
Changes of Lignin Molecular Structures in a Modification of Kraft Lignin Using Acid Catalyst


Abstract
:1. Introduction
2. Experimental
2.1. Materials
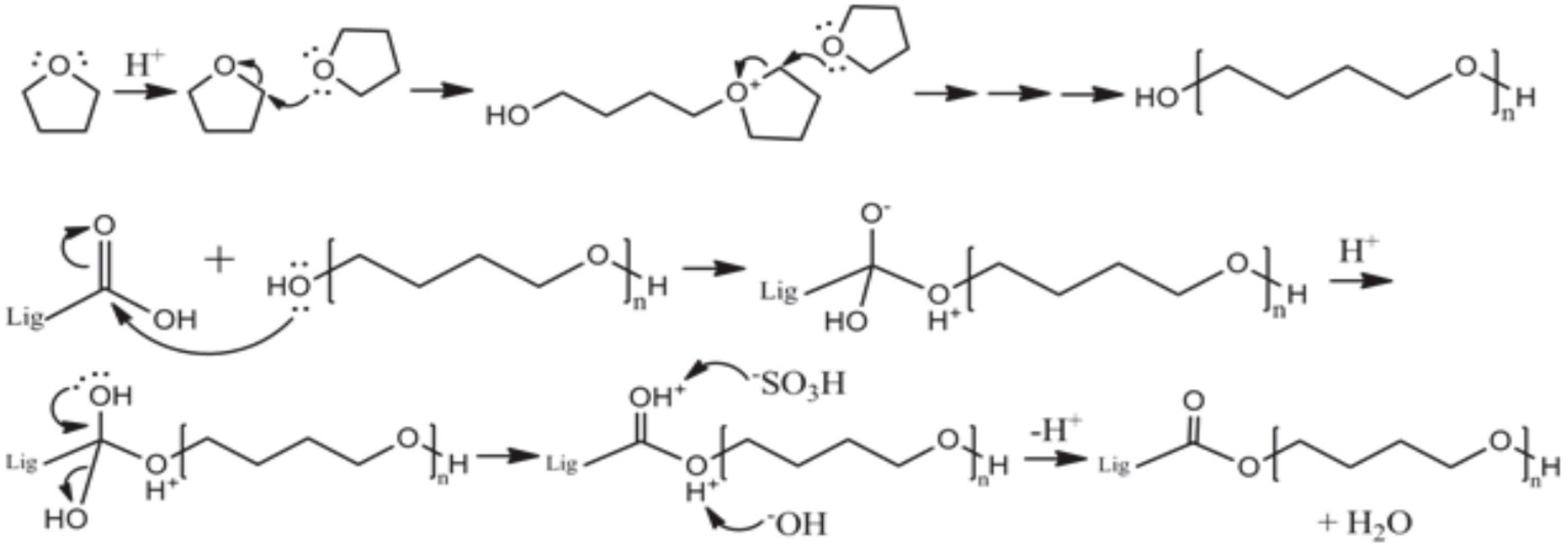
2.2. Preparation of Modified Lignin
2.3. Analyses of Modified Lignin Functional Group Using 31P-NMR and FT-IR
2.4. Analyses of Modified Lignin in Order to Investigate the Changes of Lignin Contents and Solubility
2.5. Blending of Modified Lignin and Synthetic Polymers
2.6. Tensile Test
2.7. X-ray Diffraction Analysis (XRD)
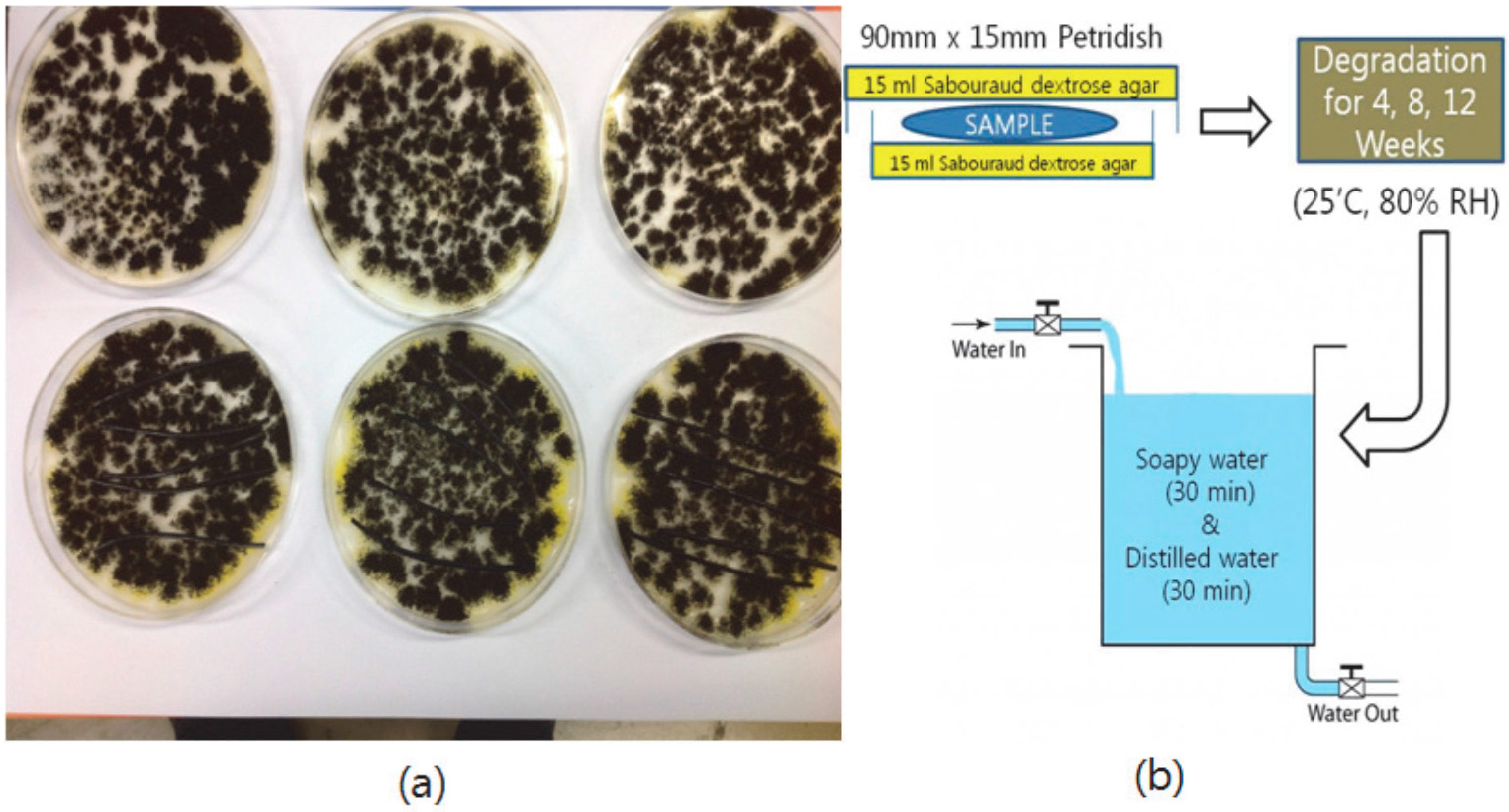
2.8. Degradation Test of Blends
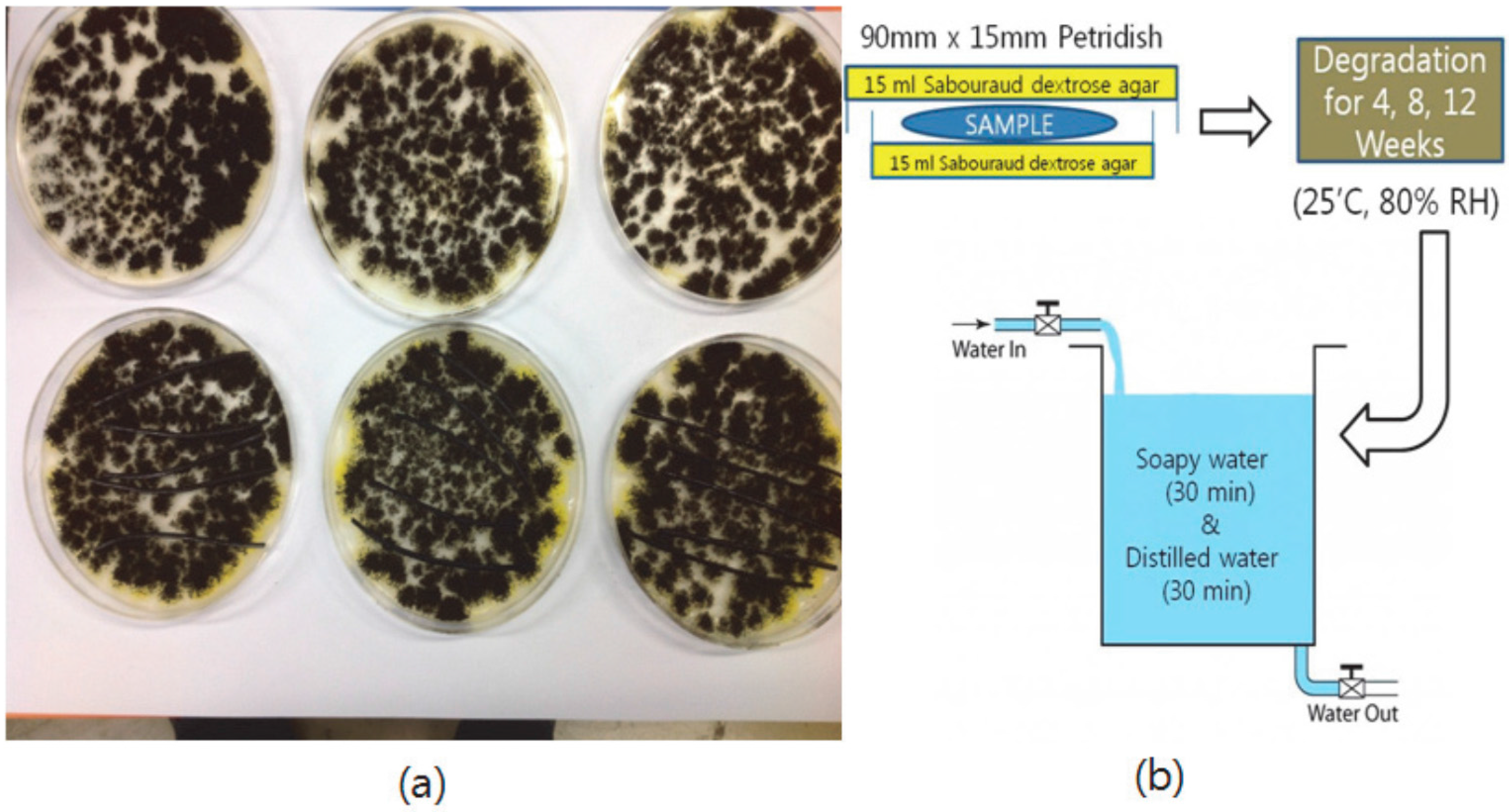
2.9. Scanning Electron Microscope (SEM) Image Analysis of Degraded Blends
3. Results and Discussion
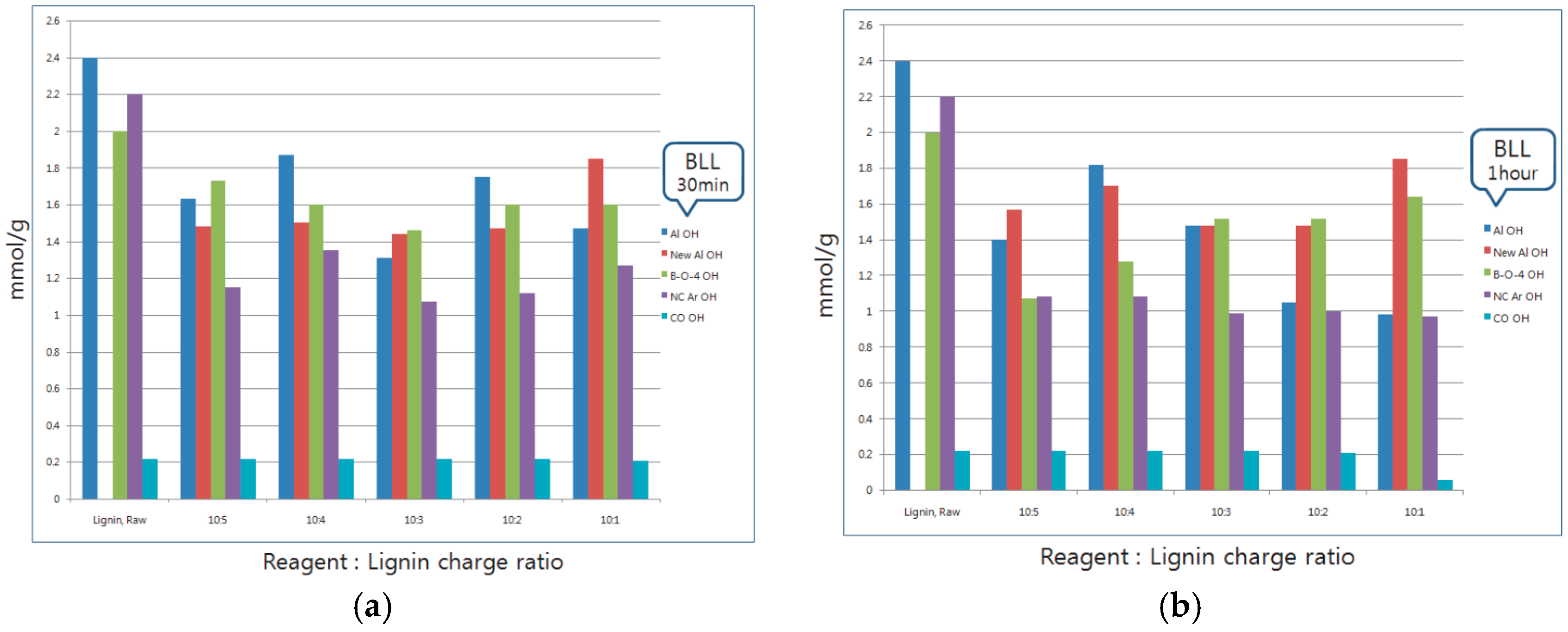
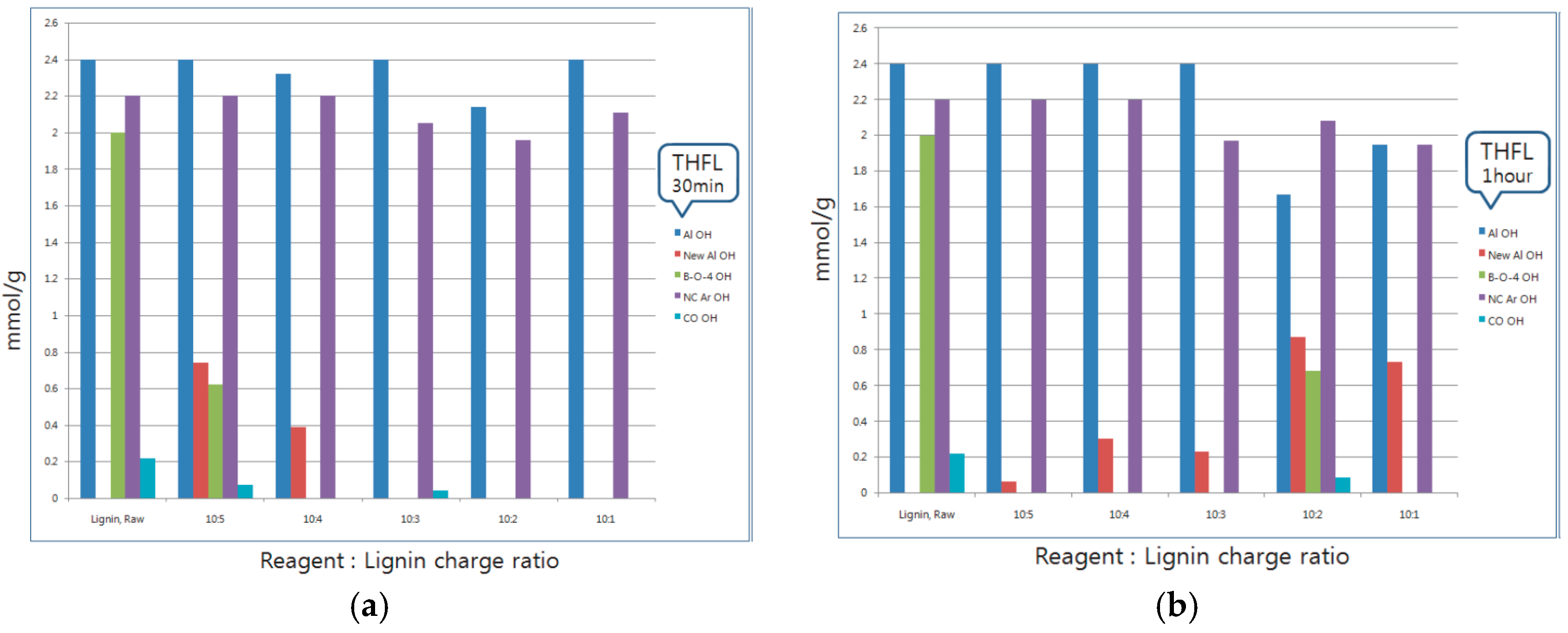
3.1. 31P-NMR
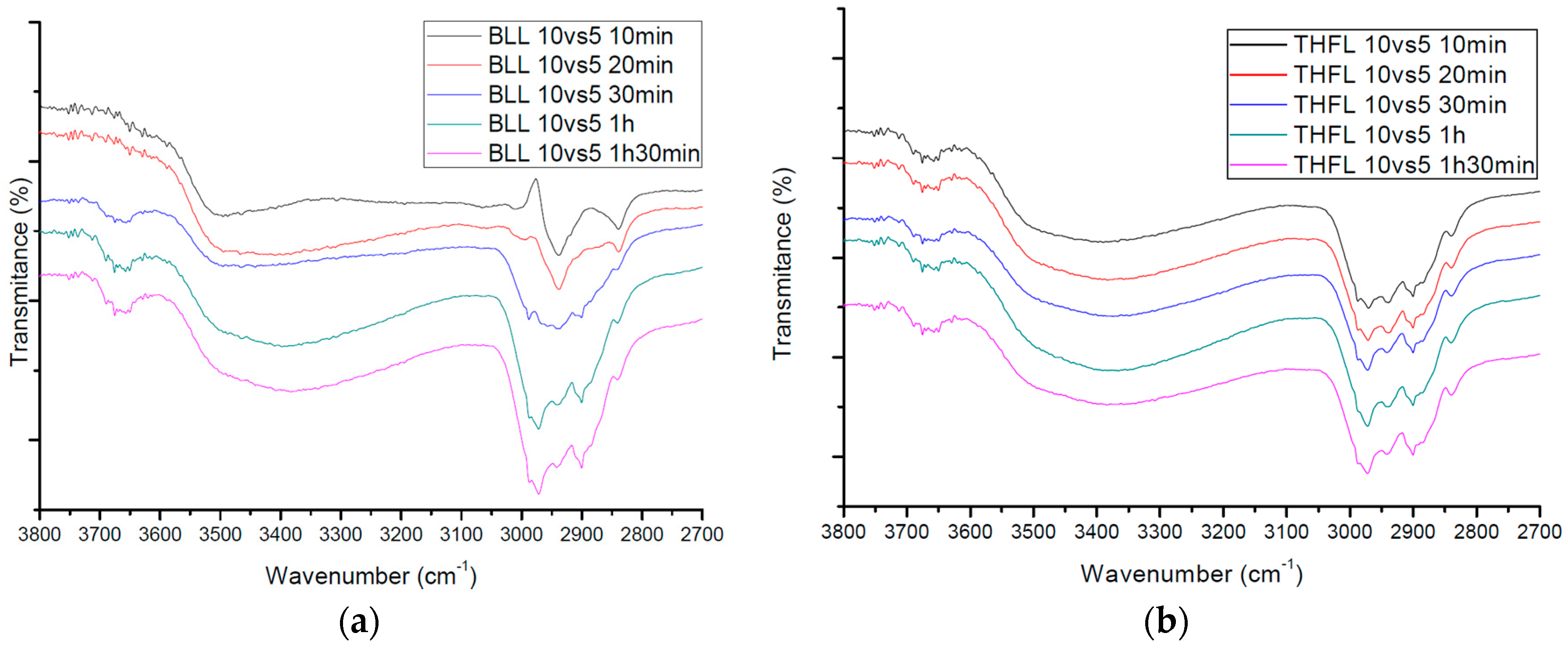
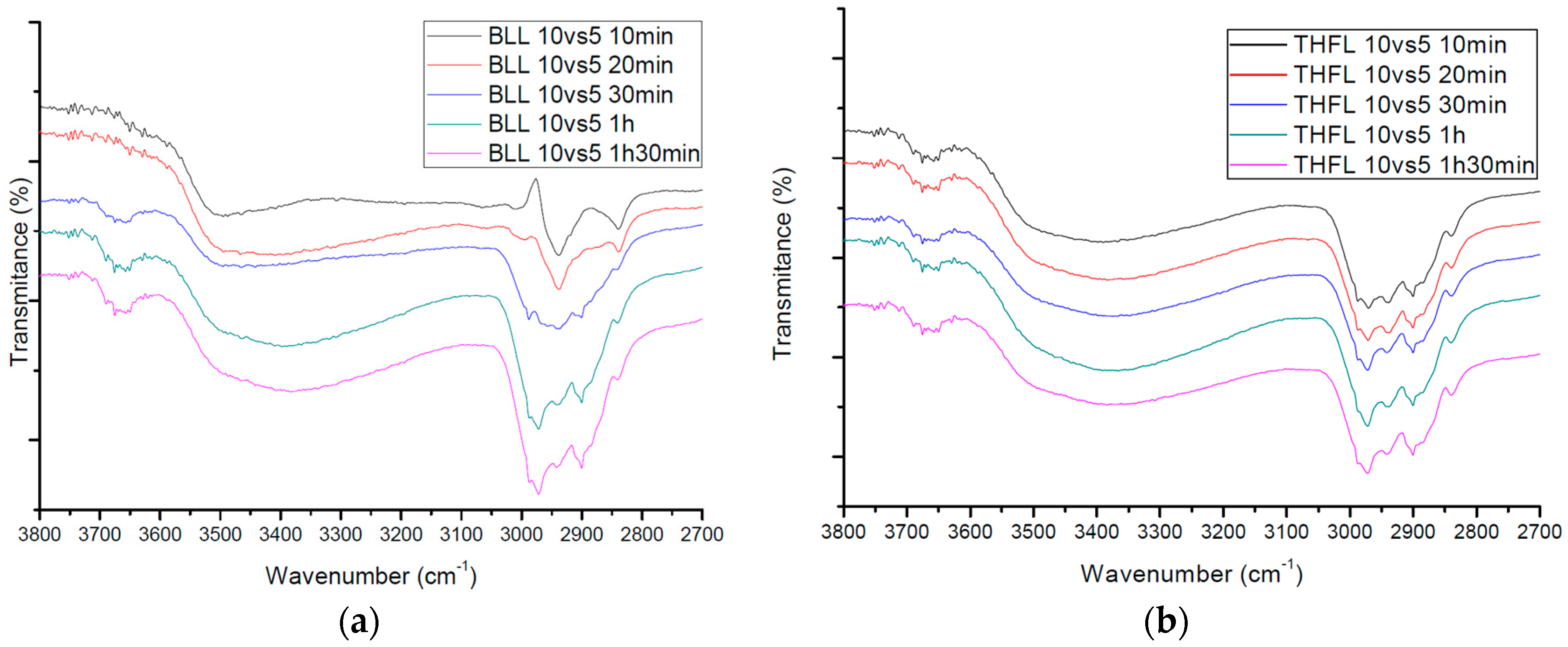
3.2. FT-IR Spectra of Modified Lignin
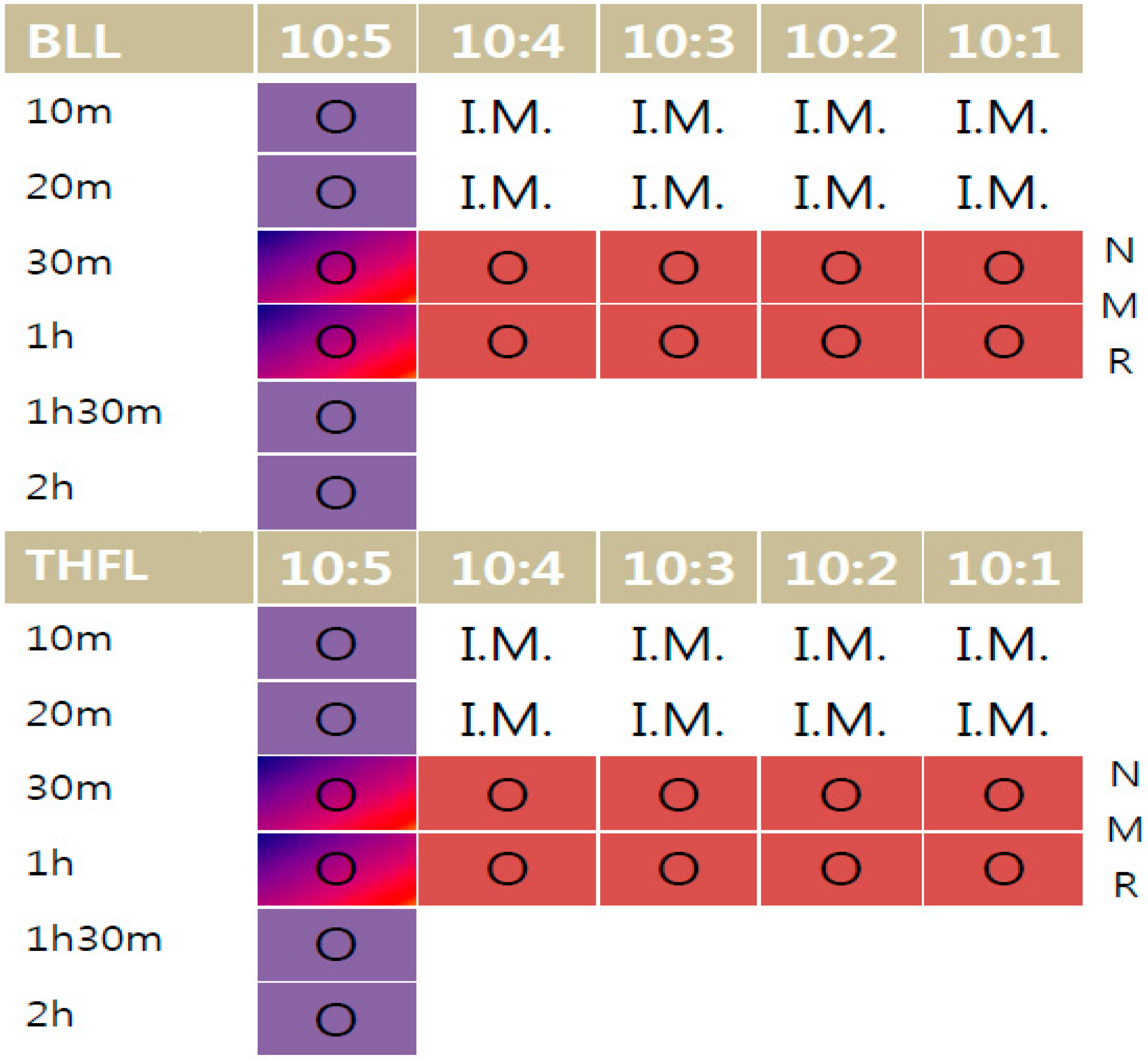
3.3. Quantification of the Insoluble Fraction in THF
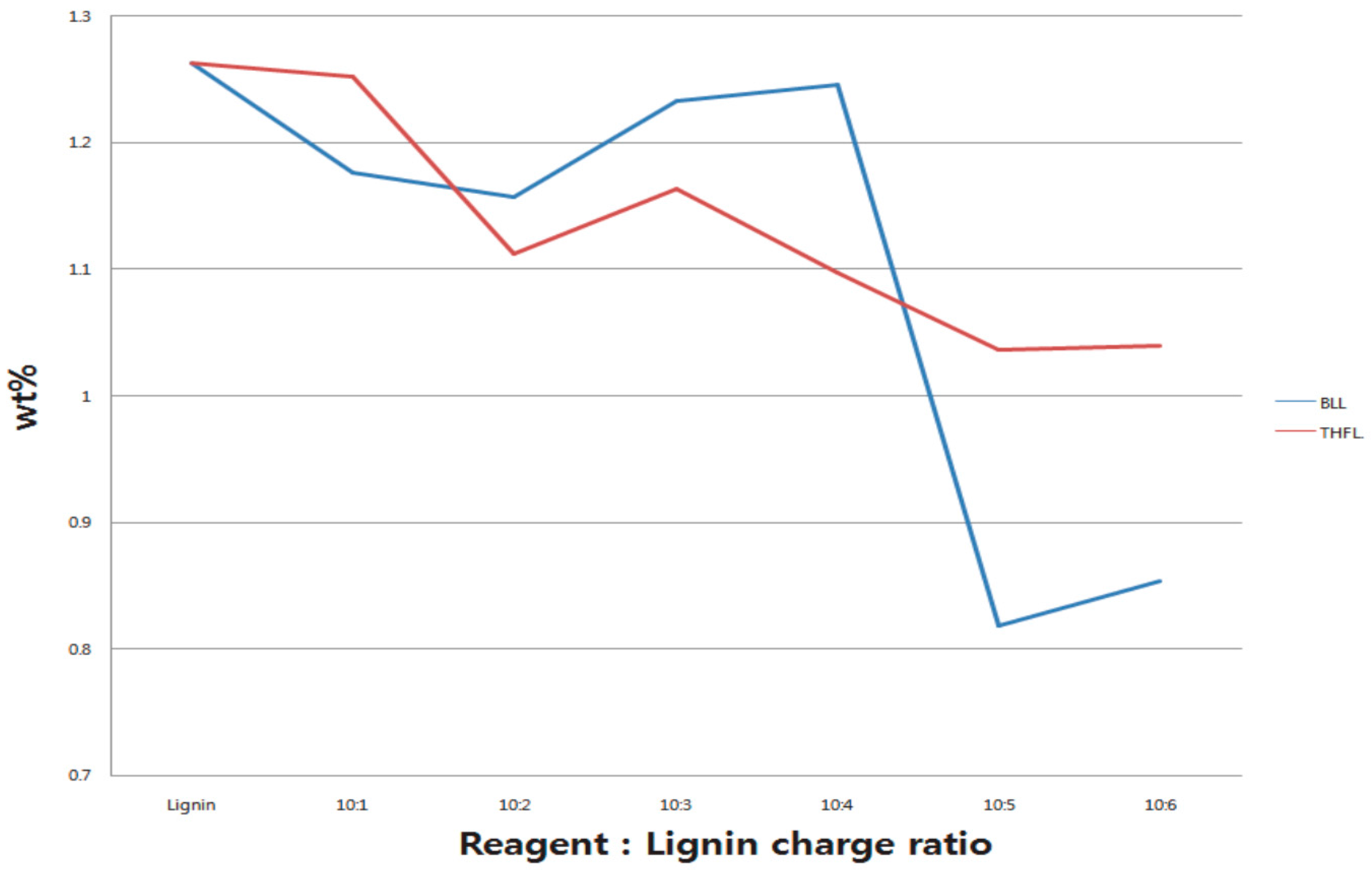
3.4. Elemental Analysis of 2 h-Reacted Lignin
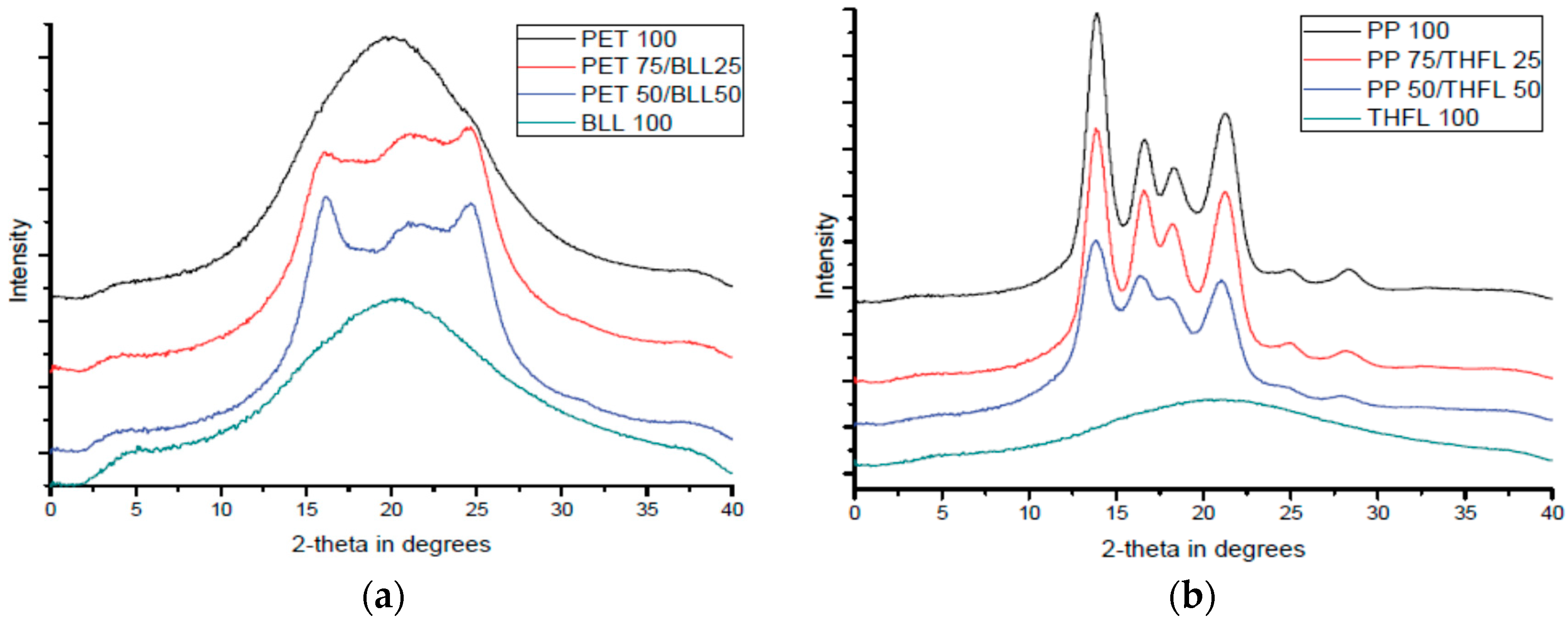
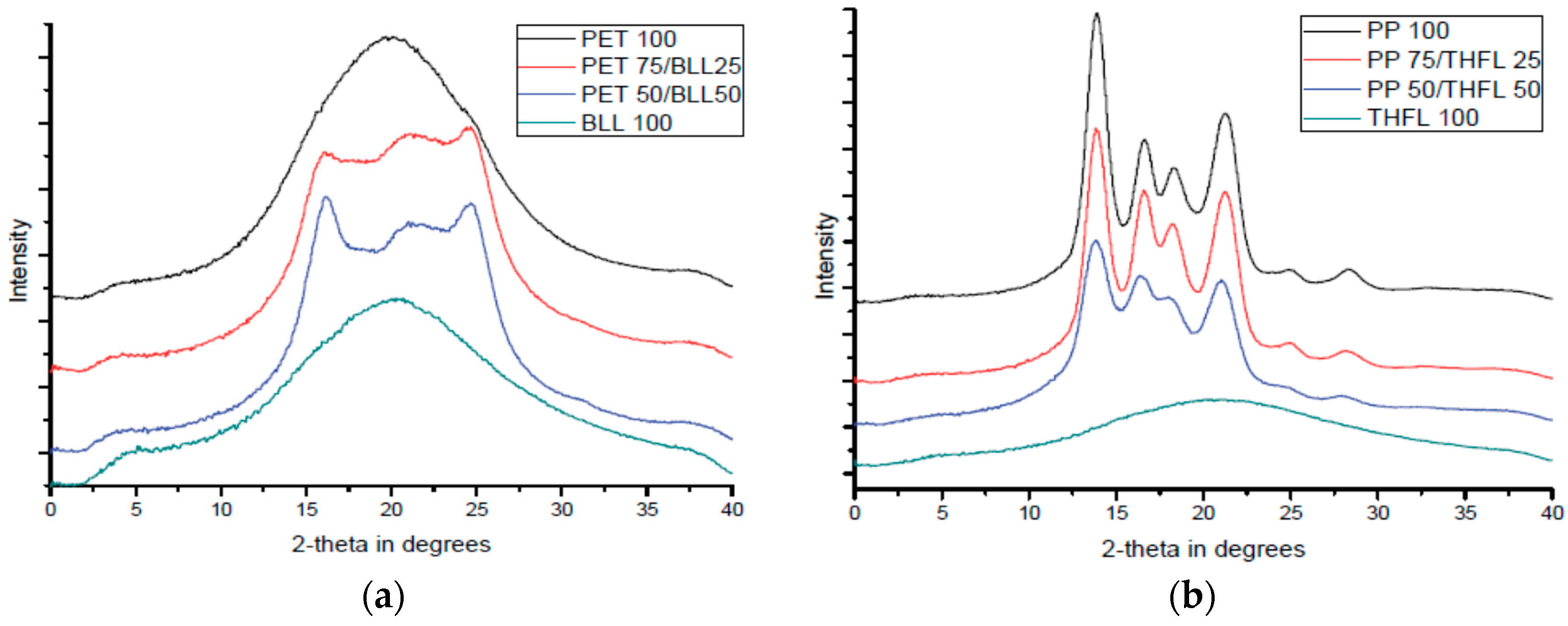
3.5. XRD Analysis of Modified Lignin/Polymer Blends
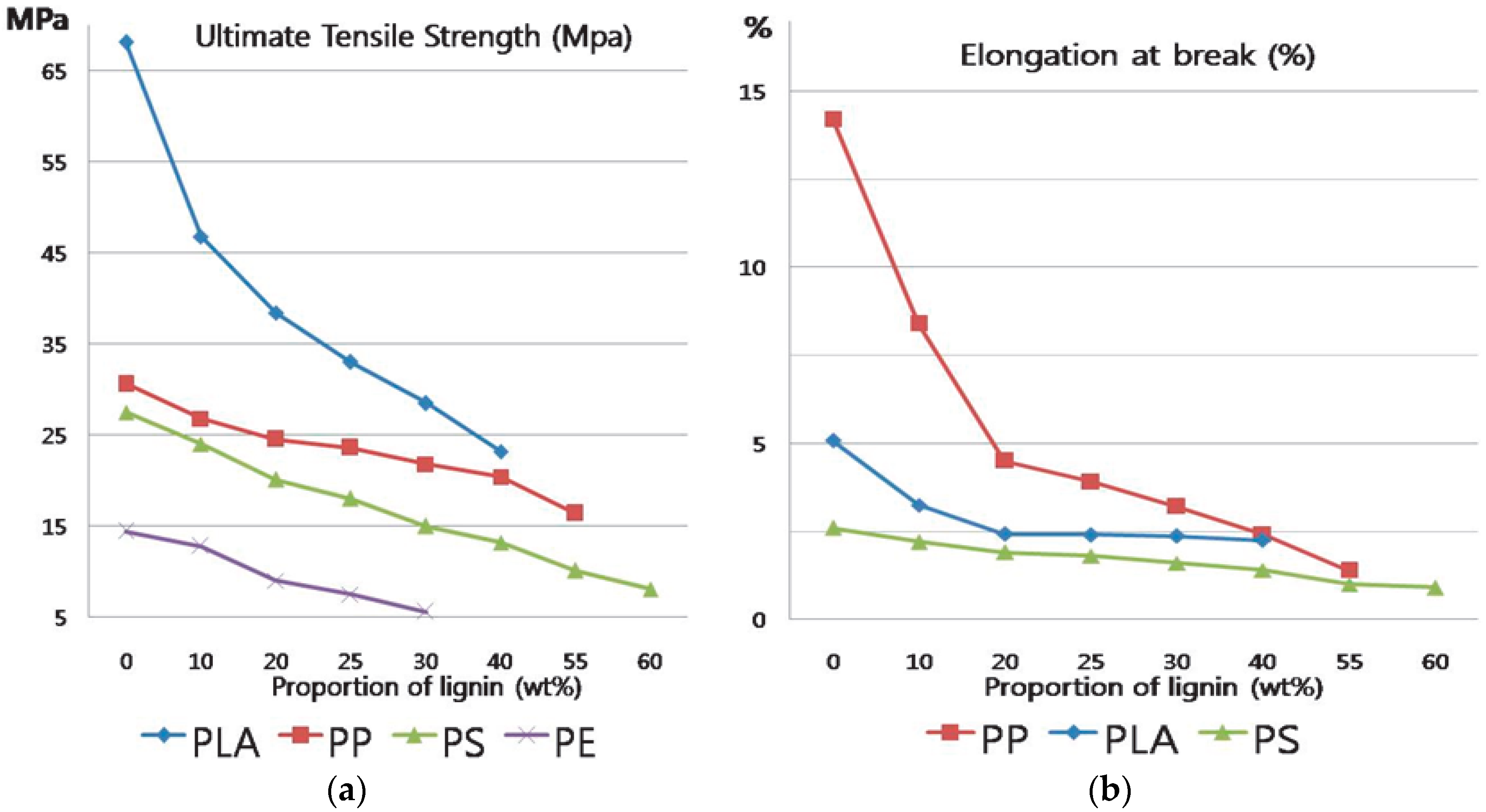
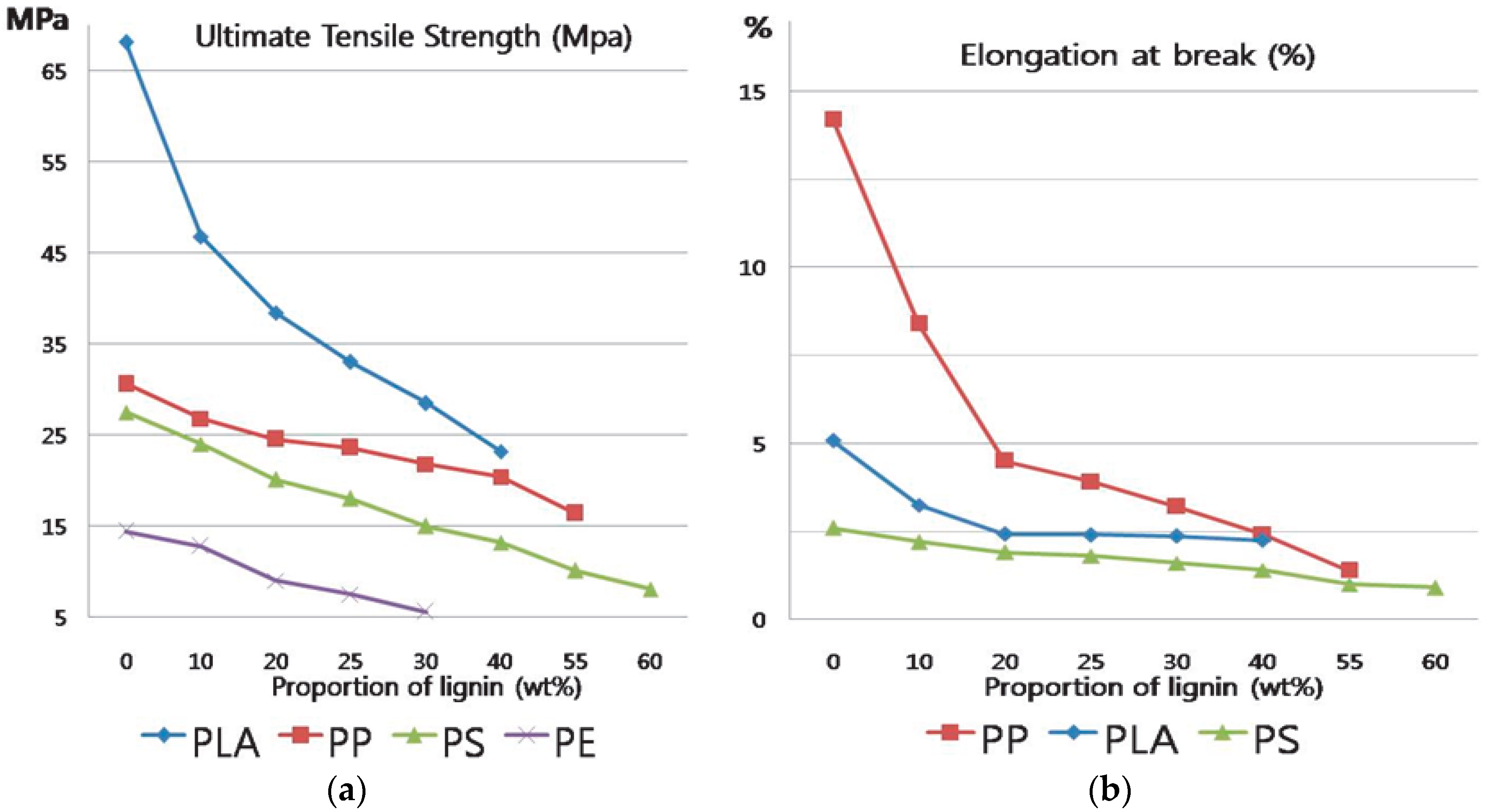
3.6. Tensile Test of 2 h-Modified Lignin and Polymer Blends
3.7. Degradation Behavior of PP/THFL and PET/BLL
4. Summary and Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Van de Ven, T.; Louis, G. Agricultural and Biological Sciences Cellulose Fundamental Aspects; InTech Books: Rijeka, Croatia, 2013; pp. 250–260. [Google Scholar]
- Kubo, S.; Gilbert, R.D.; Kadla, J.F. Natural Fibers, Biopolymers, and Biocomposites; Mohanty, A.K., Misra, M., Drzal, L.T., Eds.; Taylor and Francis: Boca Raton, FL, USA, 2005; p. 674. [Google Scholar]
- Li, H.; McDonald, A.G. Fractionation and characterization of industrial lignins. Ind. Crops Prod. 2014, 62, 67–68. [Google Scholar]
- Feldman, D. Lignin and its Polyblends—A Review. In Chemical Modification, Properties, and Usage of Lignin; Thomas, Q., Ed.; Kluwer Academic/Plenum Publishers: New York, NY, USA, 2002; pp. 81–82. [Google Scholar]
- Alexya, P.; Kosikova, B.; Podstranska, G. The effect of blending lignin with polyethylene and polypropylene on physical properties. Polymer 2000, 41, 4901–4908. [Google Scholar]
- Sanchez, C.G.; Alvarez, L.A.E. Micromechanics of lignin/polypropylene composites suitable for industrial applications. Angew. Makromol. Chem. 1999, 272, 65–70. [Google Scholar] [CrossRef]
- Barzegari, M.R.; Alemdar, A.; Zhang, Y.; Rodrigue, D. Mechanical and Rheological Behavior of Highly Filled Polystyrene with Lignin. Polym. Compos. 2012, 33, 353–361. [Google Scholar] [CrossRef]
- Li, J.; He, Y.; Inoue, Y. Thermal and mechanical properties of biodegradable blends of poly(l-lactic acid) and lignin. Polym. Int. 2003, 52, 949–955. [Google Scholar] [CrossRef]
- Fengel, D.; Przyklenk, M. Studies on Alkali Extract from Beech Holocellulose. Wood Sci. Technol. 1976, 10, 311–320. [Google Scholar]
- Alexander, L. X-ray Diffraction Methods in Polymer Science; Wiley Interscience: New York, NY, USA, 1969; pp. 165–172. [Google Scholar]
- Difco™ & BBL™ Manual, 2nd ed.; Becton, Dickinson and Company: Sparks Glencoe, MD, USA, 2009.
- Sadeghifar, H.; Cui, C.; Argyropoulos, D.S. Toward Thermoplastic Lignin Polymers. Part 1. Selective Masking of Phenolic Hydroxyl Groups in Kraft Lignins via Methylation and Oxypropylation Chemistries. Ind. Eng. Chem. Res. 2012, 51, 16713–16720. [Google Scholar] [CrossRef]
- Argyropoulos, D.S. Quantitative Phosphorus-31 NMR Analysis of Lignins, a New Tool for the Lignin Chemist. J. Wood Chem. Technol. 1994, 14, 45–63. [Google Scholar] [CrossRef]
- Cui, C.; Sadeghifar, H.; Sen, S.; Argyropoulos, D.S. Toward Thermoplastic Lignin Polymers; Part 2: Thermal & Polymer Characteristics of kraft Lignin & Derivatives. Bioresources 2013, 8, 864–886. [Google Scholar]
- Balakshin, M.; Capanema, E. On the Quantification of Lignin Hydroxyl Groups with 31P and 13C-NMR Spectroscopy. J. Wood Chem. Technol. 2015, 35, 220–237. [Google Scholar] [CrossRef]
- Lin, L.Z.; Yao, Y.G.; Yoshioka, M.; Shiraishi, N. Liquefaction mechanism of lignin in the presence of phenol at elevated temperature without catalysts: Studies on β-O-4 lignin model compound. 1. Structural characterization of the reaction products. Holzforschung 1997, 51, 316–324. [Google Scholar] [CrossRef]
- Lin, L.Z.; Yao, Y.G.; Yoshioka, M.; Shiraishi, N. Liquefaction mechanism of lignin in the presence of phenol at elevated temperature without catalysts: Studies on β-O-4 lignin model compound. 2. Reaction pathway. Holzforschung 1997, 51, 325–332. [Google Scholar] [CrossRef]
- Lin, L.Z.; Yao, Y.G.; Yoshioka, M.; Shiraishi, N. Liquefaction mechanism of lignin in the presence of phenol at elevated temperature without catalysts: Studies on β-O-4 lignin model compound. 3. Multi condensation. Holzforschung 1997, 51, 333–337. [Google Scholar] [CrossRef]
- Lin, L.Z.; Yao, Y.G.; Shiraishi, N. Liquefaction mechanism of β-O-4 lignin model compound in the presence of phenol under acid catalysis. Part 1. Identification of the reaction products. Holzforschung 2001, 55, 617–624. [Google Scholar] [CrossRef]
- Lin, L.Z.; Yao, Y.G.; Shiraishi, N. Liquefaction mechanism of β-O-4 lignin model compound in the presence of phenol under acid catalysis. Part 2. Reaction behavior and pathways. Holzforschung 2001, 55, 625–630. [Google Scholar]
- Choi, Y.S.; Singh, R.; Zhang, J.; Balasubramanian, G.; Sturgeon, M.R.; Katahira, R.; Chupka, G.; Beckham, G.T.; Shanks, B.H. Pyrolysis reaction networks for lignin model compounds: Unraveling thermal deconstruction of β-O-4 and α-O-4 compounds. Green Chem. 2016, 18, 1762–1773. [Google Scholar] [CrossRef]
- Nuyken, O.; Pask, S.D. Ring-Opening Polymerization—An Introductory Review. Polymers 2013, 5, 363–374. [Google Scholar] [CrossRef]
- Gresham, T.L.; Jansen, J.E.; Shaver, F.W.; Propiolactone, I. Polymerization Reactions. J. Am. Chem. Soc. 1948, 70, 998–999. [Google Scholar] [CrossRef]
- Foster, N.C. Sulfonation and Sulfation Processes; The Chemithon Corporation: Seattle, WA, USA, 1997. [Google Scholar]
- Whitby, R.L.D.; Smith, L.C.; Dichello, G.; Fukuda, T.; Maekawa, T.; Mikhalovsky, S.V. Cationic ring-opening polymerization of lactones onto chemically modified single layer graphene oxide. Mater. Express 2014, 4, 242–244. [Google Scholar] [CrossRef]
- Impallomeni, G.; Giuffrida, M.; Barbuzzi, T.; Musumarra, G.; Ballistreri, A. Acid Catalyzed Transesterification as a Route to Poly(3-hydroxybutyrate-co-ε-caprolactone) Copolymers from Their Homopolymers. Biomacromolecules 2002, 3, 837–839. [Google Scholar] [CrossRef]
- Cateto, C.A.; Barreiro, M.F.; Rodrigues, A.E.; Belgacem, M.N. Optimization Study of Lignin Oxypropylation in View of the Preparation of Polyurethane Rigid Foams. Ind. Eng. Chem. Res. 2009, 48, 2586–2588. [Google Scholar] [CrossRef]
- Terada, M.; Marchessault, R.H. Determination of solubility parameters for poly(3-hydroxyalkanoates). Int. J. Biol. Macromol. 1999, 25, 207–215. [Google Scholar] [CrossRef]
- Xia, W.; Binyi, Z.; Zhengyi, L.; Yunzhao, Y. Studies on Epoxy Resins Modified with Poly(butylenes terephthalate)-b-Poly(tetramethylene glycol). Chin. J. Polym. Sci. 1997, 15, 84–91. [Google Scholar]
- Niu, M.; Zhao, G.; Alma, M.H. Thermogravimetric studies on condensed wood residues in polyhydric alcohols liquefaction. Bioresources 2011, 6, 617. [Google Scholar]
- Hassan, E.M.; Shukry, N. Polyhydric alcohol liquefaction of some lignocellulosic agricultural residues. Ind. Crops Prod. 2008, 27, 33–38. [Google Scholar] [CrossRef]
- Kobayashi, M.; Asano, T.; Kajiyama, M.; Tomita, B. Analysis on residue formation during wood liquefaction with polyhydric alcohol. J. Wood Sci. 2004, 50, 408–409. [Google Scholar] [CrossRef]
- Zhang, Y.; Ikeda, A.; Hori, N.; Takemura, A.; Ono, H.; Yamada, T. Characterization of liquefied product from cellulose with phenol in the presence of sulfuric acid. Bioresour. Technol. 2006, 97, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Park, J.; Lee, J.; Roh, H.; Jeong, D.; Cho, S.; Oh, S. Potential of a Bio-disintegrable Polymer Blend Using Alkyl-chain-modified Lignin. Fibers Polym. 2015, 16, 744–751. [Google Scholar] [CrossRef]
- Jeong, H.; Park, J.; Kim, S.; Lee, J.; Jo, J. Use of Acetylated Softwood Kraft Lignin as Filler in Synthetic Polymers. Fibers Polym. 2012, 13, 1310. [Google Scholar] [CrossRef]
- Koshijima, T.; Watanabe, T.; Yaku, F. Lignin: Properties and Materials; Glasser, W.G., Sarakanen, S., Eds.; ACS Symposium Series: Washington, DC, USA, 1989; pp. 15–26. [Google Scholar]
- Martin, D.P.; Williams, S.F. Medical Applications of Poly-4-Hydroxybutyrate: A strong flexble absorbable biomaterial. Biochem. Eng. J. 2003, 16, 97–105. [Google Scholar] [CrossRef]
- Moore, T.; Adhikari, R.; Gunatillake, P. Chemosynthesis of bioresorbable poly(γ-butyrolactone) by ring-opening polymerization—A review. Biomaterials 2005, 26, 3771–3782. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.K.; Doi, Y.; Ha, C.S. Retardation Effect of Enzymatic Degradation of Microbial Polyesters at the Surface by Blending with Polystyrene. Macromol. Biosci. 2001, 1, 114–118. [Google Scholar] [CrossRef]
- Van Blitterswijk, C. Tissue Engineering; Academic Press: London, UK, 2008; pp. 205–206. [Google Scholar]
- Islam, M.S.; Pickering, K.L.; Foreman, N.J. Influence of accelerated ageing on the physico-mechanical properties of alkali-treated industrial hemp fibre reinforced poly(lactic acid) (PLA) composites. Polym. Degrad. Stab. 2010, 95, 62. [Google Scholar] [CrossRef]
- Iwata, T.; Doi, Y. Crystal structure and biodegradation of aliphatic polyester crystals. Macromol. Chem. Phys. 1999, 200, 2429–2442. [Google Scholar] [CrossRef]
- Mikulasova, M.; Kosikova, B.; Alexy, P.; Kacik, F.; Urgelova, E. Effect of blending lignin biopolymer on the biodegradability of polyyolefin plastics. World J. Microbiol. Biotechnol. 2001, 17, 603–605. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Label (wt %) | 10:1 | 10:2 | 10:3 | 10:4 | 10:5 |
|---|---|---|---|---|---|
| BLL 30 min | 23.2 | 3.4 | 3.2 | 4 | 3.1 |
| BLL 1 h | 34.1 | 3.3 | 6.4 | 4.6 | 3.5 |
| THFL 30 min | 49.7 | 30.9 | 19.7 | 16.6 | 15.2 |
| THFL 1 h | 63.8 | 31.7 | 27.7 | 28.1 | 19.6 |
| Sample Label (wt %) | 10 min | 20 min | 30 min | 1 h | 1 h 30 min |
|---|---|---|---|---|---|
| BLL 10:5 | 0.3 | 0.2 | 3.1 | 3.5 | 5 |
| THFL 10:5 | 14.2 | 12 | 15.2 | 19.6 | 20.9 |
| Sample Label (wt %) | 10:1 | 10:2 | 10:3 | 10:4 | 10:5 | 10:6 |
|---|---|---|---|---|---|---|
| BLL | 1.176 | 1.157 | 1.233 | 1.246 | 0.819 | 0.854 |
| THFL | 1.253 | 1.113 | 1.164 | 1.098 | 1.037 | 1.04 |
| Blend Type & Proportion (%) | 100:0 | 75:25 | 50:50 | 0:100 |
|---|---|---|---|---|
| PP/THFL | 51.7 | 49.5 | 40 | 3.3 |
| PET/BLL | 30.1 | 35.4 | 33.4 | 10.9 |
| Blend Type & Proportion (%) | Ultimate Tensile Strength (MPa) | Young’s Modulus (MPa) | Elongation at Break (%) |
|---|---|---|---|
| PP 100 | 32.42 ( | 711 ( | 4.39 ( |
| PP75/THFL25 | 26.37 ( | 712 ( | 8.57 ( |
| PP50/THFL50 | 24.73 ( | 959 ( | 9.56 ( |
| PET 100 | 45.49 ( | 1145 ( | 5.33 ( |
| PET75/BLL25 | 23.23 ( | 601 ( | 447 ( |
| PET50/BLL50 | 7.26 ( | 126 ( | 503 ( |
| Degradation Time (Week) | Ultimate Tensile Strength (MPa) | Elongation at Break (%) | Young’s Modulus (MPa) | Weight Loss (%) |
|---|---|---|---|---|
| PP/THFL 4W | 19.7 ( | 8.8 ( | 634.8 ( | 2 |
| PP/THFL 8W | 22.8 ( | 5.6 ( | 1031 ( | 2 |
| PP/THFL 12W | 24.8 ( | 5.5 ( | 1264 ( | +1.3 |
| PET/BLL 4W | 12.1 ( | 6.3 ( | 1524 ( | 1 |
| PET/BLL 8W | 9.8 ( | 2 ( | 1347 ( | 0.5 |
| PET/BLL 12W | 9 ( | 1.8 ( | 1408 ( | +1.5 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.; Oh, S.; Lee, J.; Roh, H.-g.; Park, J. Changes of Lignin Molecular Structures in a Modification of Kraft Lignin Using Acid Catalyst. Materials 2016, 9, 657. https://doi.org/10.3390/ma9080657
Kim S, Oh S, Lee J, Roh H-g, Park J. Changes of Lignin Molecular Structures in a Modification of Kraft Lignin Using Acid Catalyst. Materials. 2016; 9(8):657. https://doi.org/10.3390/ma9080657
Chicago/Turabian StyleKim, Sunghoon, Seungtaek Oh, Jungmin Lee, Hyun-gyoo Roh, and Jongshin Park. 2016. "Changes of Lignin Molecular Structures in a Modification of Kraft Lignin Using Acid Catalyst" Materials 9, no. 8: 657. https://doi.org/10.3390/ma9080657
APA StyleKim, S., Oh, S., Lee, J., Roh, H.-g., & Park, J. (2016). Changes of Lignin Molecular Structures in a Modification of Kraft Lignin Using Acid Catalyst. Materials, 9(8), 657. https://doi.org/10.3390/ma9080657