
Strength Development and Hydration Behavior of Self-Activation of Commercial Ground Granulated Blast-Furnace Slag Mixed with Purified Water



Abstract
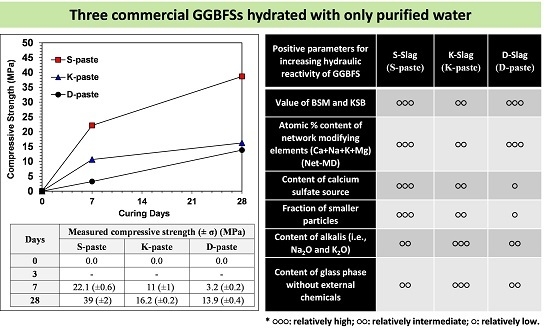
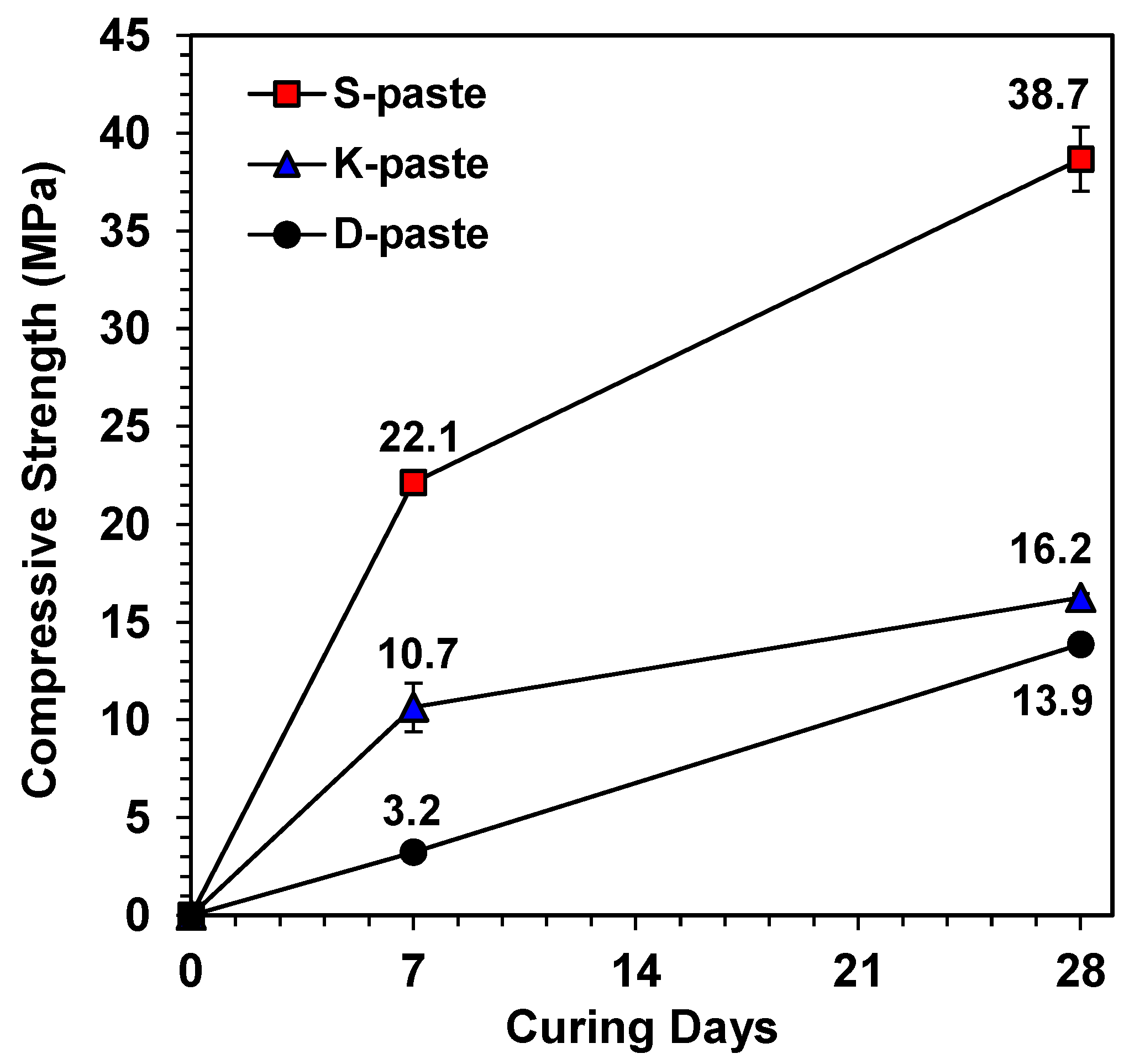
:
1. Introduction
2. Experimental Program
3. Results and Discussion
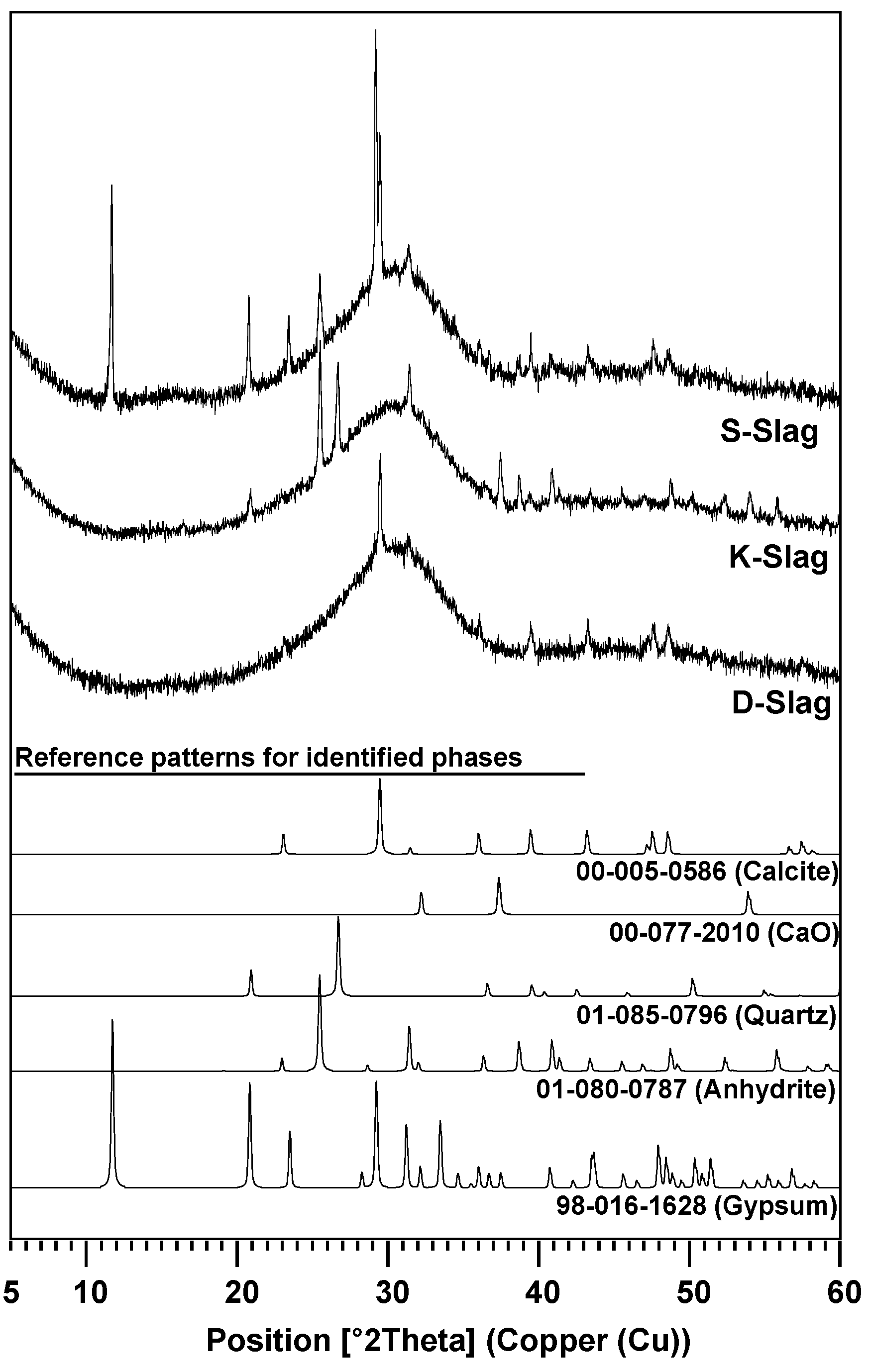
3.1. Materials Parameters of GGBFS
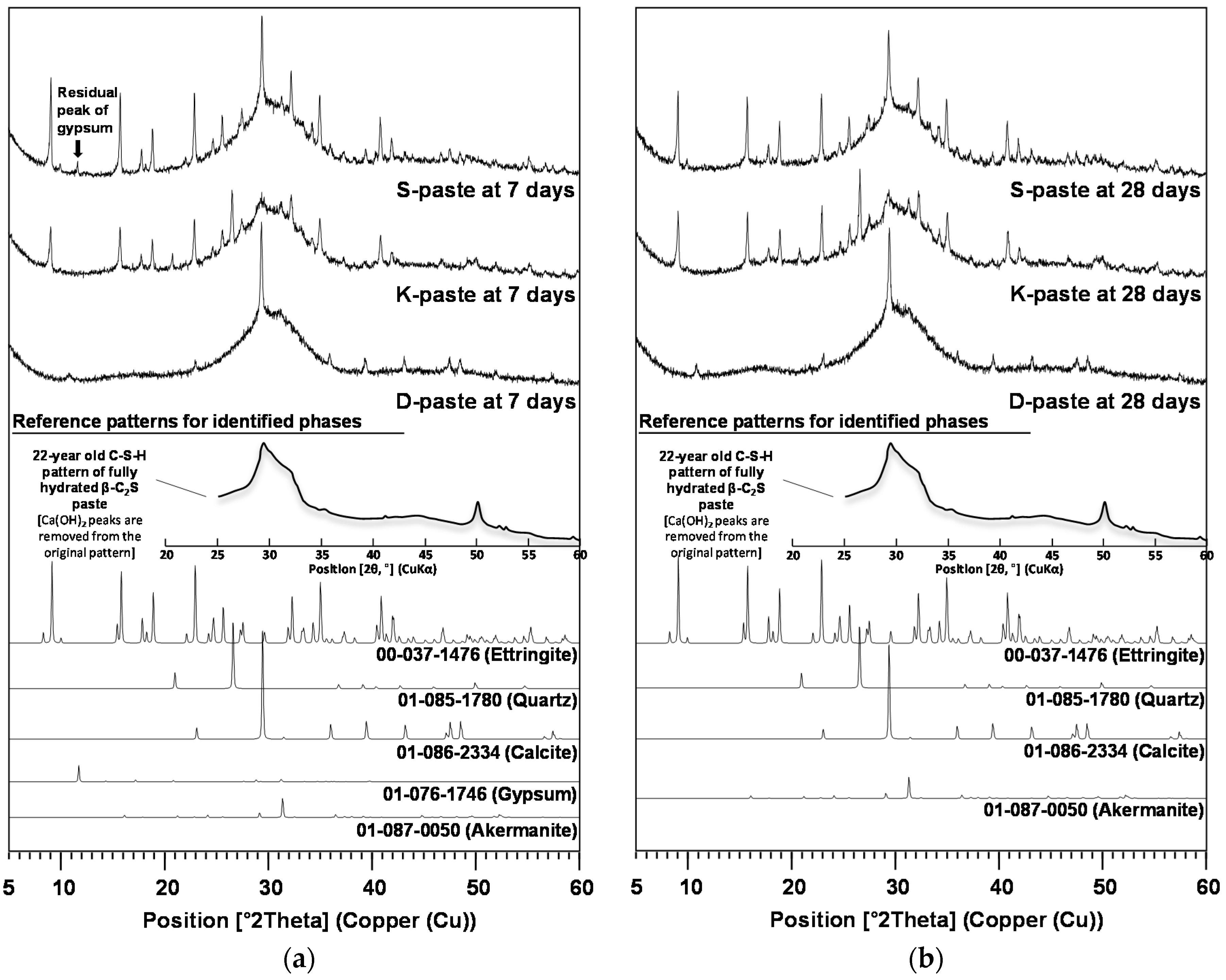
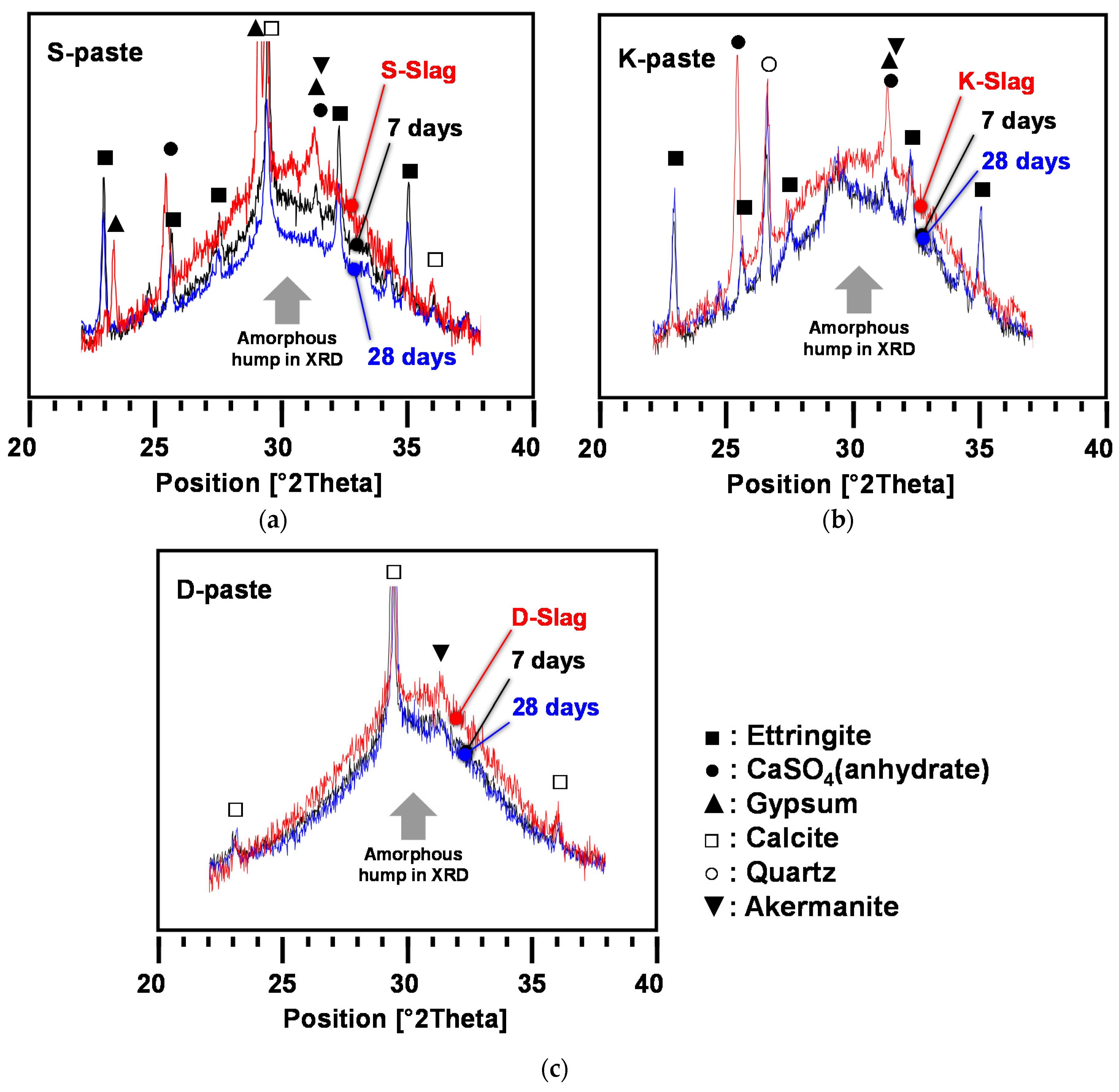
3.2. Characterization of Hardened Pastes
4. Conclusions
- (1)
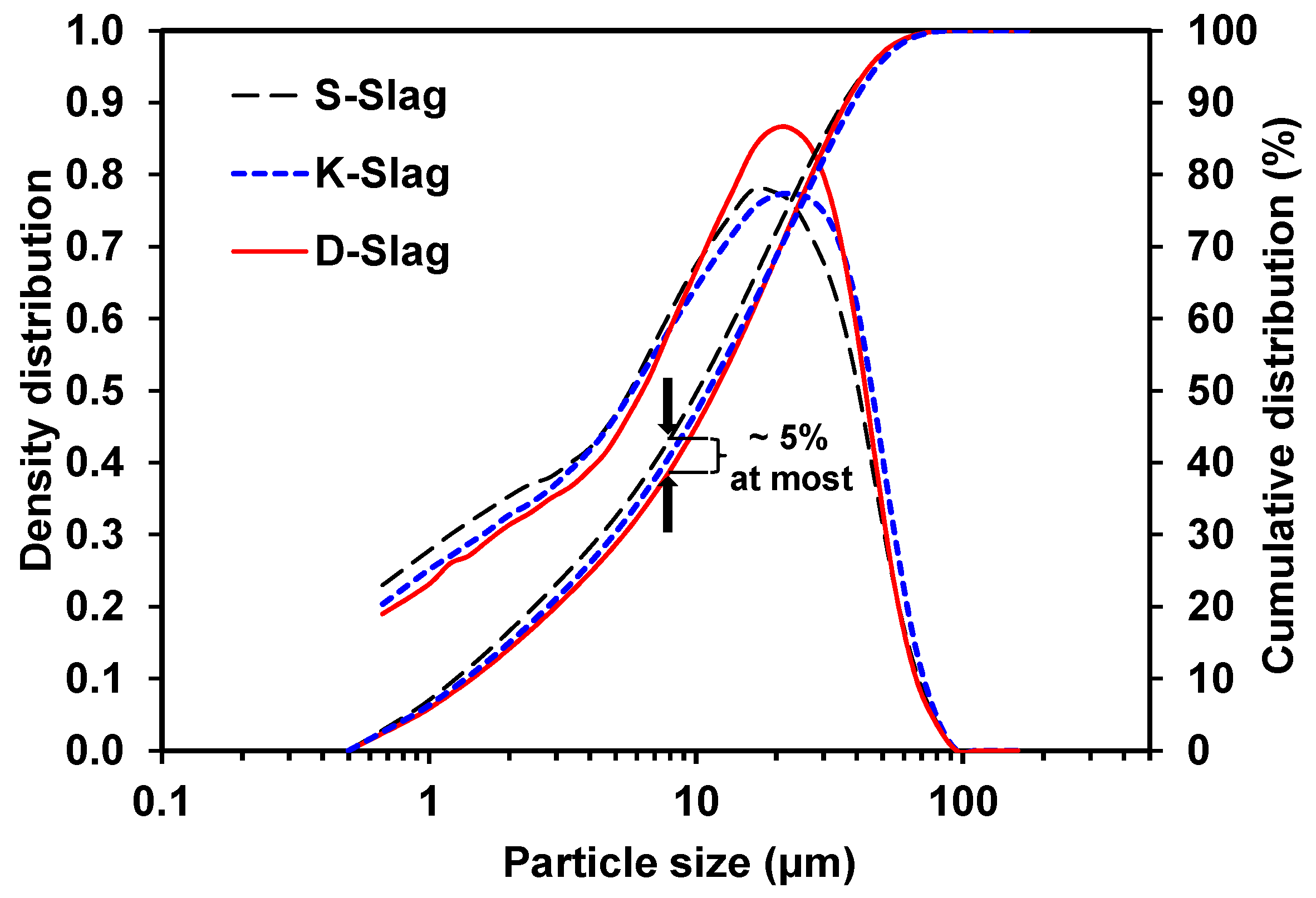
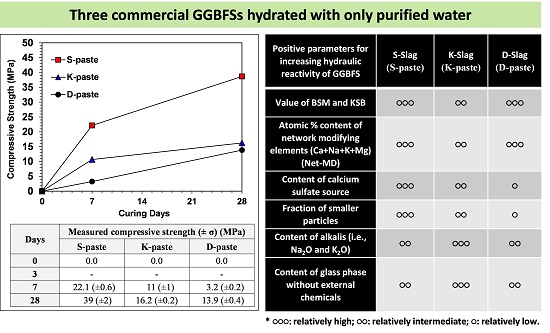
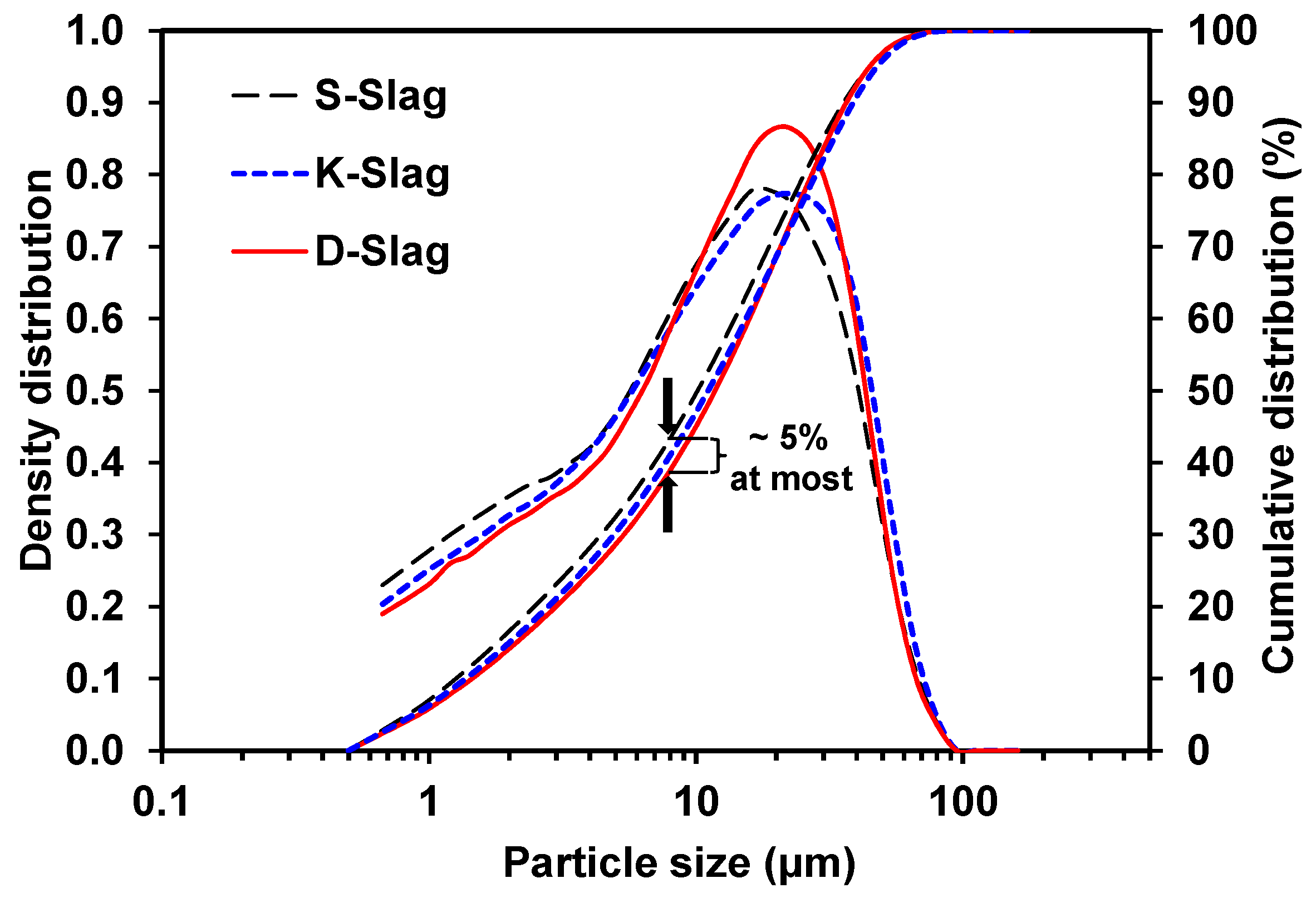
- The raw GGBFS samples showed quantitative and qualitative differences in the material parameters, which were compositional indicators (CM, KSB, and Net-MD), and particle-size distribution, as well as calcium-sulfate, alkali, and glass-phase content.
- (2)
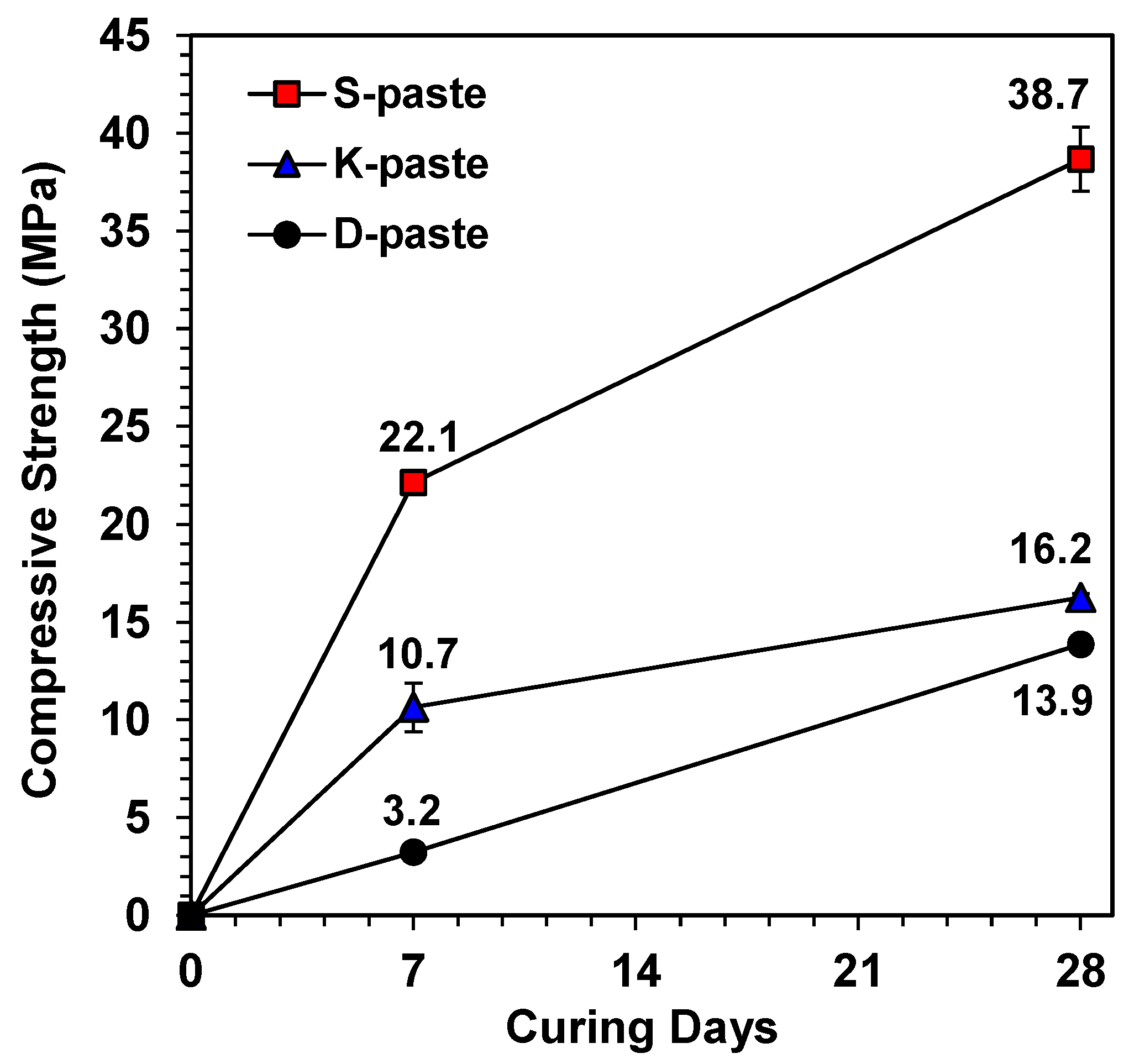
- In this study, the strength was not governed by any single dominant parameter, but was rather the comprehensive consequence of all material parameters of the raw GGBFS (summarized in Table 3); the relative comparison of these parameters predicted that the S- and D-pastes would produce the highest and the lowest strengths, respectively.
- (3)
- S-Slag produced a high strength that was comparable to that of alkali- or Ca(OH)2-activated GGBFS.
- (4)
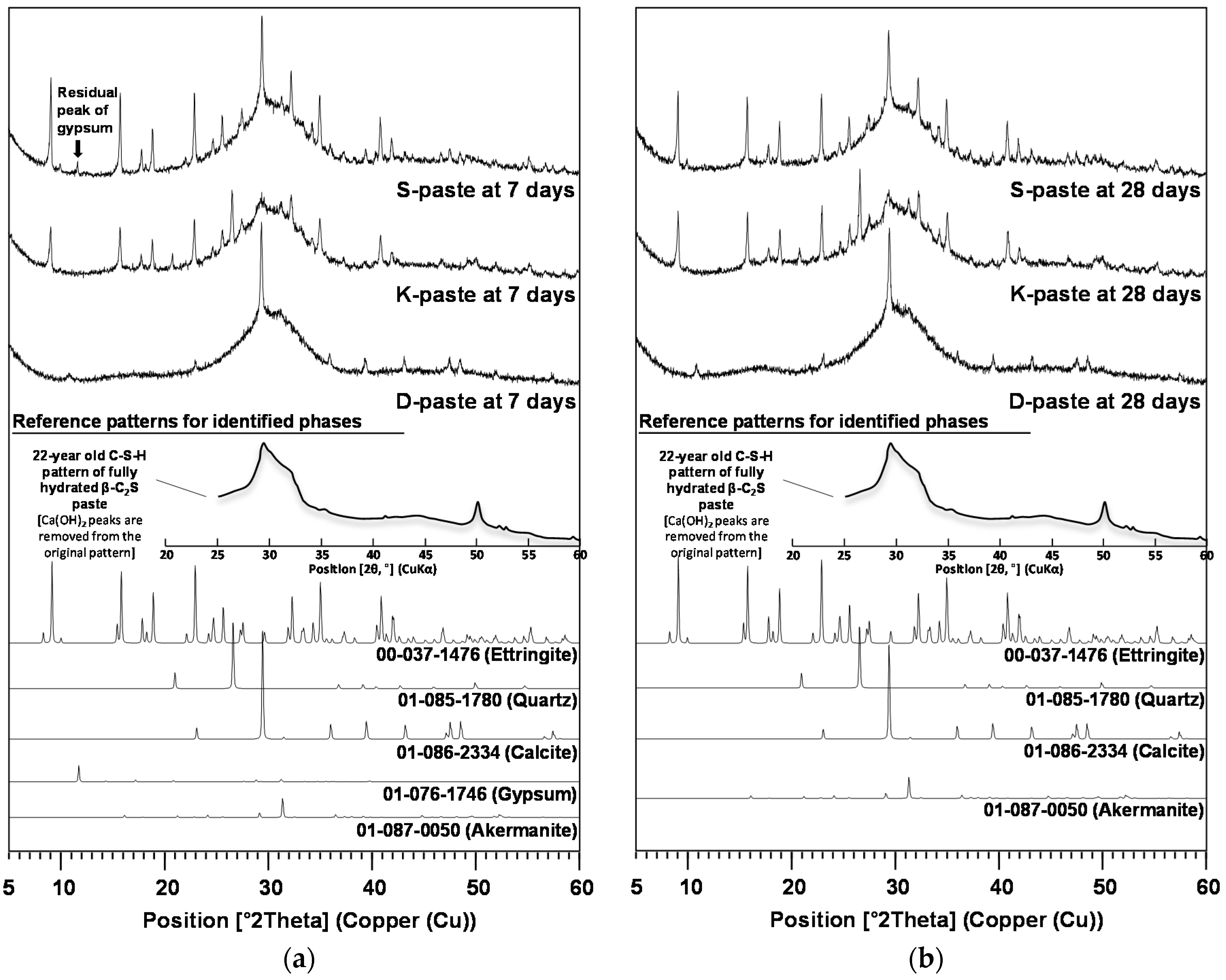
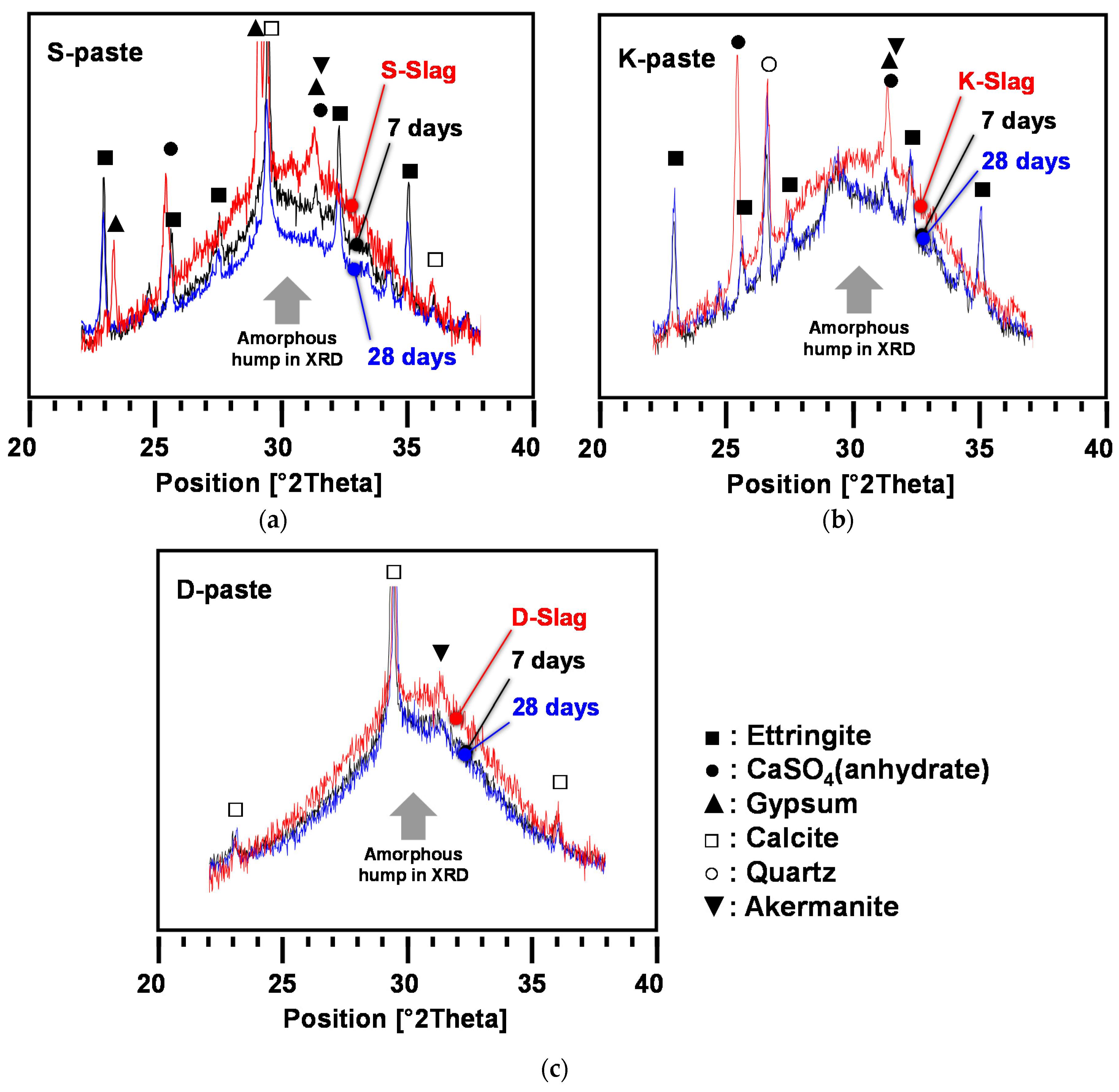
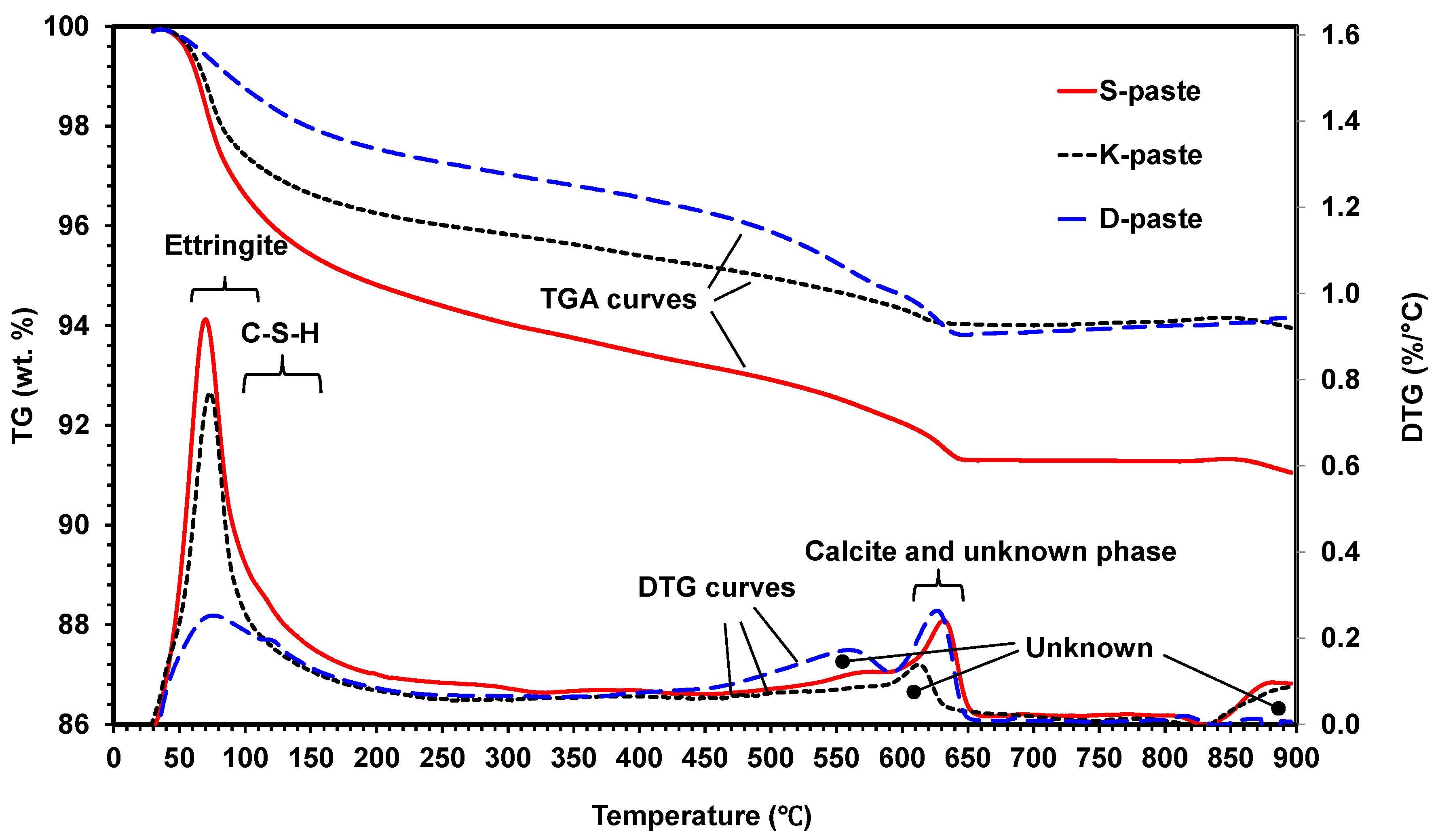
- All paste samples produced C-S-H, but only the S- and K-pastes formed ettringite as a main-reaction product; the ettringite formation improved the early strength of the S- and K-pastes.
- (5)
- The S-paste formed the largest amount of C-S-H and ettringite, which could be one of the possible causes for the best strength of the S-paste.
- (6)
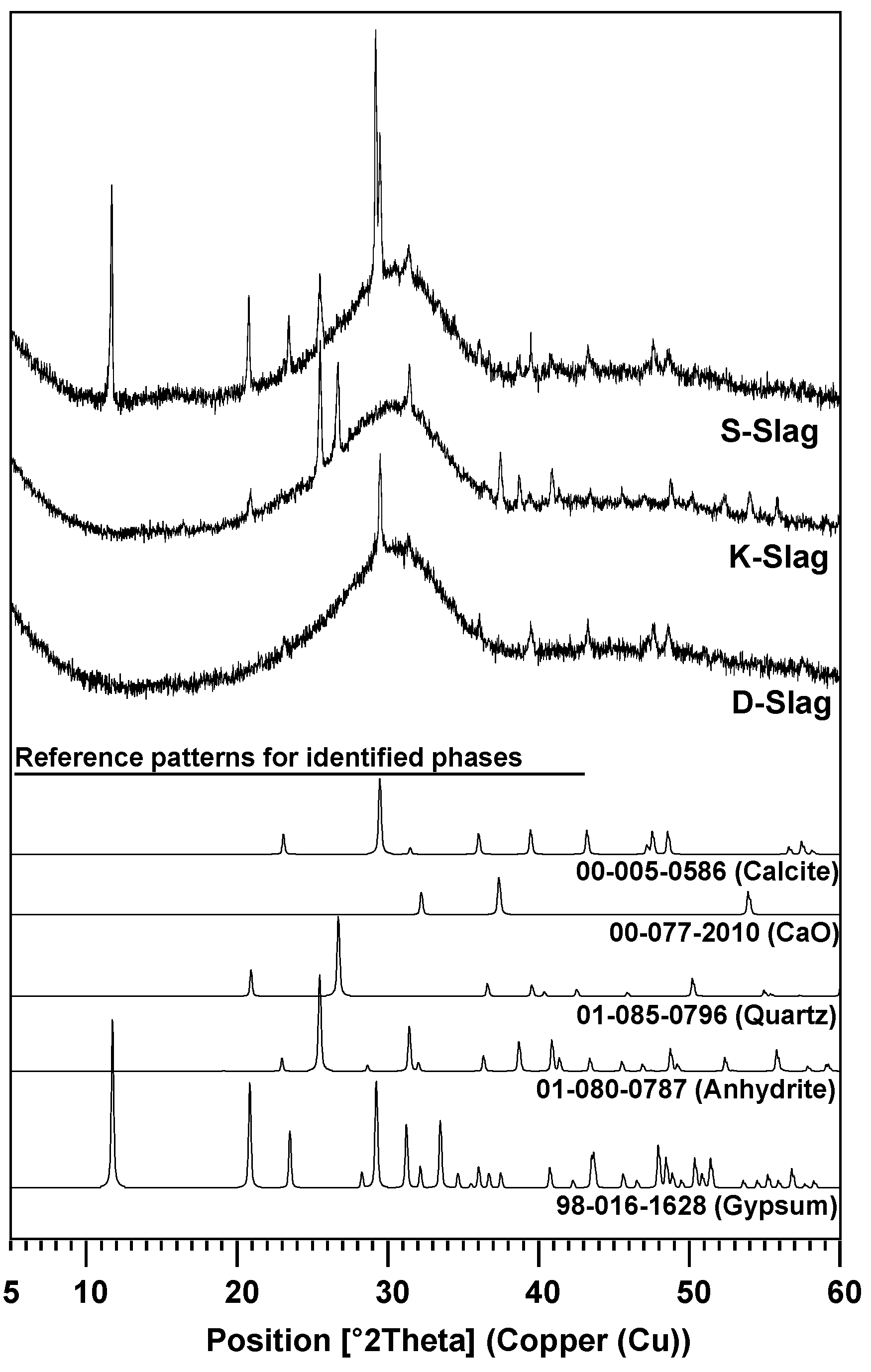
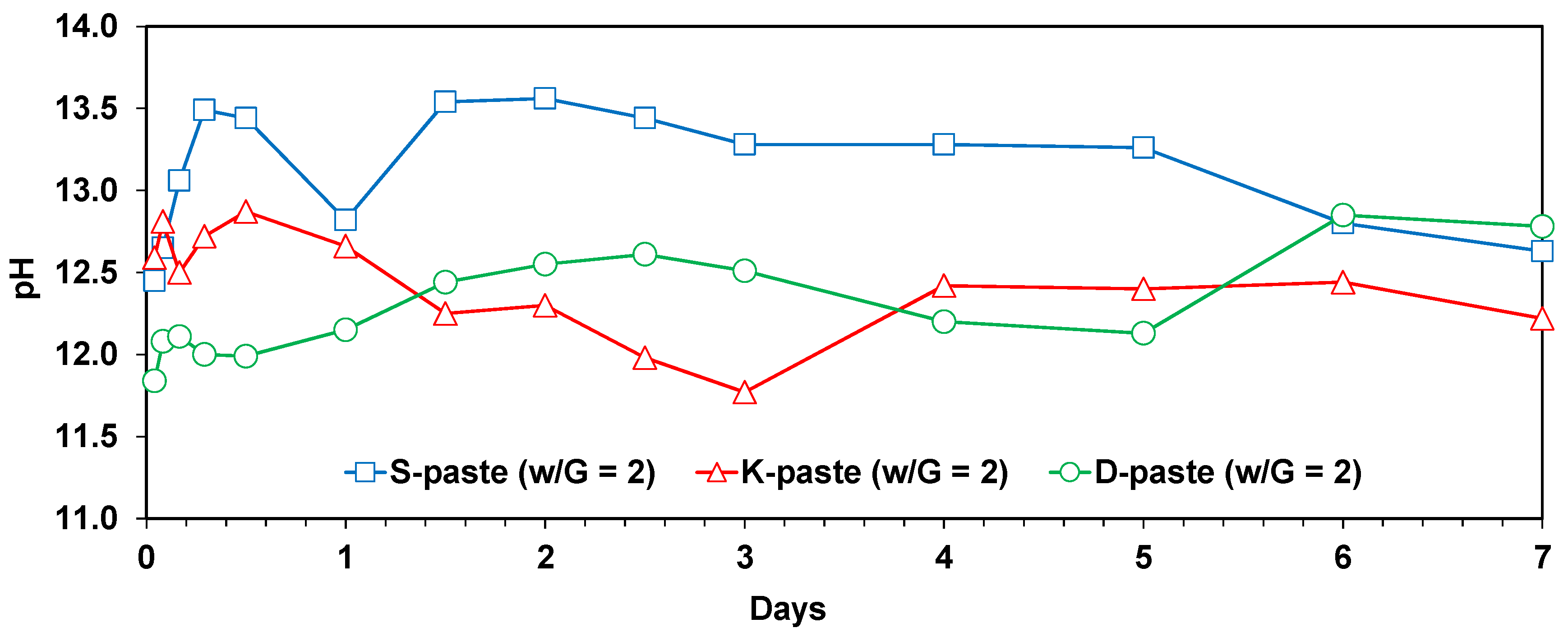
- The degree of dissolution of GGBFS with purified water was indirectly estimated through the monitoring of the pH of diluted GGBFS paste and the gradual reduction of amorphous humps in the XRD analysis, which were increased more by the smaller particle size, the higher alkali content, and the higher calcium-sulfate content of the GGBFS than by the other material parameters, such as a high glass content; in particular, the calcium-sulfate content appeared to be the most responsible for the dissolution.
- (7)
- The highest degree of dissolution of GGBFS was found in the S-paste, which also showed the extended dissolution of the glass phase after seven days, unlike the other pastes. These also explain the best strength of the S-paste.
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Slag Cement in Concrete and Mortar; ACI report 233R-03; American Concrete Institute: Farmington Hills, MI, USA, 2003.
- Standard Terminology Relating to Concrete and Concrete Aggregate; ASTM C 125; ASTM: West Conshohocken, PA, USA, 2011.
- Taylor, H.F.W. Cement Chemistry; Thomas Telford: London, UK, 1997. [Google Scholar]
- Song, S.; Jennings, H.M. Pore solution chemistry of alkali-activated ground granulated blast-furnace slag1. Cem. Concr. Res. 1999, 29, 159–170. [Google Scholar] [CrossRef]
- Bellmann, F.; Stark, J. Activation of blast furnace slag by a new method. Cem. Concr. Res. 2009, 39, 644–650. [Google Scholar] [CrossRef]
- Chen, Q.-H.; Tagnit-Hamou, A.; Sarkar, S.L. Strength and Microstructural Properties of Water Glass Activated Slag; MRS Proceedings; Cambridge University Press: Cambridge, UK, 1991; p. 49. [Google Scholar]
- Kim, M.S.; Jun, Y.; Lee, C.; Oh, J.E. Use of CaO as an activator for producing a price-competitive non-cement structural binder using ground granulated blast furnace slag. Cem. Concr. Res. 2013, 54, 208–214. [Google Scholar] [CrossRef]
- Jeong, Y.; Oh, J.E.; Jun, Y.; Park, J.; Ha, J.-H.; Sohn, S.G. Influence of four additional activators on hydrated-lime [Ca(OH)2] activated ground granulated blast-furnace slag. Cem. Concr. Comp. 2016, 65, 1–10. [Google Scholar] [CrossRef]
- Karim, M.R.; Hossain, M.M.; Khan, M.N.N.; Zain, M.F.M.; Jamil, M.; Lai, F.C. On the utilization of pozzolanic wastes as an alternative resource of cement. Materials 2014, 7, 7809–7827. [Google Scholar] [CrossRef]
- Matschei, T.; Bellmann, F.; Stark, J. Hydration behaviour of sulphate-activated slag cements. Adv. Cem. Res. 2005, 17, 167–178. [Google Scholar] [CrossRef]
- Standard Practice for Mechanical Mixing of Hydraulic Cement Pastes and Mortars of Plastic Consistency; ASTM C 305–11; American Society for Testing and Materials: West Conshohocken, PA, USA, 2011.
- Madsen, I.C.; Scarlett, N.V.Y. Quantitative Phase Analysis, Powder Diffraction: Theory and Practice; Royal Society of Chemistry: Cambridge, UK, 2008; pp. 298–331. [Google Scholar]
- PANalytical; X–Pert HighScore Plus; PANalytical: Almelo, The Netherlands, 2012.
- PDF-2; Database Sets; International Centre for Diffraction Data: Newtown Square, PA, USA, 2011.
- Allmann, R.; Hinek, R. The introduction of structure types into the Inorganic Crystal Structure Database ICSD. Acta Crystallogr. Sect. A Found. Crystallogr. 2007, 63, 412–417. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, C.R.; Snyder, R.L. RIR—Measurement and use in quantitative XRD. Powder Diffr. 1988, 3, 74–77. [Google Scholar] [CrossRef]
- Shi, C.; Roy, D.; Krivenko, P. Alkali-activated Cements and Concretes; CRC Press: New York, NY, USA, 2006. [Google Scholar]
- Ground Granulated Blast-Furnace Slag for Use in Concrete; Korean Standard Association: Seoul, Korea, 2009; Volume 2563.
- Specification for Ground Granulated Blastfurnace slag for Use with Portland Cement; The British Standards Institution: London, UK, 1992; Volume 6699.
- Wan, H.; Shui, Z.; Lin, Z. Analysis of geometric characteristics of GGBS particles and their influences on cement properties. Cem. Concr. Res. 2004, 34, 133–137. [Google Scholar] [CrossRef]
- Wang, P.Z.; Trettin, R.; Rudert, V. Effect of fineness and particle size distribution of granulated blast-furnace slag on the hydraulic reactivity in cement systems. Adv. Cem. Res. 2005, 17, 161–167. [Google Scholar] [CrossRef]
- Yang, K.-H.; Cho, A.-R.; Song, J.-K.; Nam, S.-H. Hydration products and strength development of calcium hydroxide-based alkali-activated slag mortars. Constr. Build. Mater. 2012, 29, 410–419. [Google Scholar] [CrossRef]
- Ben Haha, M.; Le Saout, G.; Winnefeld, F.; Lothenbach, B. Influence of activator type on hydration kinetics, hydrate assemblage and microstructural development of alkali activated blast-furnace slags. Cem. Concr. Res. 2011, 41, 301–310. [Google Scholar] [CrossRef]
- Mohan, K.; Taylor, H.F.W. Analytical electron microscopy of cement pastes: IV, β-dicalcium silicate pastes. J. Am. Ceram. Soc. 1981, 64, 717–719. [Google Scholar] [CrossRef]
- Bakolas, A.; Aggelakopoulou, E.; Moropoulou, A.; Anagnostopoulou, S. Evaluation of pozzolanic activity and physicomechanical characteristics in metakaolin-lime pastes. J. Therm. Anal. Calorim. 2006, 84, 157–163. [Google Scholar] [CrossRef]
- Hansen, W.; Pressler, E. Solubility of Ca(OH)2 and CaSO42H2O in dilute alkali solutions. Ind. Eng. Chem. 1947, 39, 1280–1282. [Google Scholar] [CrossRef]
- Zachariasen, W.H. The atomic arrangement in glass. J. Am. Chem. Soc. 1932, 54, 3841–3851. [Google Scholar] [CrossRef]
- Hashem, F.; Amin, M.; El-Gamal, S. Improvement of acid resistance of Portland cement pastes using rice husk ash and cement kiln dust as additives. J. Therm. Anal. Calorim. 2013, 111, 1391–1398. [Google Scholar] [CrossRef]
- Sha, W.; Pereira, G. Differential scanning calorimetry study of hydrated ground granulated blast-furnace slag. Cem. Concr. Res. 2001, 31, 327–329. [Google Scholar] [CrossRef]
- Sha, W. Differential scanning calorimetry study of the hydration products in Portland cement pastes with metakaolin replacement. In Proceedings of the International Conference on Advances in Building Technology, Hong Kong, China, 4–6 December 2002; pp. 881–888.
- Ramachandran, V.S. Applications of Differential Thermal Analysis in Cement Chemistry; Chemical Publishing Company, Inc.: New York, NY, USA, 1969. [Google Scholar]
- Yu, Q.; Brouwers, H. Gypsum: An investigation of microstructure and mechanical properties. In Proceedings 8th fib International PhD Symposium in Civil Engineering, Lyngby, Denmark, 20–23 June 2010; pp. 20–23.
- Vassileva, C.G.; Vassilev, S.V. Behaviour of inorganic matter during heating of Bulgarian coals: 1. Lignites. Fuel Process. Technol. 2005, 86, 1297–1333. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Raw GGBFS | CaSO4 (Anhydrite) | CaSO4·2H2O (Gypsum) | CaO | Calcite | Quartz | Glass Phase | Glass Content Without Anhydrite and Gypsum |
|---|---|---|---|---|---|---|---|
| S-Slag | 4.7 | 3.2 | - | 2.6 | - | 89.5 | 97.2 |
| K-Slag | 6.0 | - | 0.2 | - | 1.7 | 92.1 | 98.2 |
| D-Slag | - | - | - | 2.4 | - | 97.6 | 97.6 |
| GGBFS | Oxide Composition (wt. %) | Parameters | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CaO | SiO2 | Al2O3 | MgO | SO3 | TiO2 | K2O | Fe2O3 | MnO | Na2O | KSB 1 | CM 2 | Net-MD 3 | |
| S-Slag | 46.6 | 29.8 | 12.6 | 4.7 | 4.3 | 0.6 | 0.4 | 0.3 | 0.2 | 0.2 | 2.1 | 1.7 | 52.8 |
| K-Slag | 43.4 | 32.8 | 13.6 | 2.6 | 4.1 | 0.9 | 0.6 | 1.0 | 0.3 | 0.5 | 1.8 | 1.4 | 49.9 |
| D-Slag | 46.3 | 31.8 | 13.5 | 4.7 | 1.8 | 0.6 | 0.4 | 0.5 | 0.2 | 0.2 | 2.0 | 1.6 | 52.8 |
| Positive Parameters for Increasing Hydraulic Reactivity of GGBFS | S-Slag | K-Slag | D-Slag |
|---|---|---|---|
| Values of CM and KSB | ●●● | ●● | ●●● |
| Atomic % content of network modifying elements (Ca + Na + K + Mg) (Net-MD) in glass phase | ●●● | ●● | ●●● |
| Content of calcium sulfate source | ●●● | ●● | ● |
| Fraction of smaller particles | ●●● | ●● | ● |
| Content of alkalis (i.e., Na2O and K2O) | ●● | ●●● | ●● |
| Content of glass phase without external chemicals | ●● | ●●● | ●● |
| Phase | Temperature Range of Weight Loss |
|---|---|
| C-S-H | 105 °C [28] 120–145 °C [25] 106 ± 4 °C for first peak, 129 ± 4 °C for second peak [29] C-S-H loses its water over a broad temperature range, but the maximum loss occurs at 120–160 °C [30] 100–200 °C for C-S-H(I), 115–125 °C for C-S-H [3] |
| Ettringite | 183 ± 3 °C [29] 80–130 °C [30,31] |
| Gypsum | 80–220 °C (max. 167 °C) via transformation to anhydrite [32] 100–200 °C due to dehydration [33] |
| Calcite (CaCO3) | 720–760 °C [25] 680–760 °C [30] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, H.; Jeong, Y.; Jeong, J.-H.; Oh, J.E. Strength Development and Hydration Behavior of Self-Activation of Commercial Ground Granulated Blast-Furnace Slag Mixed with Purified Water. Materials 2016, 9, 185. https://doi.org/10.3390/ma9030185
Park H, Jeong Y, Jeong J-H, Oh JE. Strength Development and Hydration Behavior of Self-Activation of Commercial Ground Granulated Blast-Furnace Slag Mixed with Purified Water. Materials. 2016; 9(3):185. https://doi.org/10.3390/ma9030185
Chicago/Turabian StylePark, Hyeoneun, Yeonung Jeong, Jae-Hong Jeong, and Jae Eun Oh. 2016. "Strength Development and Hydration Behavior of Self-Activation of Commercial Ground Granulated Blast-Furnace Slag Mixed with Purified Water" Materials 9, no. 3: 185. https://doi.org/10.3390/ma9030185
APA StylePark, H., Jeong, Y., Jeong, J.-H., & Oh, J. E. (2016). Strength Development and Hydration Behavior of Self-Activation of Commercial Ground Granulated Blast-Furnace Slag Mixed with Purified Water. Materials, 9(3), 185. https://doi.org/10.3390/ma9030185