram illustrating the exciton transitions. All processes considering triplet excitons are visualized in red color, while singlet transitions are highlighted in blue. The diagram shows the lowest excited singlet () and lowest excited triplet state (), the singlet ground state () and charge transfer state (). Our excitonic model consists of the optical singlet exciton generation () upon the absorption of an incident photon , singlet/triplet exciton separation (), electrical generation () and decay (), triplet-triplet annihilation () and intersystem crossing (). Two triplet excitons can annihilate with a rate forming an intermediate state , which relaxes to either the lowest singlet or lowest triplet state .
Singlet excitons can be generated upon light absorption. Absorption of a photon excites one electron, resulting in a singlet exciton. The optical generation profile of singlets is modeled using a transfer matrix model (TMM) [83,84,85]. The TMM is used to calculate the generation profile in the active layer under an AM1.5 illumination spectrum. For every layer in penetration direction z, a constant generation rate is obtained. As the TMM does not provide the generation profile perpendicular to the penetration direction, the x and y position of the generated singlet is chosen randomly [10,13].
The generation of both singlets and triplets can occur by electrical excitation. The rate for electrical generation of a singlet/triplet is enabled if two charges of different polarity reside at neighboring sites and if the respective spins, a constant property of the charge carriers, show a singlet/triplet configuration. Depending on the origin of charges, we assign their spin states following a distinguished procedure. If a charge is injected, we allocate the spin following the expected singlet to triplet ratio of 1:3. If an exciton dissociates, the spin states of the charges reflect the spins of the singlet or triplet excitation. We use the convention that holes always have a spin with eigenvalue (spin-down), while the spin of the electrons is assigned such that the above criteria are fulfilled. Following Mesta et al. [23], the electrical generation rate can be calculated as a hopping process of the electron on a hole-occupied neighboring site using the Miller–Abrahams equation (Equation (11)) [74]. In comparison to Mesta et al., our model only considers electron hops for the electrical exciton generation. The energy difference for the singlet/triplet generation is given by:
where denotes the exciton binding energy. gives the charge transfer binding energy, corresponding to the Coulomb binding energy of an electron-hole pair residing at neighboring sites, and defines the LUMO energy level difference of the neighboring sites. Coulomb interactions with the surrounding charge carriers are omitted. If an electrical generation event is chosen, the electron and hole are removed, and a singlet/triplet exciton is generated at the site the former hole was located.
All excited states have a certain lifetime before they decay to the ground state. The lifetime of triplets exceeds that of singlets due to the spin selection rules by far. The rate for the exciton decay is chosen as the inverse of average lifetime . Due to the spin-orbit coupling of a phosphorescent dopant, triplets may decay due to the lifted spin-selection rules. Thus, the triplet lifetime is reduced within the radius around dopant sites.
Excitons are quasi-particles with a net charge of zero. Thus, they diffuse through the organic semiconductor by energy transfer between sites. Existing kMC models account for exciton diffusion using either Förster resonance energy transfer (FRET), Dexter type transfer or are based on a random walk [19,86,87,88]. The diffusion of singlets and triplets can be modeled using FRET [89] or by a random walk based on the Einstein–Smoluchowksi model for the Brownian motion with a diffusion coefficient D [90,91]. Förster described a non-radiative energy transfer between an exciton donor and acceptor as a dipole-dipole interaction [89]. As the Coulomb interaction causes this energy transfer, FRET is a long-distance mechanism. For the random walk (RW), the hopping rate is computed as the inverse of the mean time interval for a hopping process between neighboring sites, which gives the rate:
where l is the next-neighbor distance. Diffusion rates by FRET are modeled with:
where is the Förster radius, is the total decay rate, is the exciton energy difference between the initial and final configuration and denotes the distance between two sites. The computational effort of random walk is typically 2–3 orders of magnitude smaller compared to FRET as the number of sites considered for the FRET diffusion increases with , while RW diffusion only considers next-neighbors [19]. Especially for a low energetic disorder, RW and FRET give similar results [19], with rising energetic disorder FRET needing to be considered.
In case of the triplet diffusion, FRET is a spin-forbidden process and can only occur between phosphorescent dopants within the Förster radius . A second mechanism for the diffusion comes into play. In contrast to the Förster rate, the Dexter type energy transfer rises due to the exchange interaction and may occur between next neighbors [92]. Following [22], the Dexter type hopping is modeled as a Miller–Abrahams hopping defined by:
with the exciton energy difference of initial state i and final state j, the Dexter hop rate in the zero distance limit and the inverse localization length of donor and acceptor sites [64]. The triplet binding energy is usually assumed to be equal for every site, but can be modified by a disorder following a Gaussian distribution. The Boltzmann factor vanishes in the case of no energetic disorder in the binding energy. Both transfer mechanisms are shown schematically in Figure 6.
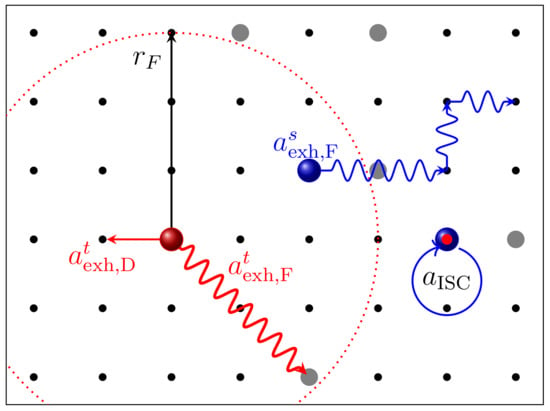
Figure 6.
Schematic diagram illustrating the hopping processes of singlet (blue spheres) and triplet (red spheres) excitons. The black dots represent the organic material, while gray dots label dopant sites. FRET is shown for a triplet exciton hopping between dopant sites within the Förster radius . The Dexter type energy transfer is possible between all adjacent sites. The intersystem crossing is visualized for a singlet exciton in proximity to the dopant as a conversion of a singlet to a triplet exciton.
To extract electrical energy from the absorbed photons in organic solar cells, singlets need to be dissociated. This process is only possible if singlets reach an acceptor-donor interface. The dissociation process is reported to happen on extremely fast timescales in BHJs [93]. A separation rate is enabled if the singlet is located at an interface. Dissociation of a triplet exciton is interpreted as an electron hopping from its current position towards an unoccupied neighboring site [21,22,23]. The separation rate is calculated with the Miller–Abrahams rate using an energy difference of . The dissociation is probable if the state following a dissociation process is energetically lower than the triplet binding energy.
Regarding high spin-orbit coupling, intersystem crossing from a singlet to a triplet state is possible. The reverse process is not considered here. In our model, a transition from singlets to triplets is only possible within a certain radius around a dopant molecule. The radius of intersystem crossing is a material dependent property. If an exciton resides at a site within , the intersystem crossing rate is enabled. The intersystem crossing rate is given by:
with an intersystem crossing frequency . The intersystem crossing decreases exponentially with the distance to a dopant site d. If the intersystem crossing occurs, the singlet is converted into a triplet exciton, residing at the corresponding site.
With rising concentration of triplets, another undesired process occurs, the triplet-triplet annihilation (TTA). If two triplet excitons meet, they either create a singlet or a triplet exciton [20,21,94,95]. The spin state of the resulting excitons depends on the spin angular momentum of the two involved triplet excitons. If a triplet exciton resides at a neighboring site or two involved triplet excitons reside at two dopant sites within the Förster radius, TTA is enabled. As explained before, TTA is interpreted as a triplet exciton hopping on a site occupied by a second triplet. If this transition is performed, at least one triplet exciton is removed. If the TTA process is Dexter-mediated, with a ratio of 1:3, the particle at the final site is either a singlet or a triplet, respectively. In the case of the Förster mediated TTA, one triplet is excited to a higher triplet level , followed by a relaxation to the lowest triplet exciton level . We assume the relaxation to happen immediately. The second triplet decays to the singlet ground state.
4. Applications
An inherent feature of the kMC method in comparison to, e.g., continuum models is the availability of the underlying time-dependent evolution of the modeled system. The kMC method allows the simulation of entire devices including effects at the nanometer scale. Single-particle dynamics, from which both the steady-state quantities and the transient behavior of the system can be extracted, are computed, and particle ensemble characteristics are evaluated.
In this section, we give two exemplary studies on organic materials using kMC. Firstly, the electrical properties of organic materials are investigated. We simulate the charge carrier mobility and current distribution in organic materials showing the effect of a correlated energetic landscape and the inclusion of polymer chains. Secondly, a numerical study of the impact of the dopant volume concentration on the exciton dynamics is presented. An analysis of the conversion efficiency between singlets and triplets is shown in single organic material, neglecting any charge transport properties.
4.1. Charge Carrier Transport through Polymer Chains
The charge carrier mobility is a crucial figure of merit for the applicability of organic semiconductors. While for some devices such as OLEDs, a low mobility is critical to optimize the performance, OSCs and OTFTs demand a high mobility. In semiconducting polymers, charge carrier mobility strongly depends on the morphology [54,55], but its interplay is not well understood [52,96]. Investigating the impact of different transport mechanisms, such as intrachain hopping, on carrier mobility can help the development of organic materials and improve device performance.
A single material cubic lattice of with periodic boundary conditions in all three spatial dimensions is used for the following investigation of the charge carrier mobility under the influence of intrachain hopping. For the energetic disorder, a Gaussian density of states with is used. The Miller–Abrahams rate given in Equation (11) is used for the hopping process with an attempt-to-escape frequency for inter-chain transitions and an inverse localization length of . Charges are allowed to hop to the six nearest neighbors on the cubic lattice. We run the simulation with four electrons placed in the simulation box resulting in a charge density of . This charge density is constant throughout the simulation because of the periodic boundary conditions. To account for the faster intrachain hopping, an attempt-to-escape frequency of , , is used to calculate the rates. Further simulation parameters are summarized in Table 1. For each electric field F, three simulation runs for a simulation time of are performed. The charge carrier mobility in the field direction is calculated by:
with the electric field in the z-direction and the average carrier velocity given by the distance traveled during the simulation time in the field direction. The results represent the mean value and the standard deviation of the mobility.

Table 1.
Simulation parameters for mobility considering polymer chains.
For this study, different morphologies with chain length are generated. We study both Gaussian distributed chains with and worm-like chains with oriented in the z-direction. The multiplication factor m is varied between one and 100. A factor of means that hopping along polymer chains is not preferred and the chain morphology does not have any impact. Thus, the blue curve in Figure 7a is a reference for comparing the impact of intrachain hopping. Because of the relatively high energetic disorder of , the carriers are slowed down due to the high energetic barriers between sites. At low fields, this slowing effect is dominant, and transport is mainly determined by diffusion. For higher fields, the impact of the energetic barriers becomes weaker, and transport is dominated by the drift field. For high fields, the carrier velocity saturates, and the mobility decreases with .
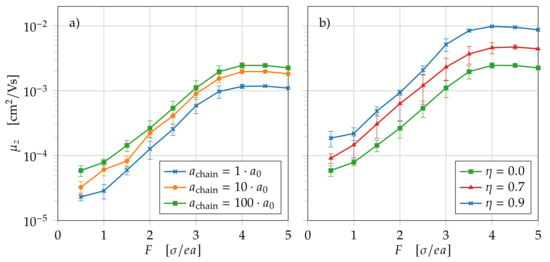
Figure 7.
Carrier mobility versus electric field F for different morphologies with chain length . (a) Fixed morphology with Gaussian chains and varying for intrachain transport; (b) morphologies with varying orientation for fixed .
Figure 7a shows the carrier mobility for completely undirected Gaussian chains. For higher intrachain hopping rates, the mobility increases slightly, but when increasing intrachain hopping rates by a factor of 100, mobility only increases by a factor of 2.2. When including very fast hopping along undirected polymer chains, only a very small amount contributes to the carrier mobility along the field direction. The limiting factor for carrier mobility is the slower inter-chain hopping. Polymer chain orientation the in field direction, on the other hand, shows a larger influence on carrier mobility. In Figure 7b, mobility curves with constant attempt-to-escape frequency and different orientation are shown. Mobility increases from Gaussian chains () to highly oriented chains () by a factor of 3.7. In total, the impact of polymer chains on carrier mobility adds up to a factor of 8.1 for highly oriented chains with and increased hopping rates of .
The combination of polymer chains and the non-cubic Voronoi set allows a realistic representation of the organic materials. In future work, the chains shall be generated from molecular dynamics simulations. With this, kMC can be used to gain insight into the device characteristics based on a realistic lattice model down to the sub-nanoscale.
4.2. Impact of Dopant Volume Concentration on Exciton Dynamics
Using the presented kMC framework, we study the impact of the dopant concentration on the exciton dynamics in an organic bulk material. We focus on the diffusion length and lifetime of excitons and analyze the number of singlets converted into triplets. With this simulation, which is rather theoretical, we aim to mimic the exciton dynamics and interplay of excitons within a single organic material without any interfaces, defects and contacts.
As for the charge transport simulation, a single material cubic lattice of with a lattice constant of and periodic boundaries in all spatial dimensions is generated. Rates for Dexter energy transfer and random walk are computed for the six nearest-neighbors, while FRET is considered for all neighbors within the Förster radius. Simulations are executed for different dopant volume concentrations varying from % up to %. As no contacts are included, charges cannot be injected or collected. Due to the lack of interfaces, exciton splitting cannot occur. Singlet excitons are generated with a constant optical generation rate . Triplet excitons can be induced upon intersystem crossing close to the phosphorescent dopants. In Table 2, the simulation parameters used are summarized.
Table 2.
Simulation parameters for the exciton dynamics.
In Figure 8, the dependence of the average lifetime and effective diffusion length on the dopant volume concentration c is illustrated for both singlet (a) and triplet excitons (b). The effective exciton diffusion length is determined using:
where is the Euclidean distance between the generation and final site of exciton i and denotes the number of either singlet or triplet excitons [82]. The presented results show the average effective diffusion lengths of eight configurations simulated for each dopant concentration, while the error bars indicate the standard deviation. For a concentration of %, an average singlet exciton lifetime of is obtained, which corresponds to an effective diffusion length of . As can be seen in Figure 8, both and decrease with rising dopant concentrations. This is caused by the increased intersystem crossing of singlets into triplets within simulation times below the decay time. Effective triplet diffusion lengths of are obtained for a low dopant concentration, whereas for increasing dopant concentrations, the diffusion length decreases down to for %. The average lifetime of triplets decreases from for % to at %. This strong decrease in both effective diffusion length and lifetime is caused by the higher decay rates of triplets within the radius around a dopant. With rising volume concentration, the influence of dopants becomes increasingly visible.
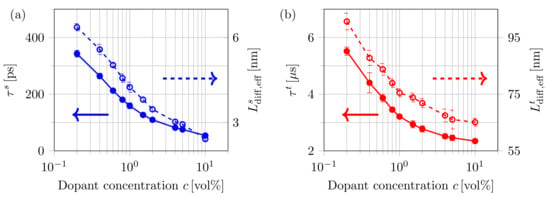
Figure 8.
Impact of the dopant volume concentration c on the average exciton lifetime (solid curves) and the effective diffusion length (dashed curves) for singlet excitons (a) and triplet excitons (b).
Figure 9 depicts the dependence of the percentage of singlets undergoing intersystem crossing and triplets annihilated by TTA on the dopant concentrations. Multiple counting of singlets back-converted from triplets upon a TTA event is omitted. For a dopant concentration of %, the proportion of converted triplets to optical generated singlets is . For higher concentrations, more than of singlets are converted into triplets before decaying. Figure 9 displays the percentage of triplets annihilated upon TTA. Starting with a ratio of at %, the proportion of triplet-triplet annihilation decreases to a value of for %. Triplet-triplet annihilation becomes unlikely for higher concentrations. The decrease in TTA can be related to the decreasing lifetime of triplets and the reduced diffusion length. Thus, the probability of triplets to meet each other decreases tremendously. This is explicitly caused by the lowering of the spin-selection rules within the strong spin-orbit coupling domains of the dopants. Higher triplet densities would significantly increase the impact of TTA. In Figure 9a, the effective exciton lifetime defined by:
is visualized. For a low dopant concentration, the increases strongly as most of the excitons are not converted into triplets. With rising c, the effective lifetime rises and reaches its maximum at %. Beyond the maximum, the effective lifetime decreases and saturates at a value of . A similar trend is observed by Gonzales et al. for doped P3HT:PCBM solar cells [24].
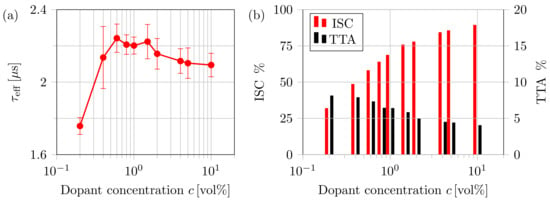
Figure 9.
Impact of the dopant volume concentration c on (a) the effective exciton lifetime and (b) intersystem crossing and triplet-triplet annihilation. The red bars show the proportion of converted triplets over the amount of optically-generated singlets, and the black bars represent the percentage of TTA over the amount of converted/generated triplets.
The results for exciton diffusion lengths and lifetime conform with the experimental values within ideal material setups, documented in [63,64,86,99]. Similar to the trend observed in [24,26], the lifetime of excitons decreases with dopant concentration. Firstly, for a small dopant concentration, the lifetime increases tremendously in comparison to singlet lifetimes caused by the conversion of singlets to the long-living triplets. However, for a higher dopant concentration, a quenching mechanism is observable due to the TTA and a higher decay rate. Simulations with higher triplet exciton decay rates within the ISC radius due to stronger spin-orbit coupling matches observed results for exciton lifetime trends [26]. Yang et al. measured the decrease of the triplet exciton lifetime by a factor of 2.8 upon doping with Ir(mppy), which is in the same order of magnitude of our simulation results [26].
5. Conclusions
To summarize, we have presented a generalized kinetic Monte Carlo framework for the study of organic semiconductors and optoelectronic devices. New developments on a realistic representation of the organic material by a generalized non-cubic Voronoi set and polymer chains have been emphasized. We have included dopant sites and improved our description of the exciton dynamics by a model of the triplet dynamics for the detailed study of OSCs and OLEDs. A simplified spatial correlated density of states accounting for a correct treatment of the energetic landscape has been presented. Our kMC framework overcomes limitations of existing kMC models due to both a generalized spatial and energetic representation of the underlying multifaceted organic materials, as well as improved treatment of the particles involved in the photocurrent generation of OSCs and the light emission of OLEDs. We presented two applications concerning electrical and optical processes inside an organic semiconductor. The impact of the intra- to inter-chain hopping rate and the directionality of the polymer chains on the charge carrier mobility has been studied. We showed that an enhanced intrachain hopping rate itself does not improve the mobility of charges, but directionality is necessary. Furthermore, the impact of the dopant volume concentration on the effective lifetime and diffusion length of excitons has been studied. The investigated exciton dynamics coincides with reported values of both singlet and triplet lifetimes and diffusion lengths.
Acknowledgments
This work was supported by the Technical University of Munich (TUM) International Graduate School of Science and Engineering (IGSSE), the excellence cluster Nanosystems Initiative Munich (NIM) of the German Research Foundation (DFG), the DFG Priority Programme FFLexCom SPP 1796 and the DFG and the TUM in the framework of the Open Access Publishing Program.
Author Contributions
A.G. proposed the idea of this paper. W.K. wrote the paper with support of M.R. and J.P. for Section 3 and Section 4 and T.A. for Section 2. W.K. implemented the Voronoi tessellation. M.R. implemented the chain algorithm and performed simulations for Section 4.1. J.P. implemented the exciton and dopant model and performed simulations for Section 4.2. T.A. and A.G. provided methodology suggestions.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Klauk, H. Organic thin-film transistors. Chem. Soc. Rev. 2010, 39, 2643–2666. [Google Scholar] [CrossRef] [PubMed]
- Dodabalapur, A. Organic light emitting diodes. Solid State Commun. 1997, 102, 259–267. [Google Scholar] [CrossRef]
- Deibel, C.; Dyakonov, V. Polymer–fullerene bulk heterojunction solar cells. Rep. Prog. Phys. 2010, 73, 096401. [Google Scholar] [CrossRef]
- Brédas, J.L.; Norton, J.E.; Cornil, J.; Coropceanu, V. Molecular understanding of organic solar cells: The challenges. Acc. Chem. Res. 2009, 42, 1691–1699. [Google Scholar] [CrossRef] [PubMed]
- Ries, B.; Bässler, H. Monte Carlo study of dispersive charge-carrier transport in spatially random systems with and without energetic disorder. Phys. Rev. B 1987, 35, 2295. [Google Scholar] [CrossRef]
- Bässler, H. Charge transport in disordered organic photoconductors a Monte Carlo simulation study. Phys. Status Solidi B 1993, 175, 15–56. [Google Scholar] [CrossRef]
- Baranovskii, S.; Zvyagin, I.; Cordes, H.; Yamasaki, S.; Thomas, P. Electronic transport in disordered organic and inorganic semiconductors. J. Non-Cryst. Solids 2002, 299, 416–419. [Google Scholar] [CrossRef]
- Köhler, A.; Bässler, H. Electronic Processes in Organic Semiconductors: An Introduction; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2015. [Google Scholar]
- Groves, C.; Marsh, R.; Greenham, N.C. Monte Carlo modeling of geminate recombination in polymer-polymer photovoltaic devices. J. Chem. Phys. 2008, 129, 114903. [Google Scholar] [CrossRef] [PubMed]
- Albes, T.; Gagliardi, A. Influence of permittivity and energetic disorder on the spatial charge carrier distribution and recombination in organic bulk-heterojunctions. Phys. Chem. Chem. Phys. 2017, 19, 20974–20983. [Google Scholar] [CrossRef] [PubMed]
- Watkins, P.K.; Walker, A.B.; Verschoor, G.L. Dynamical Monte Carlo modelling of organic solar cells: The dependence of internal quantum efficiency on morphology. Nano Lett. 2005, 5, 1814–1818. [Google Scholar] [CrossRef] [PubMed]
- Casalegno, M.; Raos, G.; Po, R. Methodological assessment of kinetic Monte Carlo simulations of organic photovoltaic devices: The treatment of electrostatic interactions. J. Chem. Phys. 2010, 132, 094705. [Google Scholar] [CrossRef] [PubMed]
- Albes, T.; Lugli, P.; Gagliardi, A. Investigation of the blend morphology in bulk-heterojunction organic solar cells. IEEE Trans. Nanotechnol. 2016, 15, 281–288. [Google Scholar] [CrossRef]
- Marsh, R.; Groves, C.; Greenham, N.C. A microscopic model for the behavior of nanostructured organic photovoltaic devices. J. Appl. Phys. 2007, 101, 083509. [Google Scholar] [CrossRef]
- Kimber, R.G.; Wright, E.N.; O’Kane, S.E.; Walker, A.B.; Blakesley, J.C. Mesoscopic kinetic Monte Carlo modeling of organic photovoltaic device characteristics. Phys. Rev. B 2012, 86, 235206. [Google Scholar] [CrossRef]
- Groves, C.; Kimber, R.G.; Walker, A.B. Simulation of loss mechanisms in organic solar cells: A description of the mesoscopic Monte Carlo technique and an evaluation of the first reaction method. J. Chem. Phys. 2010, 133, 144110. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Shang, Y.; Li, Q.; Li, Y.; Zhan, X.; Shuai, Z.; Kimber, R.G.; Walker, A.B. Dynamic Monte Carlo simulation for highly efficient polymer blend photovoltaics. J. Phys. Chem. B 2009, 114, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Maqsood, I.; Cundy, L.D.; Biesecker, M.; Kimn, J.H.; Johnson, D.; Williams, R.; Bommisetty, V. Monte Carlo Simulation of Förster Resonance Energy Transfer in 3D Nanoscale Organic Bulk Heterojunction Morphologies. J. Phys. Chem. C 2013, 117, 21086–21095. [Google Scholar] [CrossRef]
- Feron, K.; Zhou, X.; Belcher, W.; Dastoor, P. Exciton transport in organic semiconductors: Förster resonance energy transfer compared with a simple random walk. J. Appl. Phys. 2012, 111, 044510. [Google Scholar] [CrossRef]
- Van Eersel, H.; Bobbert, P.A.; Coehoorn, R. Kinetic Monte Carlo study of triplet-triplet annihilation in organic phosphorescent emitters. J. Appl. Phys. 2015, 117, 115502. [Google Scholar] [CrossRef]
- Van Eersel, H.; Bobbert, P.; Janssen, R.; Coehoorn, R. Monte Carlo study of efficiency roll-off of phosphorescent organic light-emitting diodes: Evidence for dominant role of triplet-polaron quenching. Appl. Phys. Lett. 2014, 105, 156_1. [Google Scholar] [CrossRef]
- Coehoorn, R.; van Eersel, H.; Bobbert, P.; Janssen, R. Kinetic Monte Carlo study of the sensitivity of OLED efficiency and lifetime to materials parameters. Adv. Funct. Mater. 2015, 25, 2024–2037. [Google Scholar] [CrossRef]
- Mesta, M.; Carvelli, M.; De Vries, R.J.; Van Eersel, H.; Van Der Holst, J.J.; Schober, M.; Furno, M.; Lüssem, B.; Leo, K.; Loebl, P.; et al. Molecular-scale simulation of electroluminescence in a multilayer white organic light-emitting diode. Nat. Mater. 2013, 12, 652–658. [Google Scholar] [CrossRef] [PubMed]
- González, D.M.; Körstgens, V.; Yao, Y.; Song, L.; Santoro, G.; Roth, S.V.; Müller-Buschbaum, P. Improved power conversion efficiency of P3HT:PCBM organic solar cells by strong spin–orbit coupling-induced delayed fluorescence. Adv. Energy Mater. 2015, 5, 1401770. [Google Scholar] [CrossRef]
- Luhman, W.A.; Holmes, R.J. Enhanced exciton diffusion in an organic photovoltaic cell by energy transfer using a phosphorescent sensitizer. Appl. Phys. Lett. 2009, 94, 110. [Google Scholar] [CrossRef]
- Yang, C.M.; Wu, C.H.; Liao, H.H.; Lai, K.Y.; Cheng, H.P.; Horng, S.F.; Meng, H.F.; Shy, J.T. Enhanced photovoltaic response of organic solar cell by singlet-to-triplet exciton conversion. Appl. Phys. Lett. 2007, 90, 133509. [Google Scholar] [CrossRef]
- Baranovskii, S. Theoretical description of charge transport in disordered organic semiconductors. Phys. Status Solidi B 2014, 251, 487–525. [Google Scholar] [CrossRef]
- Tessler, N.; Preezant, Y.; Rappaport, N.; Roichman, Y. Charge transport in disordered organic materials and its relevance to thin-film devices: A tutorial review. Adv. Mater. 2009, 21, 2741–2761. [Google Scholar] [CrossRef]
- Metropolis, N.; Rosenbluth, A.W.; Rosenbluth, M.N.; Teller, A.H.; Teller, E. Equation of state calculations by fast computing machines. J. Chem. Phys. 1953, 21, 1087–1092. [Google Scholar] [CrossRef]
- Gillespie, D.T. A general method for numerically simulating the stochastic time evolution of coupled chemical reactions. J. Comput. Phys. 1976, 22, 403–434. [Google Scholar] [CrossRef]
- Bortz, A.B.; Kalos, M.H.; Lebowitz, J.L. A new algorithm for Monte Carlo simulation of Ising spin systems. J. Comp. Phys. 1975, 17, 10–18. [Google Scholar] [CrossRef]
- Voter, A.F. Introduction to the kinetic Monte Carlo method. In Radiation Effects in Solids; Springer, NATO Publishing Unit: Dordrecht, The Netherlands, 2007; pp. 1–13. [Google Scholar]
- Schwabl, F. Statistische Physik; Springer: Berlin, Germany, 2006. [Google Scholar]
- Auf der Maur, M.; Albes, T.; Gagliardi, A. Thin-film Solar Cells. In Handbook of Optoelectronic Device Modeling and Simulation: Lasers, Modulators, Photodetectors, Solar Cells, and Numerical Methods—Volume Two, Series: Series in Optics and Optoelectronics, VIII. Solar Cells, 43. 2017-09; Joachim Piprek, NUSOD Institute LLC, N.D.U., Ed.; CRC Press, Taylor & Francis Group: Boca Raton, FL, USA, 2017. [Google Scholar]
- Van Der Holst, J.; Uijttewaal, M.; Ramachandhran, B.; Coehoorn, R.; Bobbert, P.; De Wijs, G.; De Groot, R. Modeling and analysis of the three-dimensional current density in sandwich-type single-carrier devices of disordered organic semiconductors. Phys. Rev. B 2009, 79. [Google Scholar] [CrossRef]
- Bouhassoune, M.; Van Mensfoort, S.; Bobbert, P.; Coehoorn, R. Carrier-density and field-dependent charge-carrier mobility in organic semiconductors with correlated Gaussian disorder. Org. Electron. 2009, 10, 437–445. [Google Scholar] [CrossRef]
- Szymański, M.Z.; Łuszczyńska, B.; Djurado, D. Modeling the transient space-charge-limited current response of organic semiconductor diodes using the master equation approach. IEEE J. Sel. Top. Quantum Electron. 2013, 19, 1–7. [Google Scholar] [CrossRef]
- Groves, C. Developing understanding of organic photovoltaic devices: Kinetic Monte Carlo models of geminate and non-geminate recombination, charge transport and charge extraction. Energy Environ. Sci. 2013, 6, 3202–3217. [Google Scholar] [CrossRef]
- Kaiser, W.; Albes, T.; Gagliardi, A. Charge carrier mobility of disordered organic semiconductors with correlated energetic and spatial disorder. Phys. Chem. Chem. Phys. 2018. [Google Scholar] [CrossRef] [PubMed]
- Troisi, A.; Orlandi, G. Dynamics of the intermolecular transfer integral in crystalline organic semiconductors. J. Phys. Chem. A 2006, 110, 4065–4070. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Vukmirović, N.; Wang, L.W. Nonadiabatic molecular dynamics simulation for carrier transport in a pentathiophene butyric acid monolayer. Phys. Rev. B 2013, 87, 205117. [Google Scholar] [CrossRef]
- Vukmirovic, N.; Wang, L.W. Charge carrier motion in disordered conjugated polymers: A multiscale ab-initio study. Nano Lett. 2009, 9, 3996–4000. [Google Scholar] [CrossRef] [PubMed]
- Kordt, P.; Stodtmann, S.; Badinski, A.; Al Helwi, M.; Lennartz, C.; Andrienko, D. Parameter-free continuous drift–diffusion models of amorphous organic semiconductors. Phys. Chem. Chem. Phys. 2015, 17, 22778–22783. [Google Scholar] [CrossRef] [PubMed]
- Mollinger, S.A.; Salleo, A.; Spakowitz, A.J. Anomalous charge transport in conjugated polymers reveals underlying mechanisms of trapping and percolation. ACS Cent. Sci. 2016, 2, 910–915. [Google Scholar] [CrossRef] [PubMed]
- Nenashev, A.; Oelerich, J.; Dvurechenskii, A.; Gebhard, F.; Baranovskii, S. Fundamental characteristic length scale for the field dependence of hopping charge transport in disordered organic semiconductors. Phys. Rev. B 2017, 96. [Google Scholar] [CrossRef]
- Voronoi, G. Nouvelles applications des paramètres continus à la théorie des formes quadratiques. Deuxième mémoire. Recherches sur les parallélloèdres primitifs. Journal Für Die Reine und Angewandte Mathematik (Crelles Journal) 1908, 134, 198–287. (In French) [Google Scholar]
- Rycroft, C. Voro++: A three-dimensional Voronoi cell library in C++. Chaos 2009, 19, 041111. [Google Scholar] [CrossRef] [PubMed]
- Kordt, P.; van der Holst, J.J.M.; Al Helwi, M.; Kowalsky, W.; May, F.; Badinski, A.; Lennartz, C.; Andrienko, D. Modeling of Organic Light Emitting Diodes: From Molecular to Device Properties. Adv. Funct. Mater. 2015, 25, 1955–1971. [Google Scholar] [CrossRef]
- Baumeier, B.; Stenzel, O.; Poelking, C.; Andrienko, D.; Schmidt, V. Stochastic modeling of molecular charge transport networks. Phys. Rev. B 2012, 86, 184202. [Google Scholar] [CrossRef]
- Sirringhaus, H.; Wilson, R.; Friend, R.; Inbasekaran, M.; Wu, W.; Woo, E.; Grell, M.; Bradley, D. Mobility enhancement in conjugated polymer field-effect transistors through chain alignment in a liquid-crystalline phase. Appl. Phys. Lett. 2000, 77, 406–408. [Google Scholar] [CrossRef]
- Carbone, P.; Troisi, A. Charge diffusion in semiconducting polymers: Analytical relation between polymer rigidity and time scales for intrachain and inter-chain hopping. J. Phys. Chem. Lett. 2014, 5, 2637–2641. [Google Scholar] [CrossRef] [PubMed]
- Conchuir, B.O.; Tarantini, C.; McNeill, C.R.; Hüttner, S.; Zaccone, A. Chain-assisted charge transport in semicrystalline conjugated polymers. J. Phys. Chem. C 2016, 120, 14539–14548. [Google Scholar] [CrossRef][Green Version]
- Fornari, R.P.; Troisi, A. Theory of charge hopping along a disordered polymer chain. Phys. Chem. Chem. Phys. 2014, 16, 9997–10007. [Google Scholar] [CrossRef] [PubMed]
- Vanlaeke, P.; Swinnen, A.; Haeldermans, I.; Vanhoyland, G.; Aernouts, T.; Cheyns, D.; Deibel, C.; D’Haen, J.; Heremans, P.; Poortmans, J.; et al. P3HT/PCBM bulk heterojunction solar cells: Relation between morphology and electro-optical characteristics. Sol. Energy Mater. Sol. Cells 2006, 90, 2150–2158. [Google Scholar] [CrossRef]
- Singh, C.R.; Gupta, G.; Lohwasser, R.; Engmann, S.; Balko, J.; Thelakkat, M.; Thurn-Albrecht, T.; Hoppe, H. Correlation of charge transport with structural order in highly ordered melt-crystallized poly (3-hexylthiophene) thin films. J. Polym. Sci., Part B: Polym. Phys. 2013, 51, 943–951. [Google Scholar] [CrossRef]
- Sirringhaus, H.; Brown, P.; Friend, R.; Nielsen, M.M.; Bechgaard, K.; Langeveld-Voss, B.; Spiering, A.; Janssen, R.A.; Meijer, E.; Herwig, P.; et al. Two-dimensional charge transport in self-organized, high-mobility conjugated polymers. Nature 1999, 401, 685–688. [Google Scholar] [CrossRef]
- Pröller, S.; Liu, F.; Zhu, C.; Wang, C.; Russell, T.P.; Hexemer, A.; Müller-Buschbaum, P.; Herzig, E.M. Following the morphology formation in situ in printed active layers for organic solar cells. Adv. Energy Mater. 2016, 6. [Google Scholar] [CrossRef]
- Pasveer, W.; Cottaar, J.; Tanase, C.; Coehoorn, R.; Bobbert, P.; Blom, P.; De Leeuw, D.; Michels, M. Unified description of charge-carrier mobilities in disordered semiconducting polymers. Phys. Rev. Lett. 2005, 94, 206601. [Google Scholar] [CrossRef] [PubMed]
- Landau, R.H.; Paez, M.J.; Bordeianu, C.C. Computational Physics: Problem Solving with Python; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2015. [Google Scholar]
- Moore, J. Gel permeation chromatography. I. A new method for molecular weight distribution of high polymers. J. Polym. Sci. Part A Polym. Chem. 1964, 2, 835–843. [Google Scholar] [CrossRef]
- Förster, S.; Schmidt, M.; Antonietti, M. Static and dynamic light scattering by aqueous polyelectrolyte solutions: Effect of molecular weight, charge density and added salt. Polymer 1990, 31, 781–792. [Google Scholar] [CrossRef]
- Yu, G.; Gao, J.; Hummelen, J.C.; Wudl, F.; Heeger, A.J. Polymer photovoltiac cells: Enhanced efficiencies via a network of internal donor-acceptor heterojunctions. Science 1995, 270, 1789. [Google Scholar] [CrossRef]
- Xu, B.; Holdcroft, S. Phosphorescence and delayed fluorescence of poly (3-hexylthiophene) films. Thin Solid Films 1994, 242, 174–177. [Google Scholar] [CrossRef]
- Köhler, A.; Bässler, H. Triplet states in organic semiconductors. Mater. Sci. Eng. R Rep. 2009, 66, 71–109. [Google Scholar] [CrossRef]
- Zhang, W.; Xu, Y.; Wang, H.; Xu, C.; Yang, S. Fe3O4 nanoparticles induced magnetic field effect on efficiency enhancement of P3HT: PCBM bulk heterojunction polymer solar cells. Sol. Energy Mater. Sol. Cells 2011, 95, 2880–2885. [Google Scholar] [CrossRef]
- Zhang, W.; Nguyen, N.A.; Murray, R.; Xin, J.; Mackay, M.E. A comparative study on the morphology of P3HT: PCBM solar cells with the addition of Fe3O4 nanoparticles by spin and rod coating methods. J. Nanopart. Res. 2017, 19, 315. [Google Scholar] [CrossRef]
- Baldo, M.A.; O’brien, D.; You, Y.; Shoustikov, A.; Sibley, S.; Thompson, M.; Forrest, S. Highly efficient phosphorescent emission from organic electroluminescent devices. Nature 1998, 395, 151–154. [Google Scholar] [CrossRef]
- Baldo, M.; Thompson, M.; Forrest, S. High-efficiency fluorescent organic light-emitting devices using a phosphorescent sensitizer. Nature 2000, 403, 750–753. [Google Scholar] [CrossRef] [PubMed]
- Sousa, L.; Volpi, R.; da Silva Filho, D.A.; Linares, M. Mobility field and mobility temperature dependence in PC61BM: A kinetic Monte-Carlo study. Chem. Phys. Lett. 2017, 689, 74–81. [Google Scholar] [CrossRef]
- Tanase, C.; Meijer, E.; Blom, P.; De Leeuw, D. Unification of the hole transport in polymeric field-effect transistors and light-emitting diodes. Phys. Rev. Lett. 2003, 91, 216601. [Google Scholar] [CrossRef] [PubMed]
- Gartstein, Y.N.; Conwell, E. High-field hopping mobility in molecular systems with spatially correlated energetic disorder. Chem. Phys. Lett. 1995, 245, 351–358. [Google Scholar] [CrossRef]
- Cordes, H.; Baranovskii, S.; Kohary, K.; Thomas, P.; Yamasaki, S.; Hensel, F.; Wendorff, J.H. One-dimensional hopping transport in disordered organic solids. I. Analytic calculations. Phys. Rev. B 2001, 63, 094201. [Google Scholar] [CrossRef]
- Kordt, P.; Andrienko, D. Modeling of spatially correlated energetic disorder in organic semiconductors. J. Chem. Theory Comput. 2016, 12, 36. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.; Abrahams, E. Impurity conduction at low concentrations. Phys. Rev. 1960, 120, 745. [Google Scholar] [CrossRef]
- Coehoorn, R.; Pasveer, W.; Bobbert, P.; Michels, M. Charge-carrier concentration dependence of the hopping mobility in organic materials with Gaussian disorder. Phys. Rev. B 2005, 72, 155206. [Google Scholar] [CrossRef]
- Coropceanu, V.; Cornil, J.; da Silva Filho, D.A.; Olivier, Y.; Silbey, R.; Brédas, J.L. Charge transport in organic semiconductors. Chem. Rev. 2007, 107, 926–952. [Google Scholar] [CrossRef] [PubMed]
- Marcus, R.A. Electron transfer reactions in chemistry. Theory and experiment. Rev. Mod. Phys. 1993, 65, 599. [Google Scholar] [CrossRef]
- Ewald, P.P. Die Berechnung optischer und elektrostatischer Gitterpotentiale. Ann. Phys. 1921, 369, 253–287. [Google Scholar] [CrossRef]
- Stehr, V.; Pfister, J.; Fink, R.; Engels, B.; Deibel, C. First-principles calculations of anisotropic charge-carrier mobilities in organic semiconductor crystals. Phys. Rev. B 2011, 83, 155208. [Google Scholar] [CrossRef]
- Kirkpatrick, J.; Marcon, V.; Nelson, J.; Kremer, K.; Andrienko, D. Charge mobility of discotic mesophases: A multiscale quantum and classical study. Phys. Rev. Lett. 2007, 98, 227402. [Google Scholar] [CrossRef] [PubMed]
- Scholes, G.D.; Rumbles, G. Excitons in nanoscale systems. Nat. Mater. 2006, 5, 683–696. [Google Scholar] [CrossRef] [PubMed]
- Mikhnenko, O.V.; Blom, P.W.; Nguyen, T.Q. Exciton diffusion in organic semiconductors. Energy Environ. Sci. 2015, 8, 1867–1888. [Google Scholar] [CrossRef]
- Pettersson, L.A.; Roman, L.S.; Inganäs, O. Modeling photocurrent action spectra of photovoltaic devices based on organic thin films. J. Appl. Phys. 1999, 86, 487–496. [Google Scholar] [CrossRef]
- Peumans, P.; Yakimov, A.; Forrest, S.R. Small molecular weight organic thin-film photodetectors and solar cells. J. Appl. Phys. 2003, 93, 3693–3723. [Google Scholar] [CrossRef]
- Burkhard, G.F.; Hoke, E.T.; McGehee, M.D. Accounting for interference, scattering, and electrode absorption to make accurate internal quantum efficiency measurements in organic and other thin solar cells. Adv. Mater. 2010, 22, 3293–3297. [Google Scholar] [CrossRef] [PubMed]
- Mikhnenko, O.V.; Lin, J.; Shu, Y.; Anthony, J.E.; Blom, P.W.; Nguyen, T.Q.; Loi, M.A. Effect of thermal annealing on exciton diffusion in a diketopyrrolopyrrole derivative. Phys. Chem. Chem. Phys. 2012, 14, 14196–14201. [Google Scholar] [CrossRef] [PubMed]
- Athanasopoulos, S.; Emelianova, E.V.; Walker, A.B.; Beljonne, D. Exciton diffusion in energetically disordered organic materials. Phys. Rev. B 2009, 80, 195209. [Google Scholar] [CrossRef]
- Hoffmann, S.T.; Athanasopoulos, S.; Beljonne, D.; Bässler, H.; Köhler, A. How do triplets and charges move in disordered organic semiconductors? A Monte Carlo study comprising the equilibrium and nonequilibrium regime. J. Phys. Chem. C 2012, 116, 16371–16383. [Google Scholar] [CrossRef]
- Forster, T. Energiewanderung und Fluoreszenz. Naturwissenschaften 1946, 33, 166–175. [Google Scholar] [CrossRef]
- Einstein, A. Über die von der molekularkinetischen Theorie der Wärme geforderte Bewegung von in ruhenden Flüssigkeiten suspendierten Teilchen. Ann. Phys. 1905, 322, 549–560. (In German) [Google Scholar] [CrossRef]
- Von Smoluchowski, M. Zur kinetischen Theorie der Brownschen Molekularbewegung und der Suspensionen. Ann. Phys. 1906, 326, 756–780. (In German) [Google Scholar] [CrossRef]
- Dexter, D.L. A theory of sensitized luminescence in solids. J. Chem. Phys. 1953, 21, 836–850. [Google Scholar] [CrossRef]
- Hwang, I.W.; Moses, D.; Heeger, A.J. Photoinduced carrier generation in P3HT/PCBM bulk heterojunction materials. J. Phys. Chem. C 2008, 112, 4350–4354. [Google Scholar] [CrossRef]
- Scheidler, M.; Cleve, B.; Bässler, H.; Thomas, P. Monte Carlo simulation of bimolecular exciton annihilation in an energetically random hopping system. Chem. Phys. Lett. 1994, 225, 431–436. [Google Scholar] [CrossRef]
- Murawski, C.; Leo, K.; Gather, M.C. Efficiency roll-off in organic light-emitting diodes. Adv. Mater. 2013, 25, 6801–6827. [Google Scholar] [CrossRef] [PubMed]
- Cheung, D.L.; McMahon, D.P.; Troisi, A. Computational Study of the Structure and Charge-Transfer Parameters in Low-Molecular-Mass P3HT. J. Phys. Chem. B 2009, 113, 9393–9401. [Google Scholar] [CrossRef] [PubMed]
- Kraabel, B.; Moses, D.; Heeger, A. Direct observation of the intersystem crossing in poly (3-octylthiophene). J. Chem. Phys. 1995, 103, 5102–5108. [Google Scholar] [CrossRef]
- Kawamura, Y.; Brooks, J.; Brown, J.J.; Sasabe, H.; Adachi, C. Intermolecular interaction and a concentration-quenching mechanism of phosphorescent Ir (III) complexes in a solid film. Phys. Rev. Lett. 2006, 96. [Google Scholar] [CrossRef] [PubMed]
- Mikhnenko, O.V.; Ruiter, R.; Blom, P.W.; Loi, M.A. Direct measurement of the triplet exciton diffusion length in organic semiconductors. Phys. Rev. Lett. 2012, 108, 137401. [Google Scholar] [CrossRef] [PubMed]









© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).