
Effects of Al-Impurity Type on Formation Energy, Crystal Structure, Electronic Structure, and Optical Properties of ZnO by Using Density Functional Theory and the Hubbard-U Method


Abstract
:1. Introduction
2. Calculation Methods
3. Results and Discussion
3.1. Formation Energy
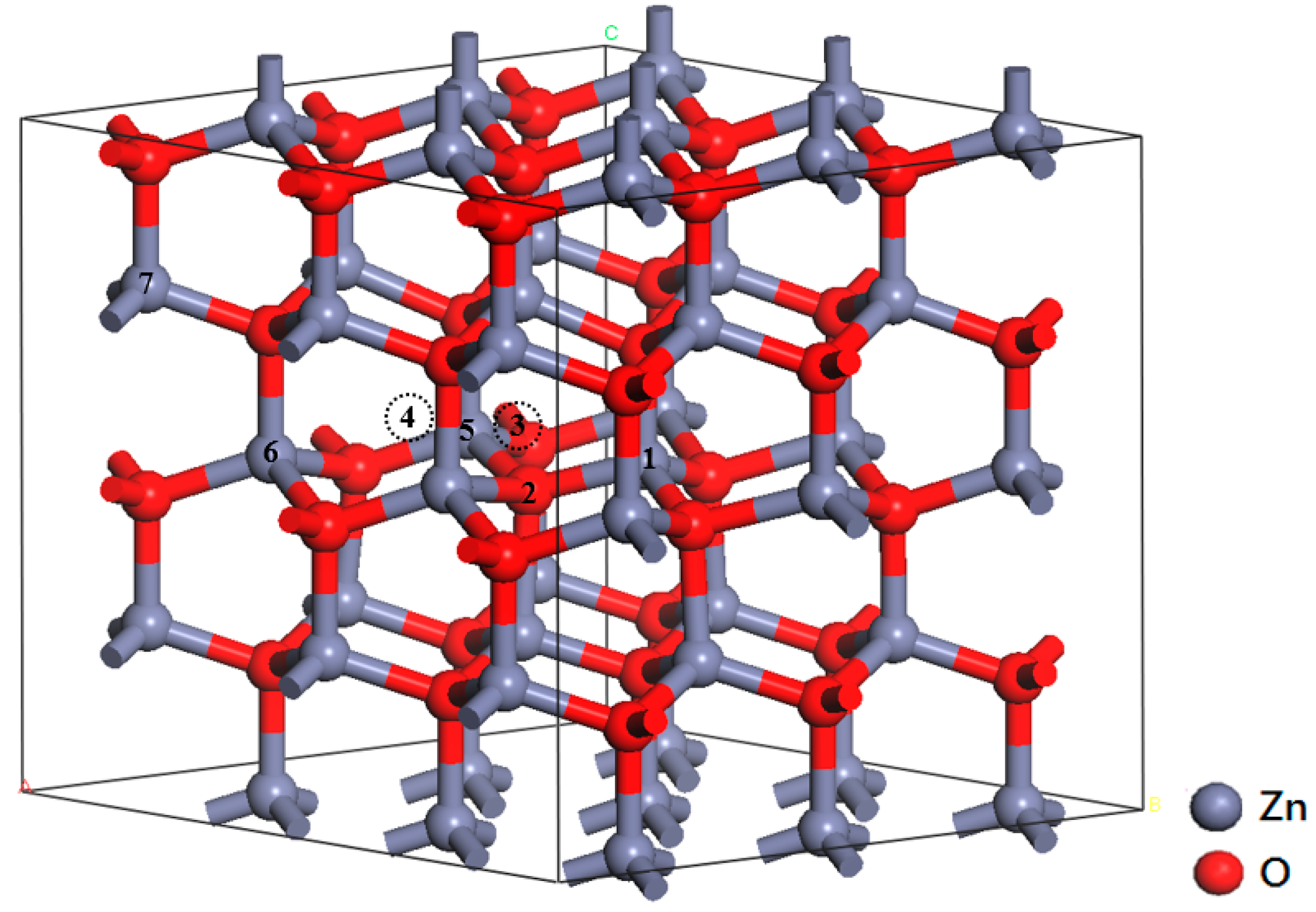
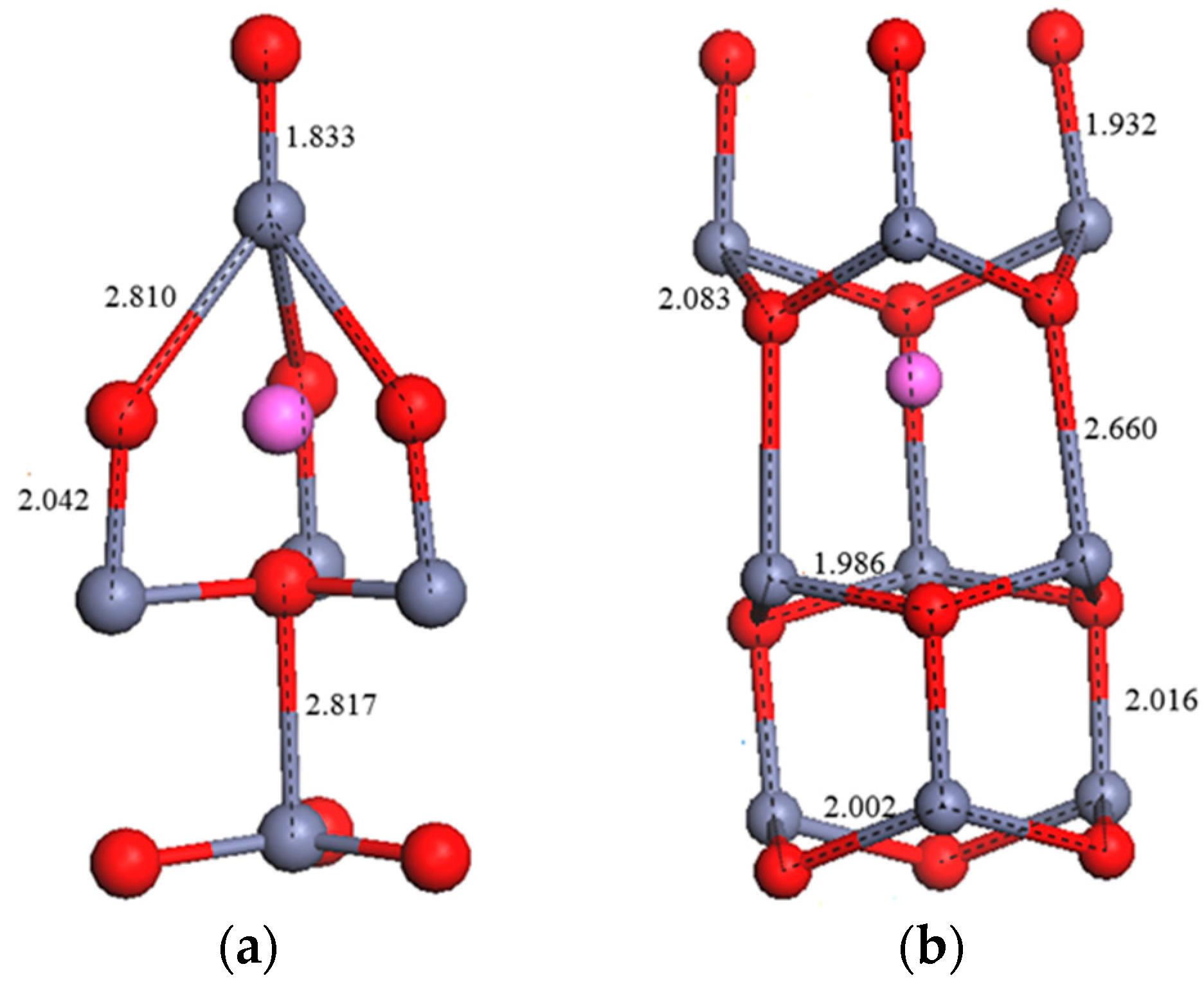
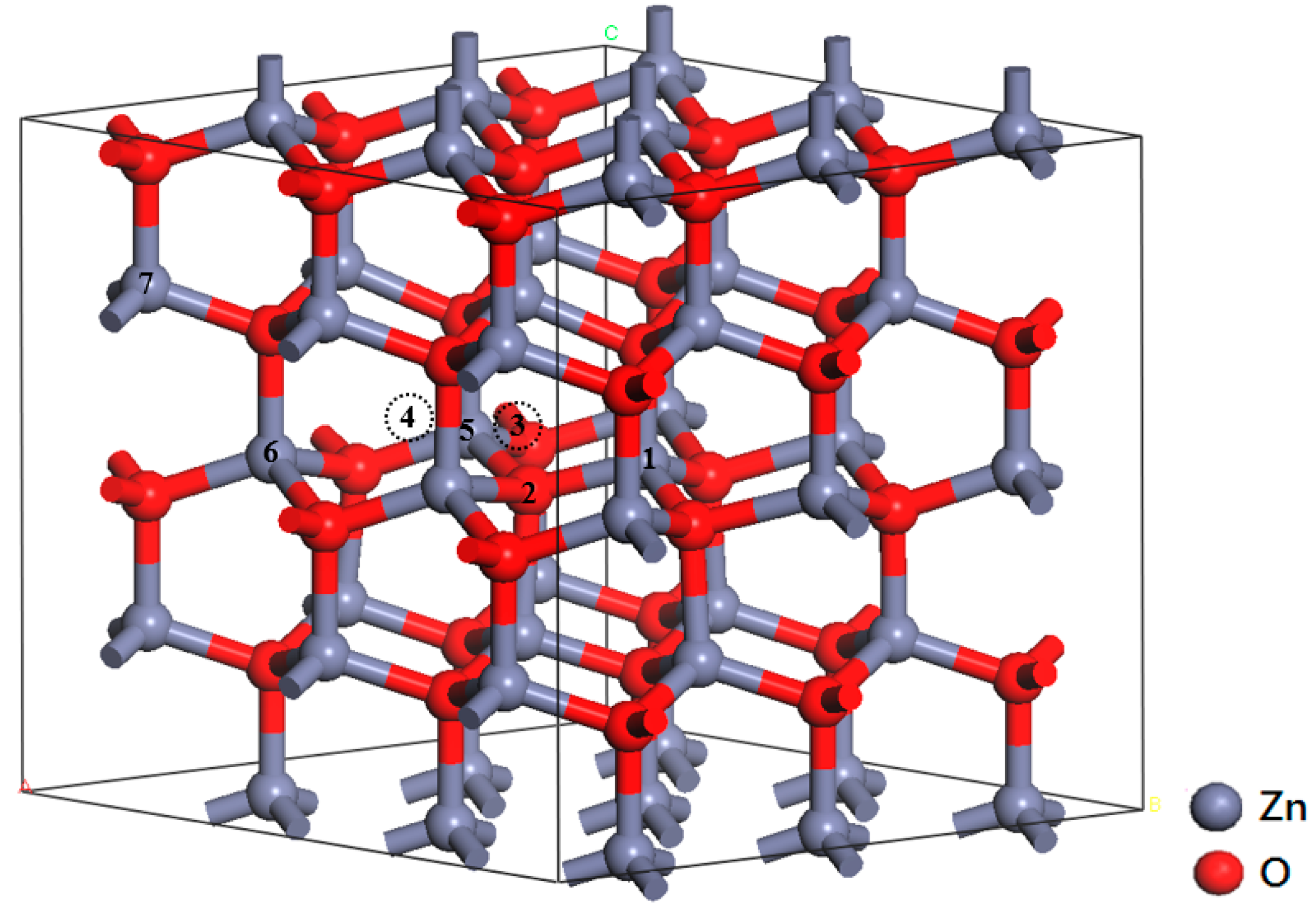
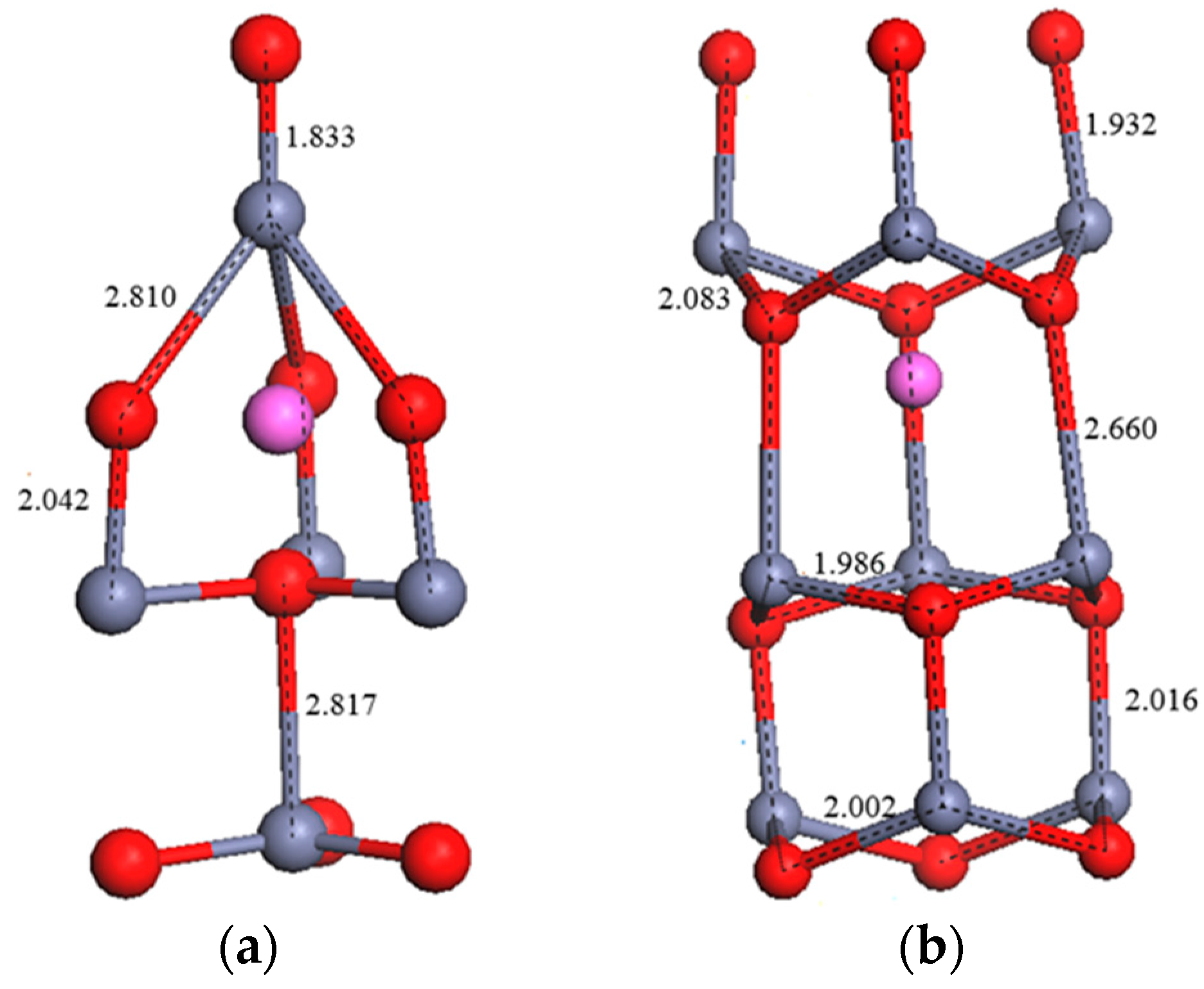
3.2. Crystal Structure
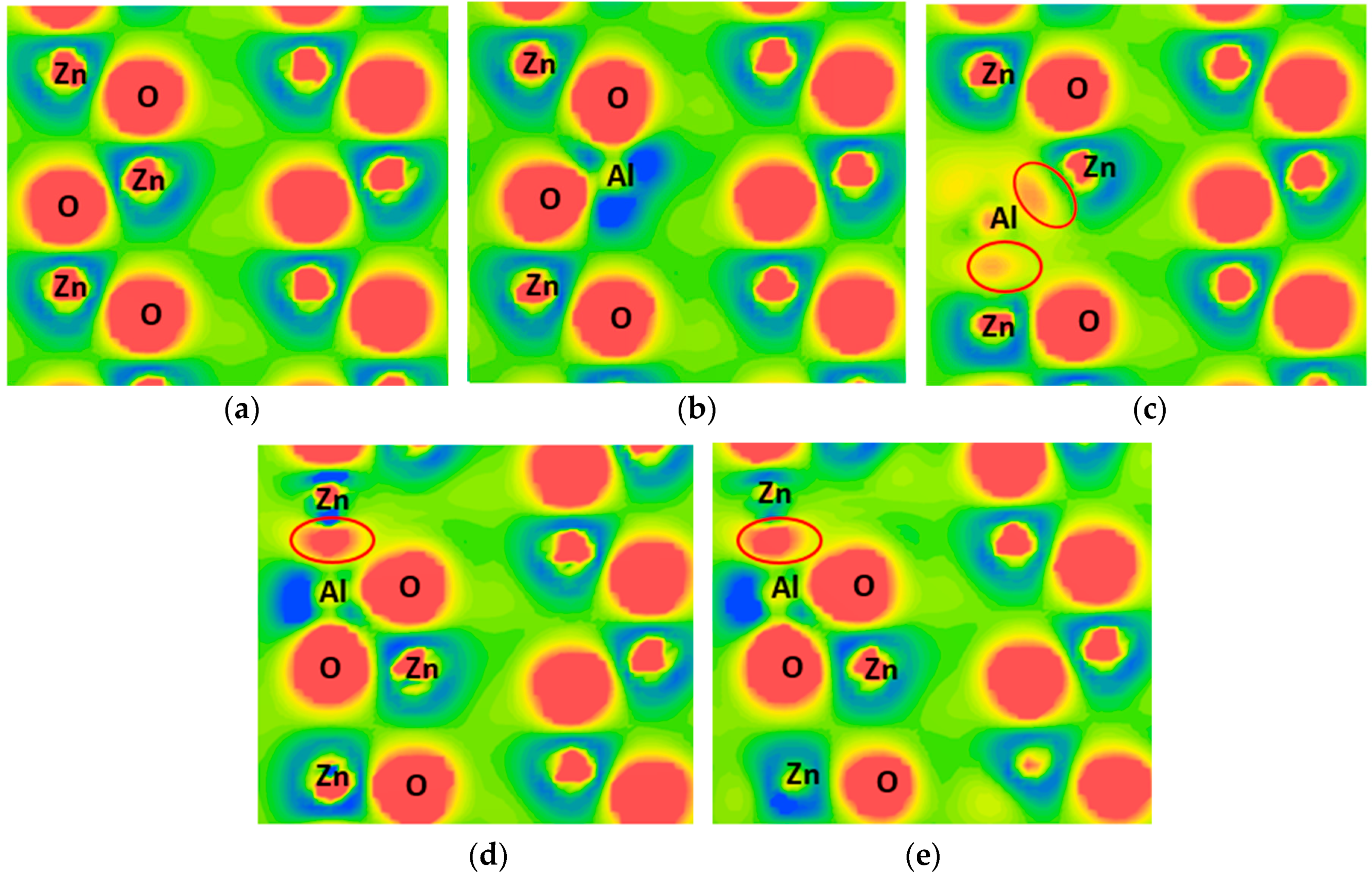
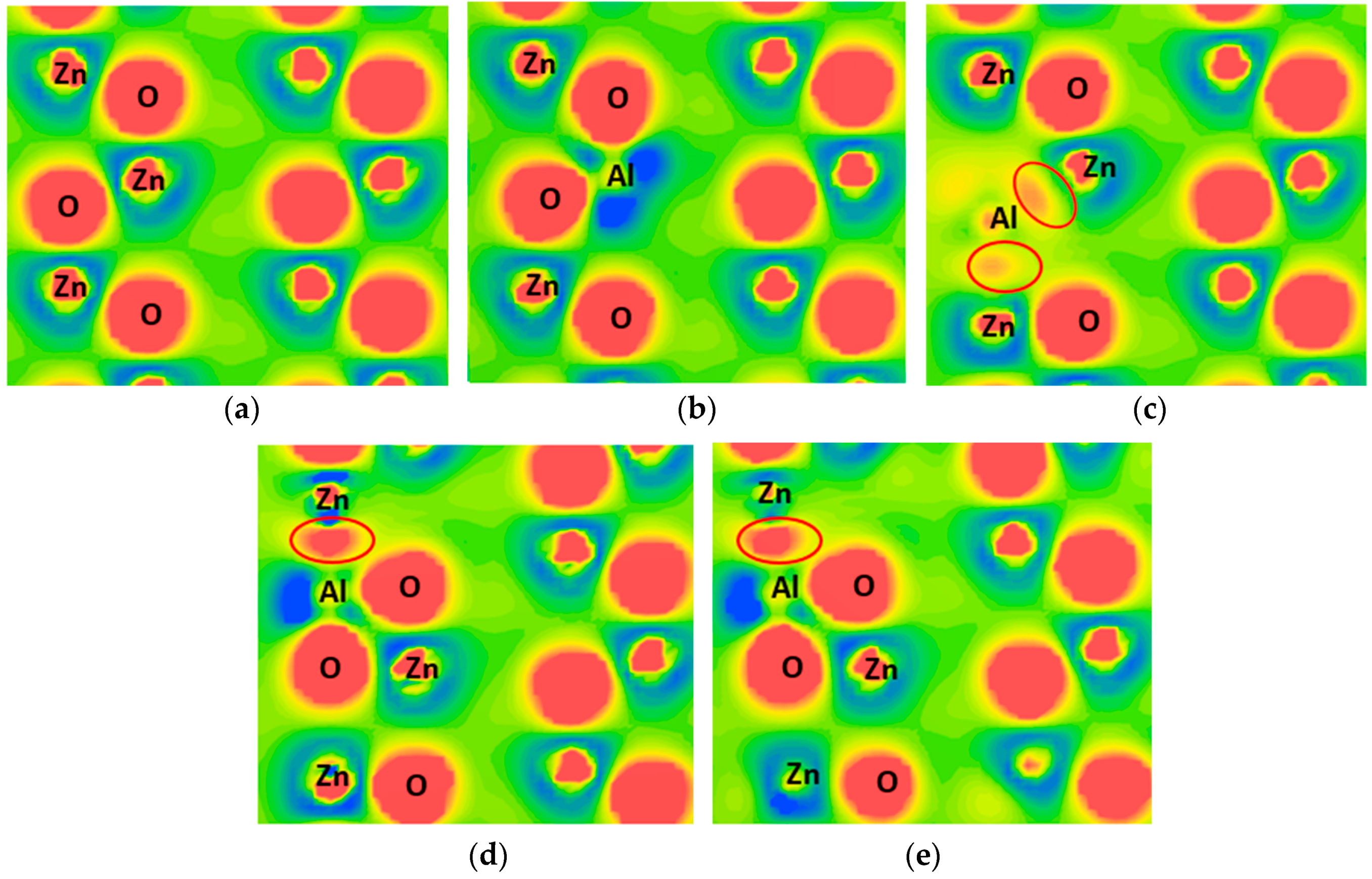
3.3. Charge Density
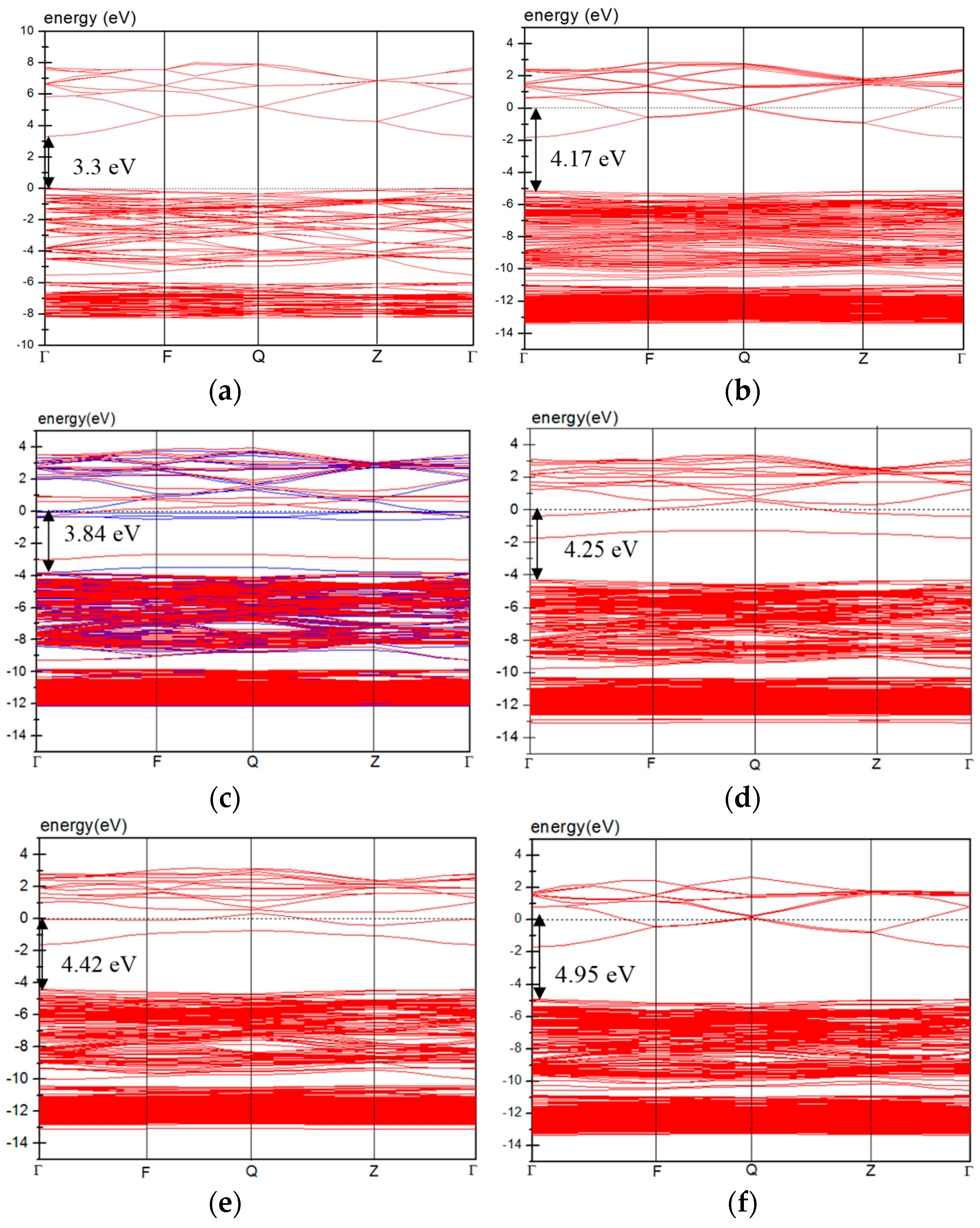
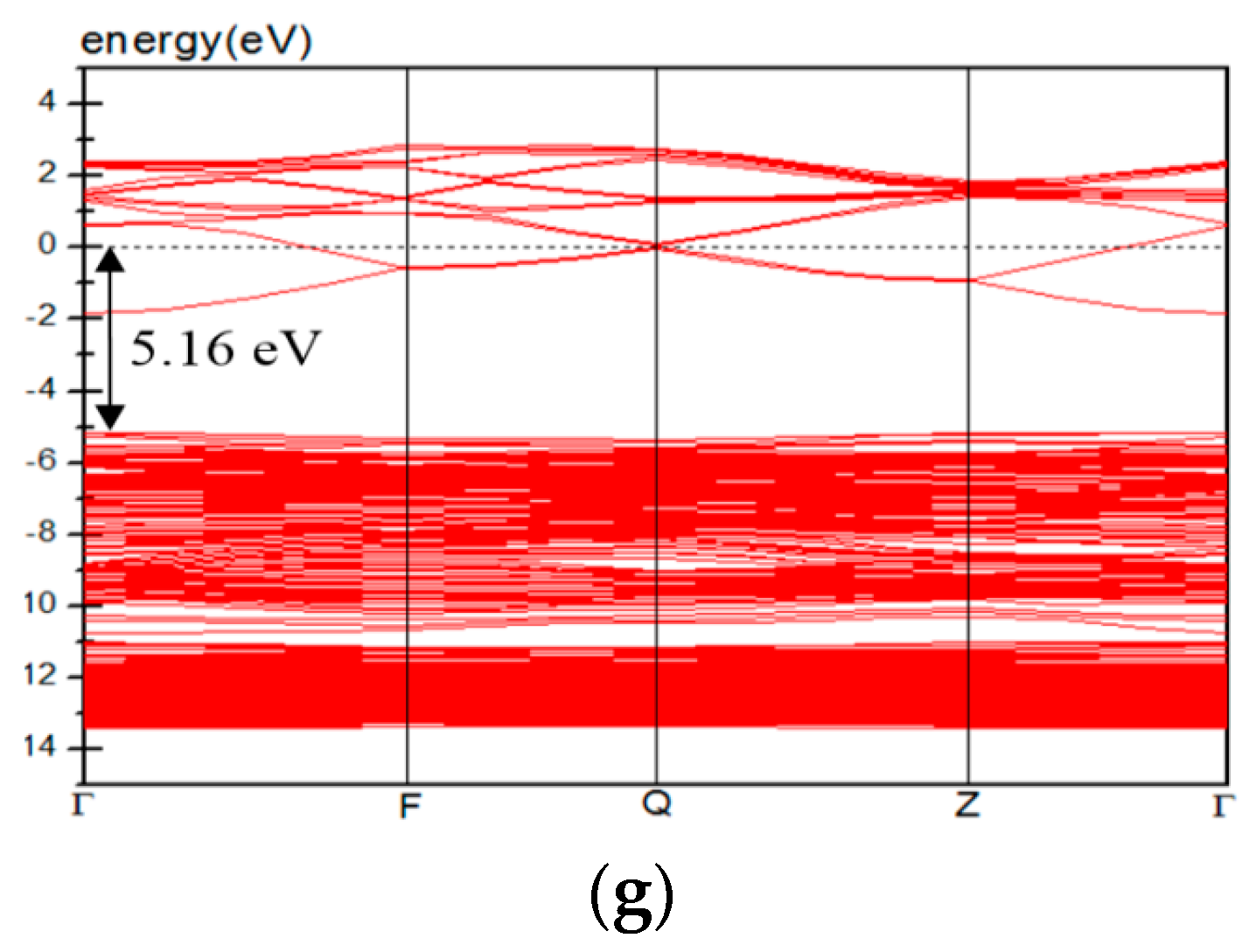
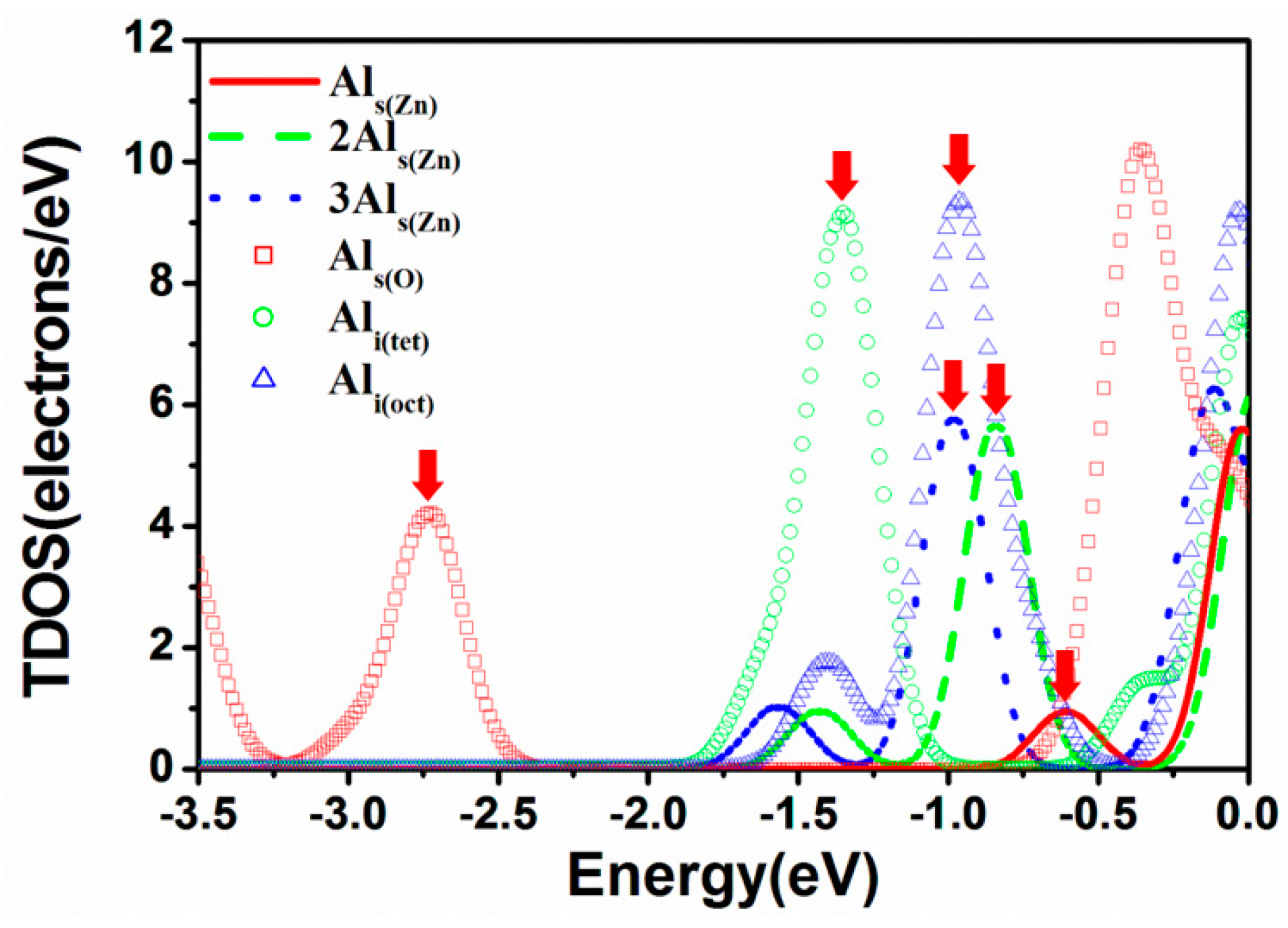
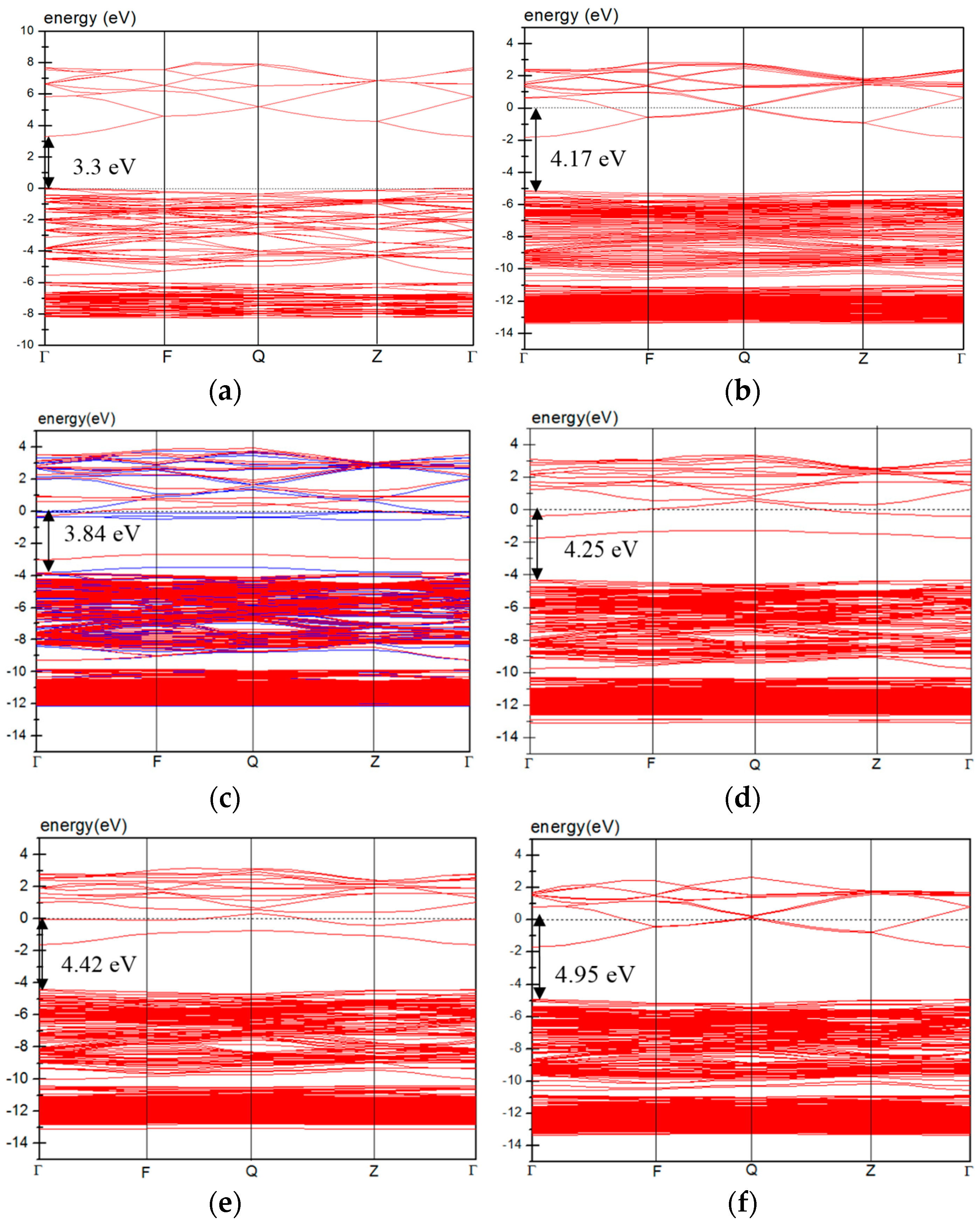
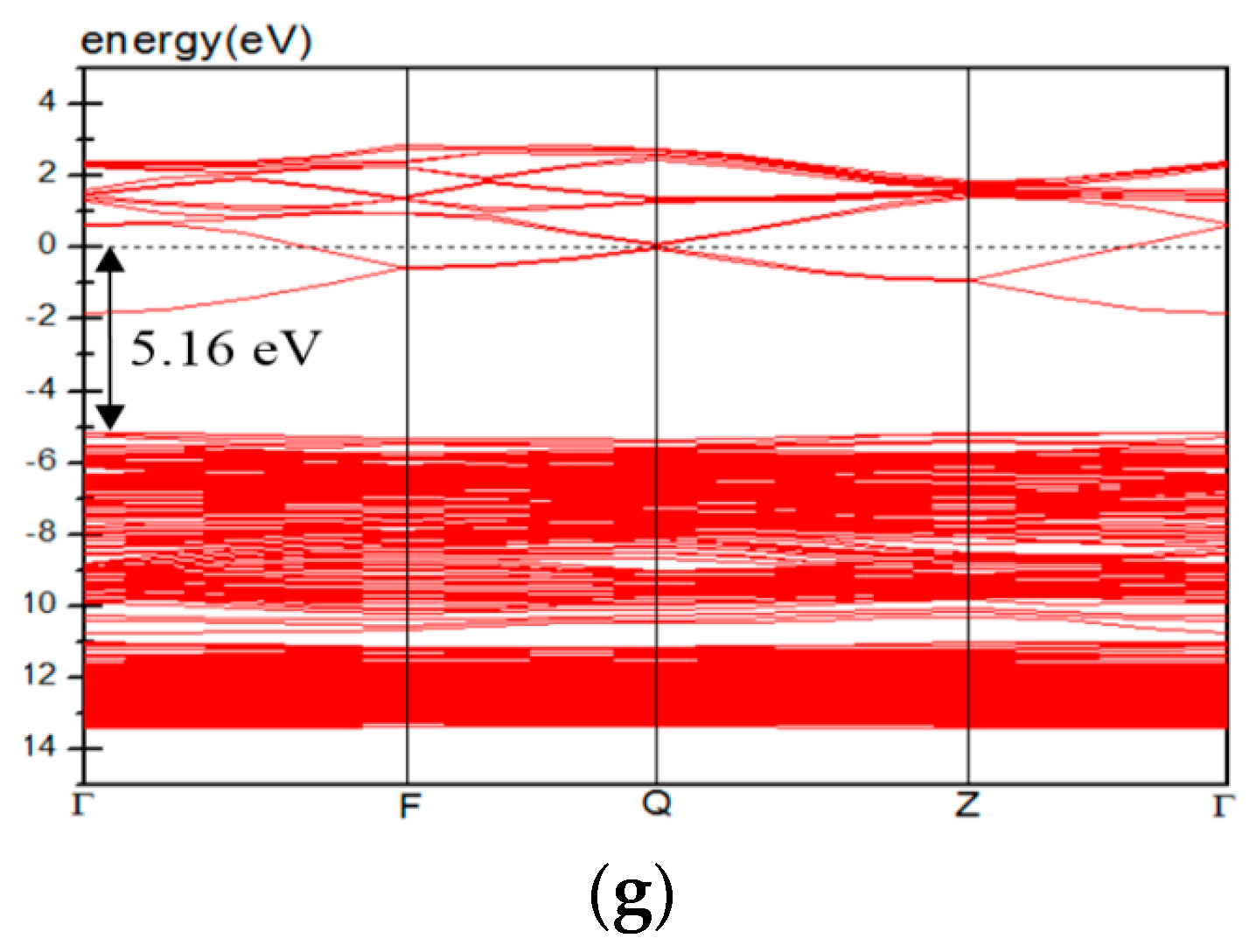
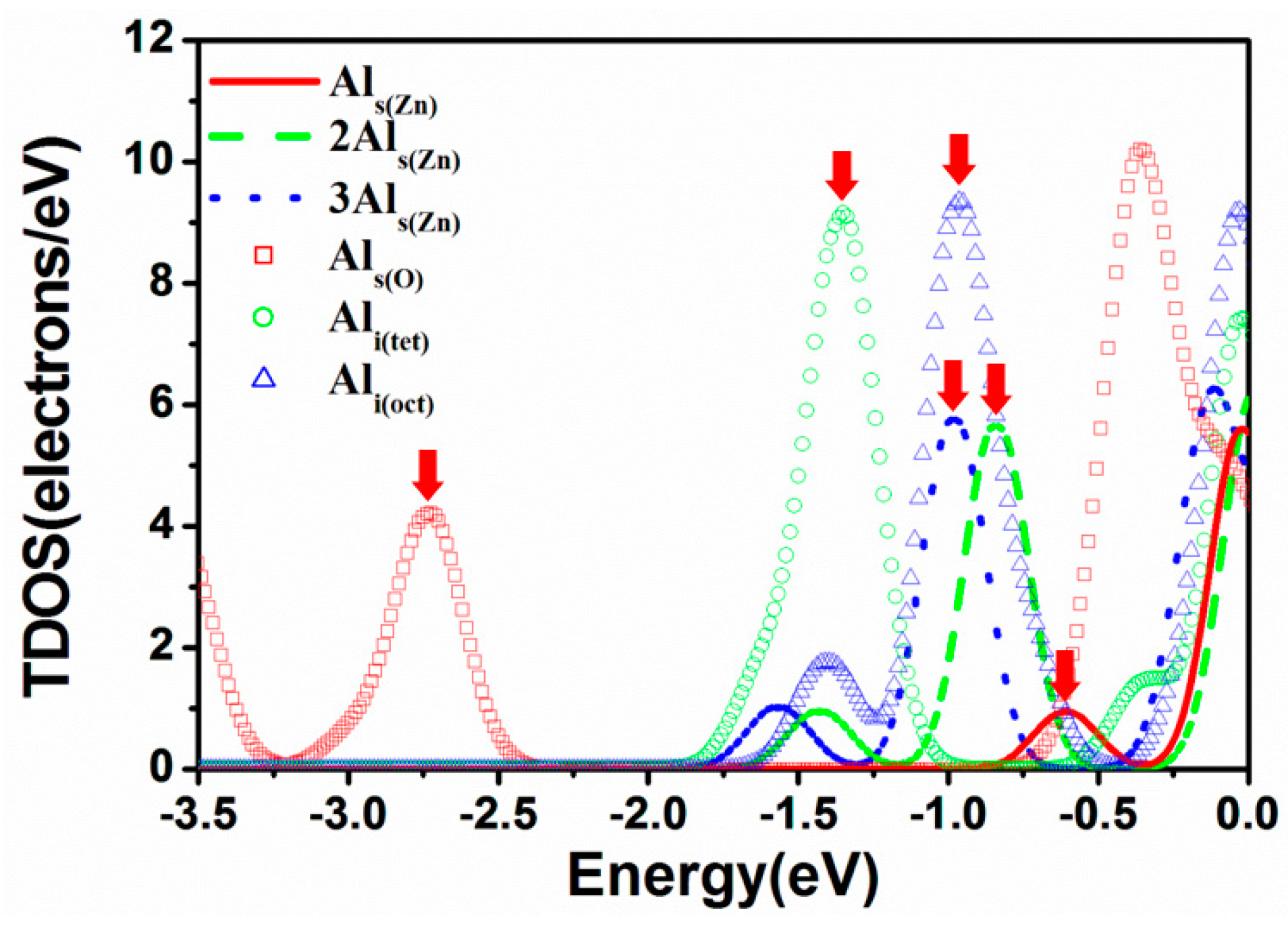
3.4. Electric Properties
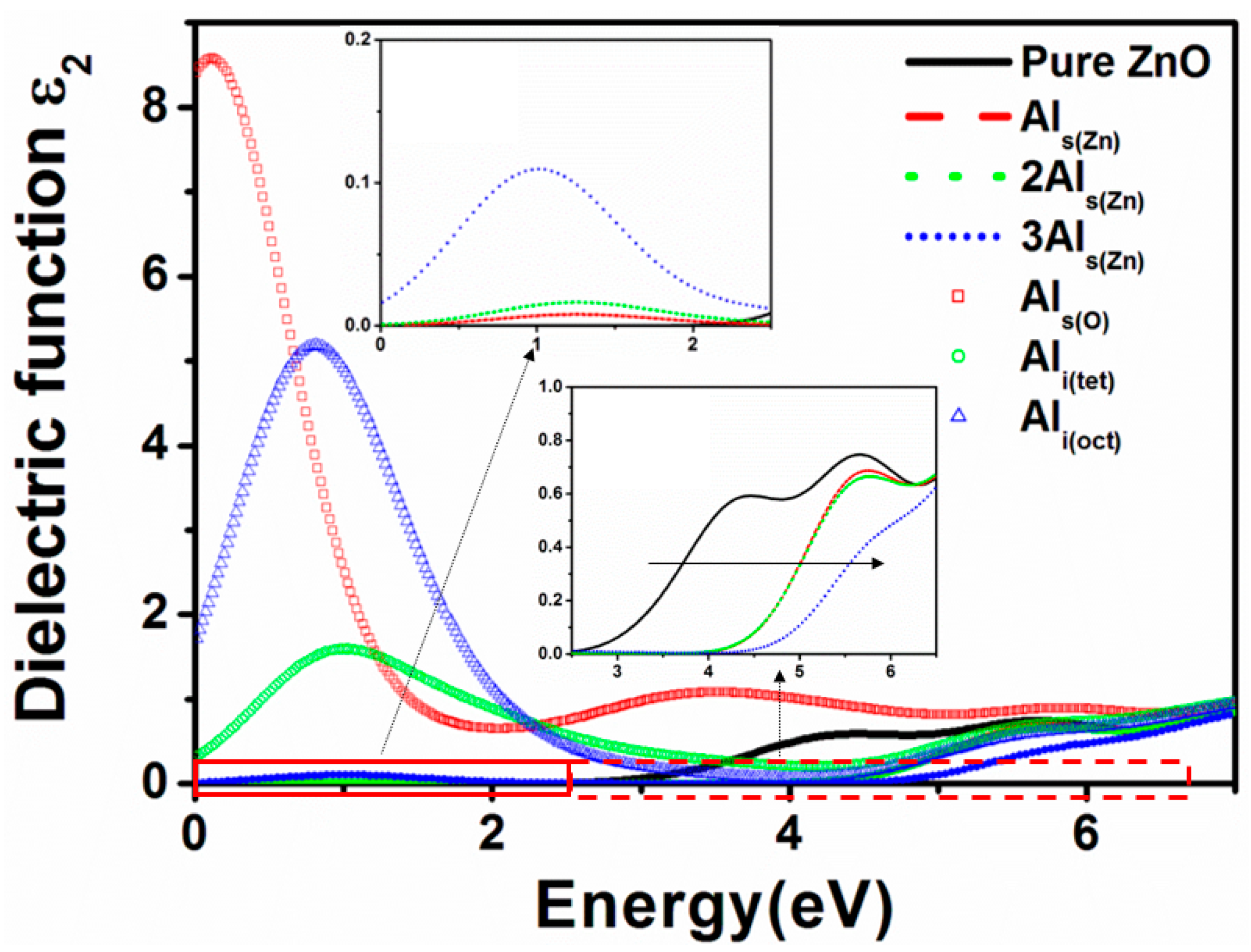
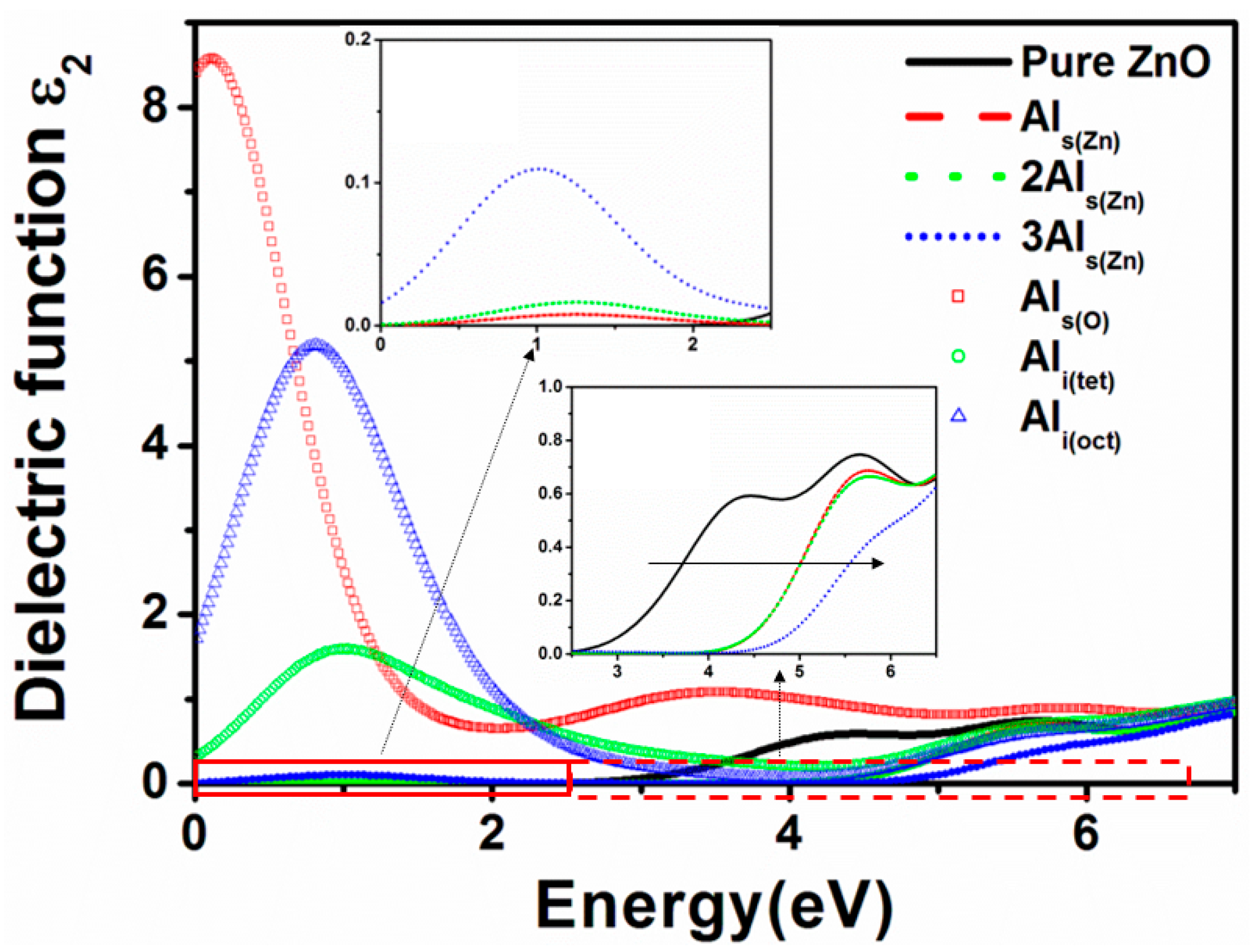
3.5. Optical Properties
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kim, K.; Park, S.; Seon, J.B.; Lim, K.H.; Char, K.; Shin, K.; Kim, Y.S. Patterning of flexible transparent thin-film transistors with solution-processed ZnO using the binary solvent mixture. Adv. Funct. Mater. 2011, 21, 3546–3553. [Google Scholar] [CrossRef]
- Jiang, X.; Wong, F.L.; Fung, M.K.; Lee, S.T. Aluminum-doped zinc oxide films as transparent conductive electrode for organic light-emitting devices. Appl. Phys. Lett. 2003, 83, 1875–1877. [Google Scholar] [CrossRef]
- Verma, A.; Khan, F.; Kumar, D.; Kar, M.; Chakravarty, B.C.; Singh, S.N.; Husain, M. Sol-gel derived aluminum doped zinc oxide for application as anti-reflection coating in terrestrial silicon solar cells. Thin Solid Films 2010, 518, 2649–2653. [Google Scholar] [CrossRef]
- Lee, K.I.; Kang, H.I.; Lee, T.Y.; Lee, J.H.; Song, J.T. Structural and electrical properties of Al-doped ZnO and Al, B-codoped ZnO films deposited on flexible substrate. J. Korean Chem. Phys. Soc. 2008, 53, 2407–2410. [Google Scholar]
- Youn, C.J.; Jeong, T.S.; Han, M.S.; Kim, J.H. Optical properties of Zn-terminated ZnO bulk. J. Cryst. Growth 2004, 261, 526–532. [Google Scholar] [CrossRef]
- Lee, Y.S.; Peng, Y.C.; Lu, J.H.; Zhu, Y.R.; Wu, H.C. Electronic and optical properties of Ga-doped ZnO. Thin Solid Films 2014, 570, 464–470. [Google Scholar] [CrossRef]
- Shinde, S.D.; Deshmukh, A.V.; Date, S.K.; Sathe, V.G.; Adhi, K.P. Effect of Ga doping on micro/structural, electrical and optical properties of pulsed laser deposited ZnO thin films. Thin Solid Films 2011, 520, 1212–1217. [Google Scholar] [CrossRef]
- Hafdallah, A.; Yanineb, F.; Aida, M.S.; Attaf, N. In doped ZnO thin films. J. Alloys Compd. 2011, 509, 7267–7270. [Google Scholar] [CrossRef]
- Fuchs, P.; Hagendorfer, H.; Romanyuk, Y.E.; Tiwari, A.N. Doping strategies for highly conductive Al-doped ZnO films grown from aqueous solution. Phys. Status Solidi A 2015, 212, 51–55. [Google Scholar] [CrossRef]
- Manthina, V.; Agrios, A.G. Single-pot ZnO nanostructure synthesis by chemical bath deposition and their applications. Nano-Struct. Nano-Objects 2016, 7, 1–11. [Google Scholar] [CrossRef]
- Agura, H.; Suzuki, A.; Matsushita, T.; Aoki, T.; Okuda, M. Low resistivity transparent conducting Al-doped ZnO films prepared by pulsed laser deposition. Thin Solid Films 2003, 445, 263–267. [Google Scholar] [CrossRef]
- Fragala, M.E.; Malandrino, G.; Giangregorio, M.M.; Losurdo, M.; Bruno, G. Structural, optical, and electrical characterization of ZnO and Al-doped ZnO thin films deposited by MOCVD. Chem. Vap. Depos. 2009, 15, 327–333. [Google Scholar] [CrossRef]
- Maeng, W.J.; Lee, J.W.; Lee, J.H.; Chung, K.B.; Park, J.S. Studies on optical, structural and electrical properties of atomic layer deposited Al-doped ZnO thin films with various Al concentrations and deposition temperatures. J. Phys. D Appl. Phys. 2011, 44, 445–305. [Google Scholar] [CrossRef]
- Blagoev, B.S.; Dimitrov, D.Z.; Mehandzhiev, V.B.; Kovacheva, D.; Terziyska, P.; Pavlic, J.; Lovchinov, K.; Mateev, E.; Leclercq, J.; Sveshtarov, P. Electron transport in Al-doped ZnO nanolayers obtained by atomic layer deposition. J. Phys. Conf. Ser. 2016, 700, 012040. [Google Scholar] [CrossRef]
- Singh, C.; Panda, E. Variation of electrical properties in thickening Al-doped ZnO films: Role of defect chemistry. RSC Adv. 2016, 6, 48910–48918. [Google Scholar] [CrossRef]
- Ganesh, T.; Rajesh, S.; Xavier, F.P. Effect of Al dopant concentration on structural, optical and photoconducting properties in nanostructured zinc oxide thin films. Mater. Sci. Semiconduct. Process. 2013, 16, 295–302. [Google Scholar] [CrossRef]
- Periasamy, C.; Chakrabart, P. Tailoring the structural and optoelectronic properties of Al-doped nanocrystalline ZnO thin films. J. Electron. Mater. 2011, 40, 259–266. [Google Scholar] [CrossRef]
- Qu, X.; Wang, W.; Lv, S.; Jia, D. Thermoelectric properties and electronic structure of Al-doped ZnO. Solid State Commun. 2011, 151, 332–336. [Google Scholar] [CrossRef]
- Palacios, P.; Sánchez, K.; Wahnón, P. Ab-initio valence band spectra of Al, In doped ZnO. Thin Solid Films 2009, 517, 2448–2451. [Google Scholar] [CrossRef]
- Gabás, M.; Torelli, P.; Barrett, N.T.; Sacchi, M.; Bruneval, F.; Cui, Y.; Simonelli, L.; Díaz-Carrasco, P.; Barrado, J.R.R. Direct observation of Al-doping-induced electronic states in the valence band and band gap of ZnO films. Phys. Rev. B Condens. Matter 2011, 84, 153303. [Google Scholar] [CrossRef]
- Li, P.; Deng, S.H.; Li, Y.B.; Huang, J.; Liu, G.H.; Zhang, L. Aluminum and nitrogen impurities in wurtzite ZnO: First-principles studies. Physica B 2011, 406, 3125–3129. [Google Scholar] [CrossRef]
- Lee, M.H.; Peng, Y.C.; Wu, H.C. Effects of intrinsic defects on electronic structure and optical properties of Ga-doped ZnO. J. Alloys Compd. 2014, 616, 122–127. [Google Scholar] [CrossRef]
- Segall, M.D.; Lindan, P.J.D.; Probert, M.J.; Pickard, C.J.; Hasnip, P.J.; Clark, S.J.; Payne, M.C. First-principles simulation: Ideas, illustrations and the CASTEP code. J. Phys. Condens. Matter 2002, 14, 2717–2744. [Google Scholar] [CrossRef]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892–7895. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillonin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Ma, X.; Lu, B.; Li, D.; Shi, R.; Pan, C.; Zhu, Y. Origin of photocatalytic activation of silver orthophosphate from first-principles. J. Phys. Chem. C 2011, 115, 4680–4687. [Google Scholar] [CrossRef]
- Wu, H.C.; Peng, Y.C.; Shen, T.P. Electronic and optical properties of substitutional and interstitial Si-doped ZnO. Materials 2012, 5, 2088–2100. [Google Scholar] [CrossRef]
- Fan, Q.; Yang, J.; Yu, Y.; Zhang, J.; Cao, J. Electronic structure and optical properties of Al-doped ZnO from hybrid functional calculations. Chem. Eng. Trans. 2015, 46, 985–990. [Google Scholar]
- Chowdhury, R.; Adhikari, S.; Rees, P. Optical properties of silicon doped ZnO. Physisca B 2010, 405, 4763–4767. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Models | Formation Energy (eV) | |
|---|---|---|
| O-Rich | Zn-Rich | |
| Als(Zn) | −6.69 | −3.45 |
| Als(O) | 9.19 | 5.95 |
| Ali(tet) | 1.81 | 1.81 |
| Ali(oct) | 2.97 | 2.97 |
| Models | Lattice Constants | Volume | Bond Length (Å) | ||||
|---|---|---|---|---|---|---|---|
| a (Å) | c (Å) | c/a | unit cell (Å)3 | Zn–O | Al–O | Al–Zn | |
| Pure ZnO | 3.282 | 5.265 | 1.60 | 49.01 | 1.997 | --- | --- |
| Als(Zn) | 3.280 | 5.269 | 1.61 | 48.98 | 2.002 | 1.797 | --- |
| Als(O) | 3.304 | 5.329 | 1.61 | 50.21 | 2.009 | --- | 2.451 |
| Ali(tet) | 3.283 | 5.392 | 1.64 | 50.56 | 2.015 | 1.855 | 2.286 |
| Ali(oct) | 3.286 | 5.418 | 1.65 | 50.27 | 2.023 | 1.803 | 2.695 |
| 2Als(Zn) | 3.278 | 5.271 | 1.61 | 48.92 | 2.009 | 1.797 | --- |
| 3Als(Zn) | 3.274 | 5.275 | 1.61 | 48.87 | 2.015 | 1.796 | --- |
| Models | Atomic Population (|e|) | Bond Population (|e|) | ||||
|---|---|---|---|---|---|---|
| Zn | O | Al | Zn–O | Al–O | Al–Zn | |
| Pure ZnO | 0.94 | −0.94 | --- | 0.4 | --- | --- |
| Als(Zn) | 0.94 | −1.02 | 1.62 | 0.39 | 0.5 | --- |
| Als(O) | 0.92 | −0.93 | −0.44 | 0.39 | --- | 0.94 |
| Ali(tet) | 0.90 | −0.94 | 1.41 | 0.38 | 0.44 | 0.46 |
| Ali(oct) | 0.91 | −0.93 | 0.90 | 0.38 | 0.39 | 0.42 |
| 2Als(Zn) | 0.93 | −1.02 | 1.62 | 0.38 | 0.49 | --- |
| 3Als(Zn) | 0.91 | −1.02 | 1.63 | 0.37 | 0.50 | --- |
| Models | Carrier Concentration (1020/cm3) | ||
|---|---|---|---|
| Total | Close to EF | Far from EF | |
| Als(Zn) | 12.5 | 9.7 | 2.8 |
| Als(O) | 46.9 | 32.8 | 14.1 |
| Ali(tet) | 32.7 | 10.8 | 21.9 |
| Ali(oct) | 32.8 | 10.9 | 21.9 |
| 2Als(Zn) | 25.1 | 7.9 | 17.2 |
| 3Als(Zn) | 33.5 | 15.9 | 17.6 |
| Models | Transmittance (%) | |
|---|---|---|
| UV | Visible Light | |
| Pure ZnO | 64.4 | 88.8 |
| Als(Zn) | 75.2 | 91.1 |
| Als(O) | 50.9 | 70.4 |
| Ali(tet) | 69.7 | 73.5 |
| Ali(oct) | 75.3 | 68.5 |
| 2Als(Zn) | 75.5 | 91.1 |
| 3Als(Zn) | 81.1 | 91.2 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, H.-C.; Chen, H.-H.; Zhu, Y.-R. Effects of Al-Impurity Type on Formation Energy, Crystal Structure, Electronic Structure, and Optical Properties of ZnO by Using Density Functional Theory and the Hubbard-U Method. Materials 2016, 9, 647. https://doi.org/10.3390/ma9080647
Wu H-C, Chen H-H, Zhu Y-R. Effects of Al-Impurity Type on Formation Energy, Crystal Structure, Electronic Structure, and Optical Properties of ZnO by Using Density Functional Theory and the Hubbard-U Method. Materials. 2016; 9(8):647. https://doi.org/10.3390/ma9080647
Chicago/Turabian StyleWu, Hsuan-Chung, Hsing-Hao Chen, and Yu-Ren Zhu. 2016. "Effects of Al-Impurity Type on Formation Energy, Crystal Structure, Electronic Structure, and Optical Properties of ZnO by Using Density Functional Theory and the Hubbard-U Method" Materials 9, no. 8: 647. https://doi.org/10.3390/ma9080647
APA StyleWu, H.-C., Chen, H.-H., & Zhu, Y.-R. (2016). Effects of Al-Impurity Type on Formation Energy, Crystal Structure, Electronic Structure, and Optical Properties of ZnO by Using Density Functional Theory and the Hubbard-U Method. Materials, 9(8), 647. https://doi.org/10.3390/ma9080647