
Reactivation of a Retarded Suspension of Ground Granulated Blast-Furnace Slag


Abstract
:
1. Introduction
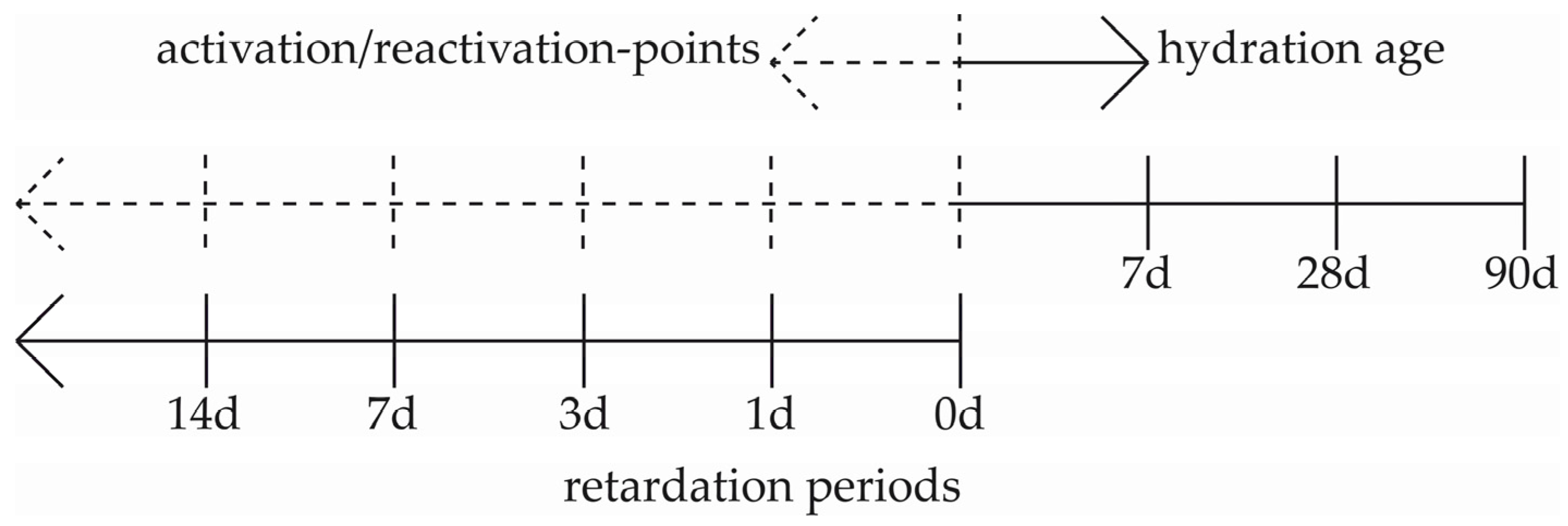
2. Materials and Methods
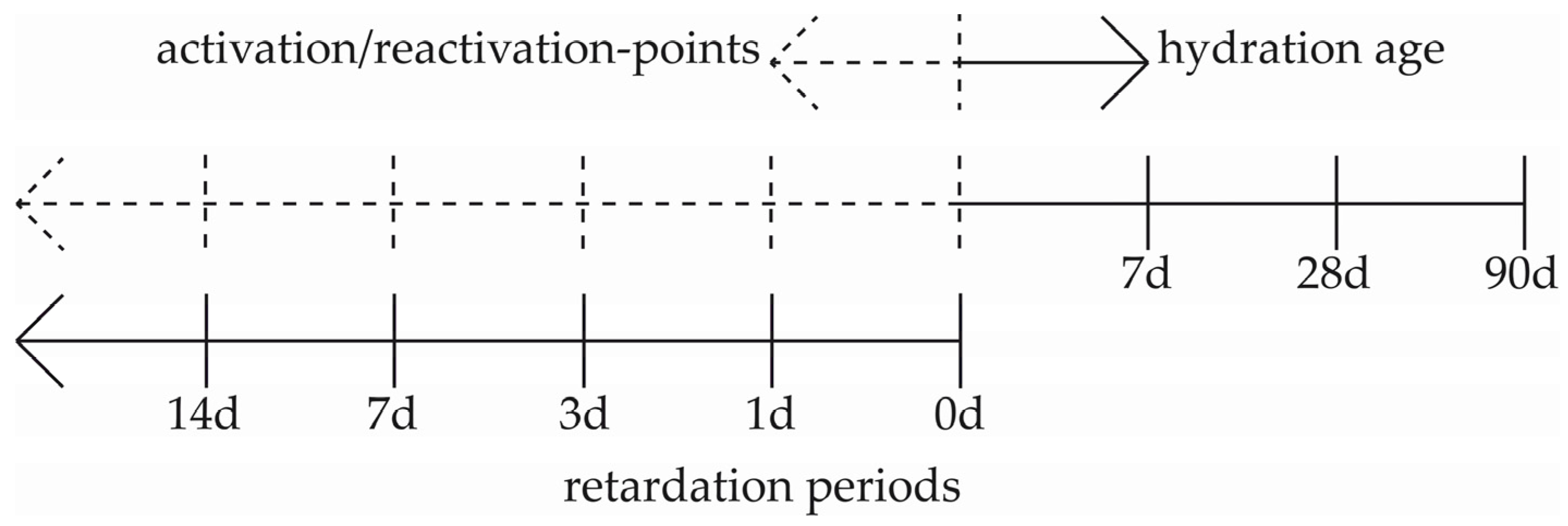
2.1. Slump Test and the Measurement of US-Velocity
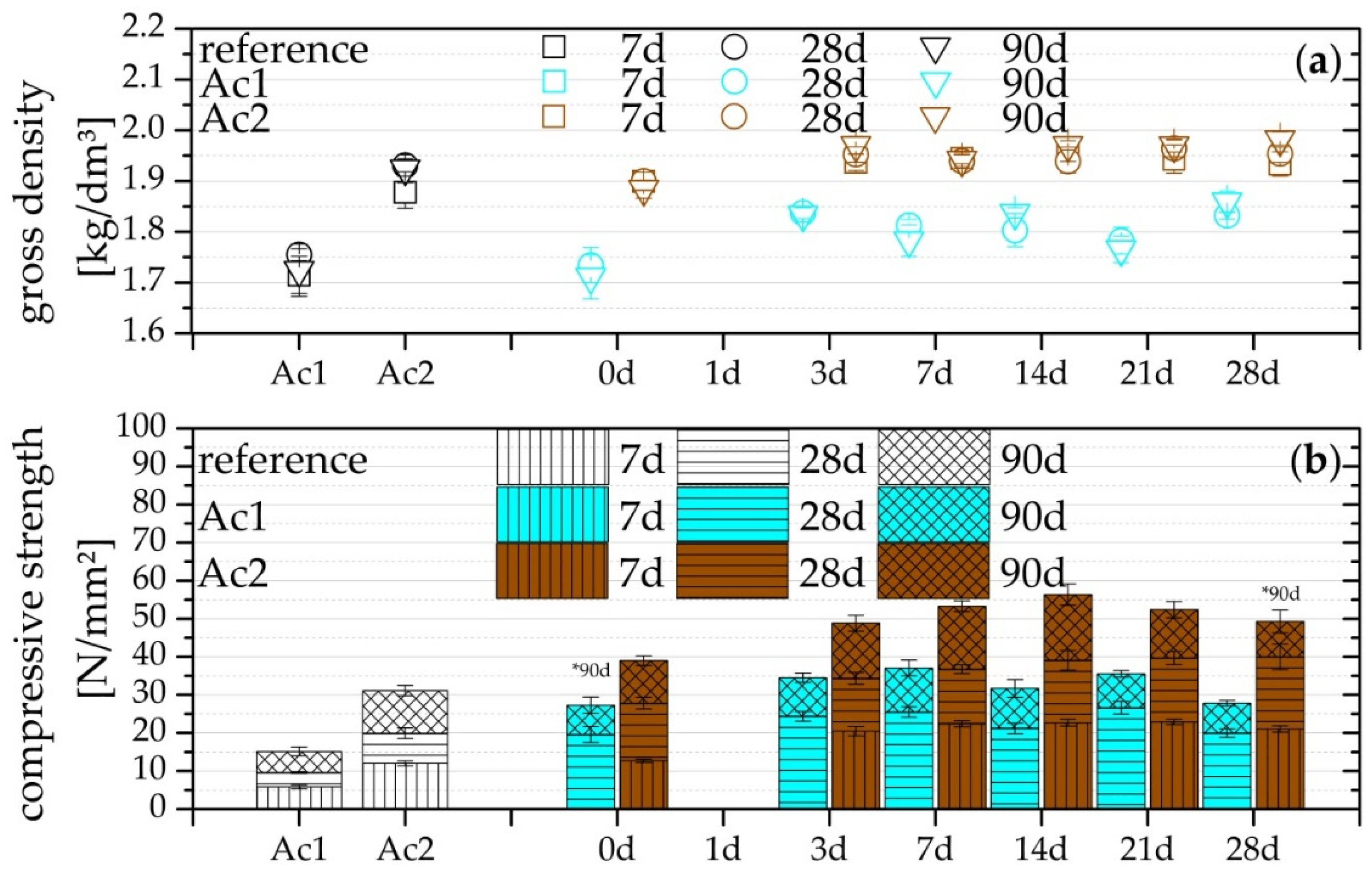
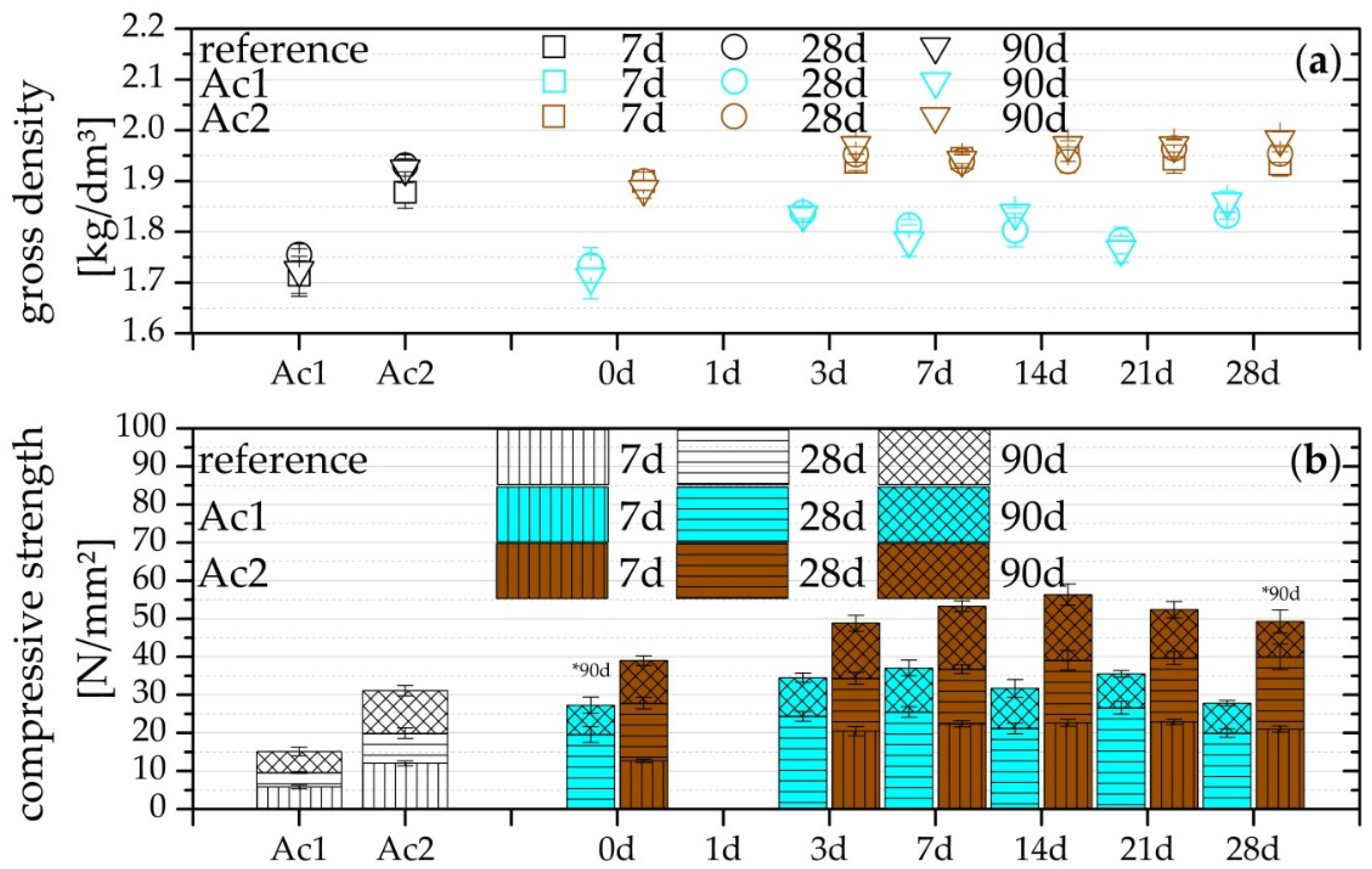
2.2. Compressive Strength and Gross Density
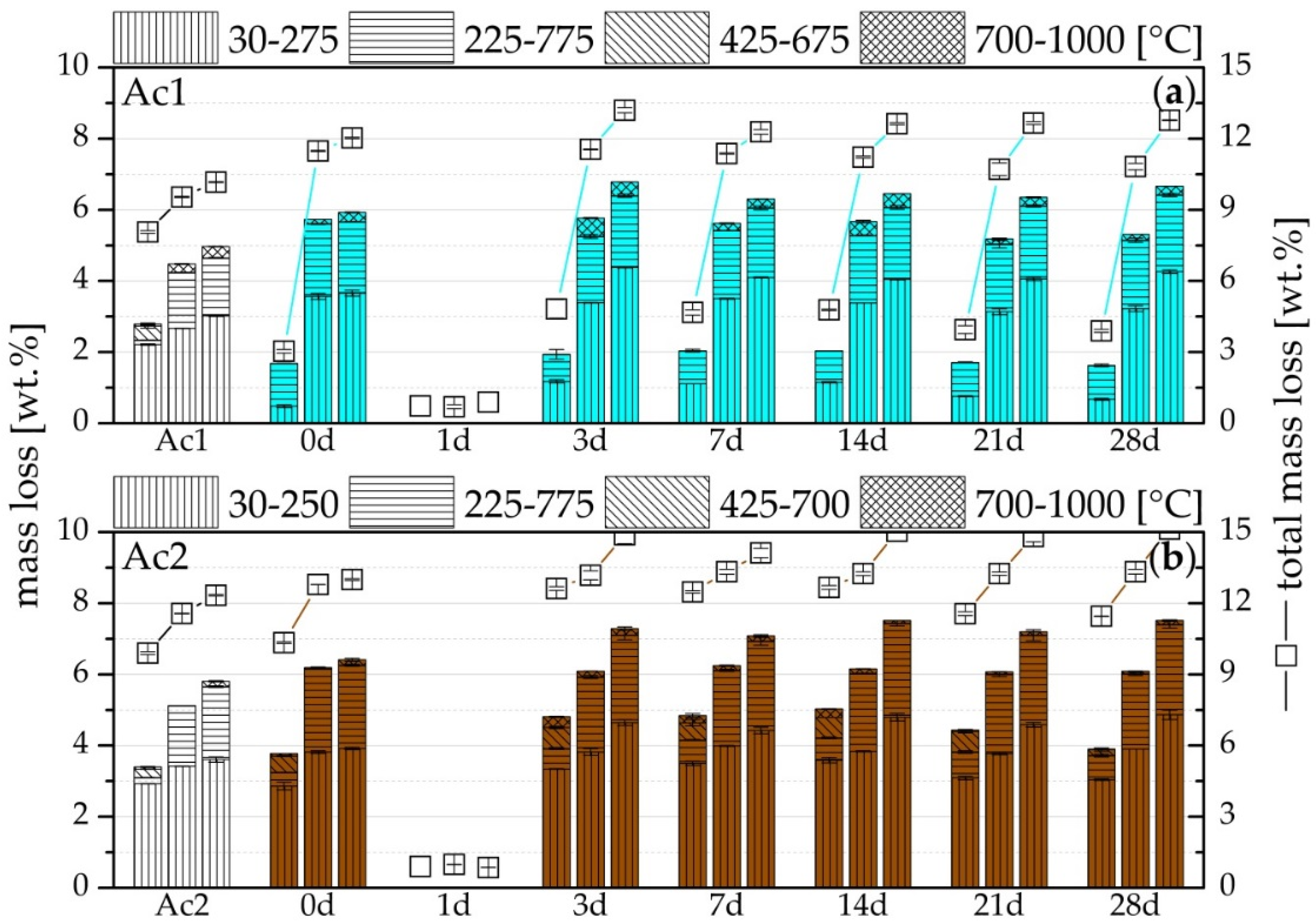
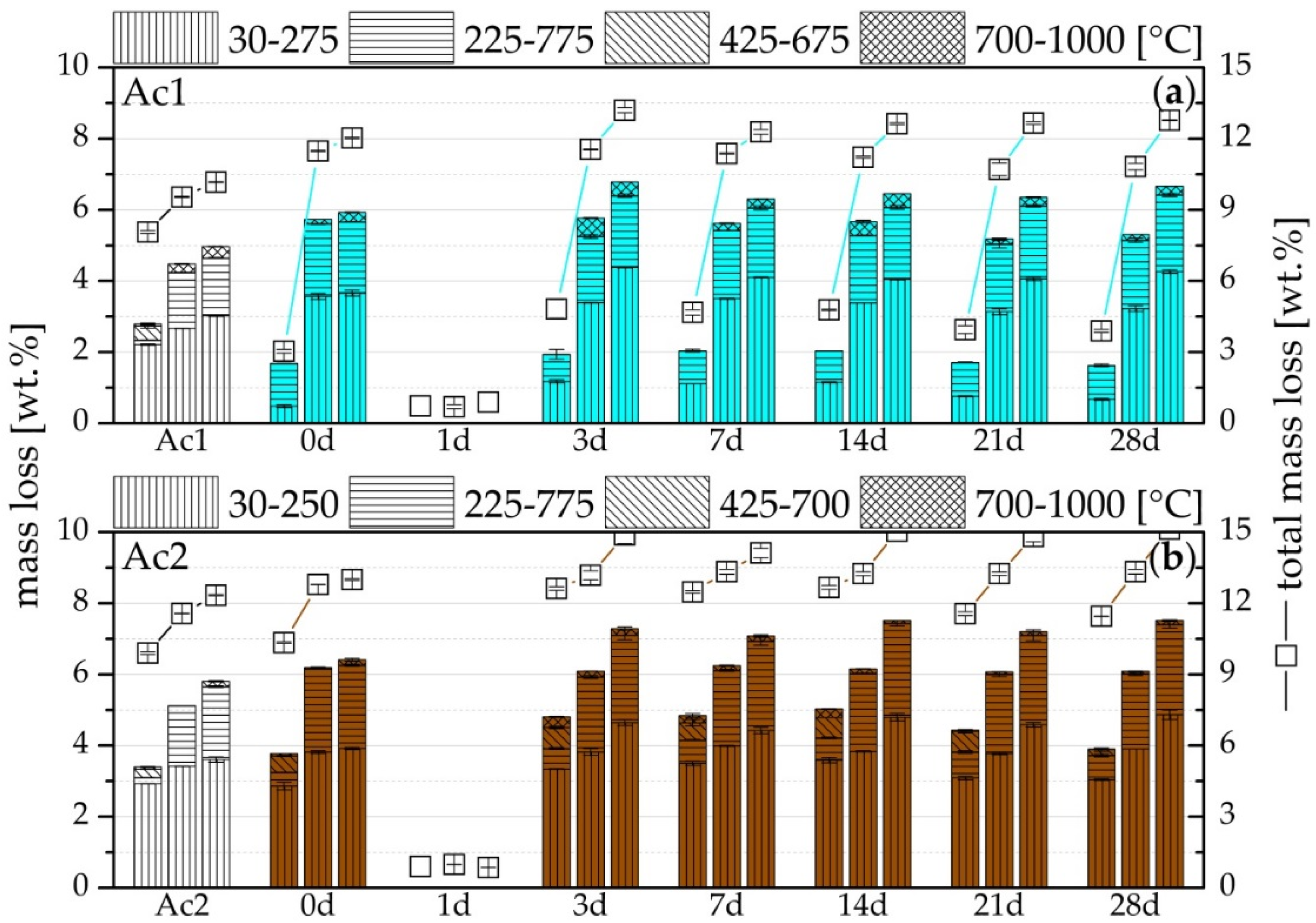
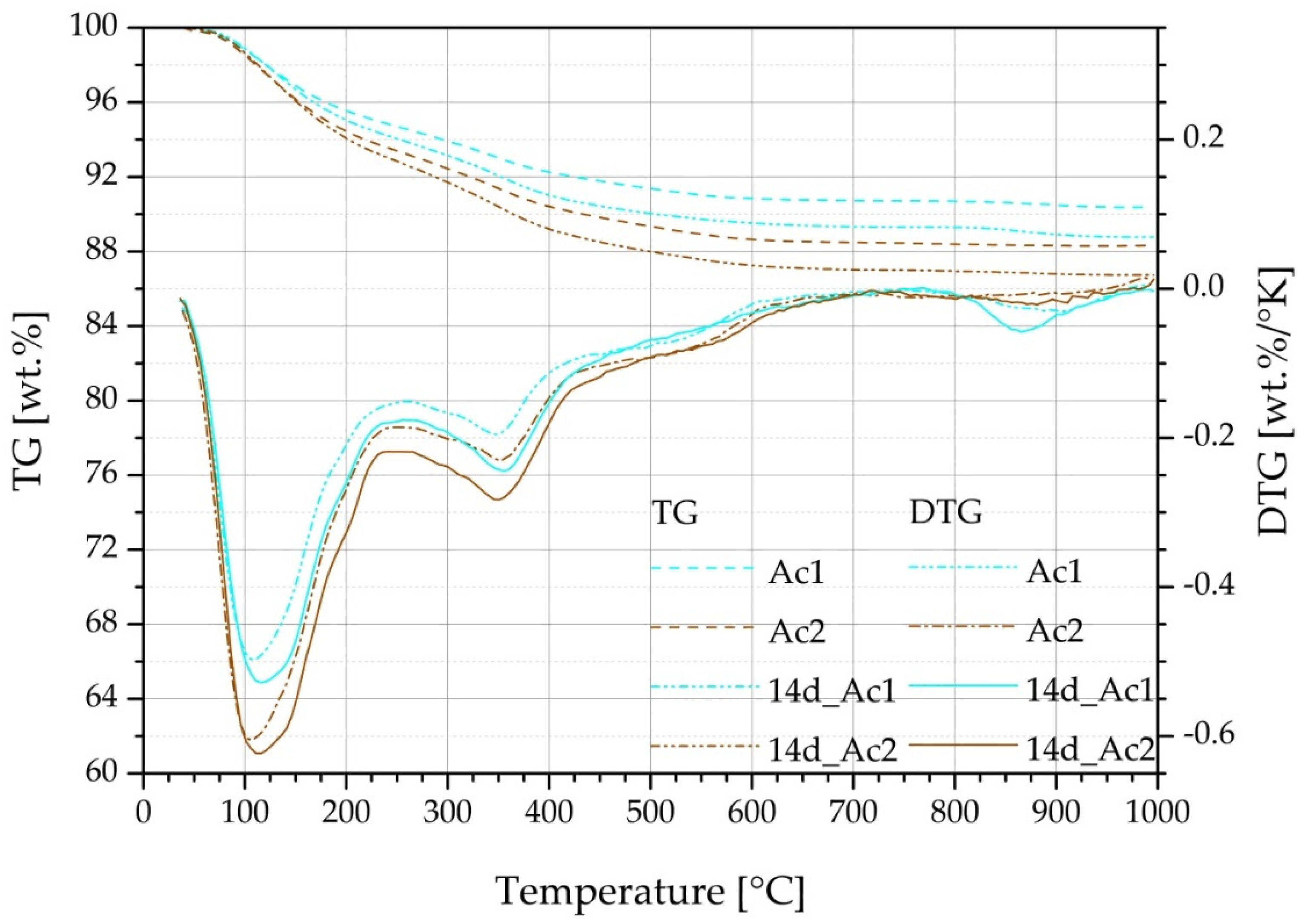
2.3. Thermogravimetry (TG) and SEM
3. Results and Discussion
3.1. Slump Test
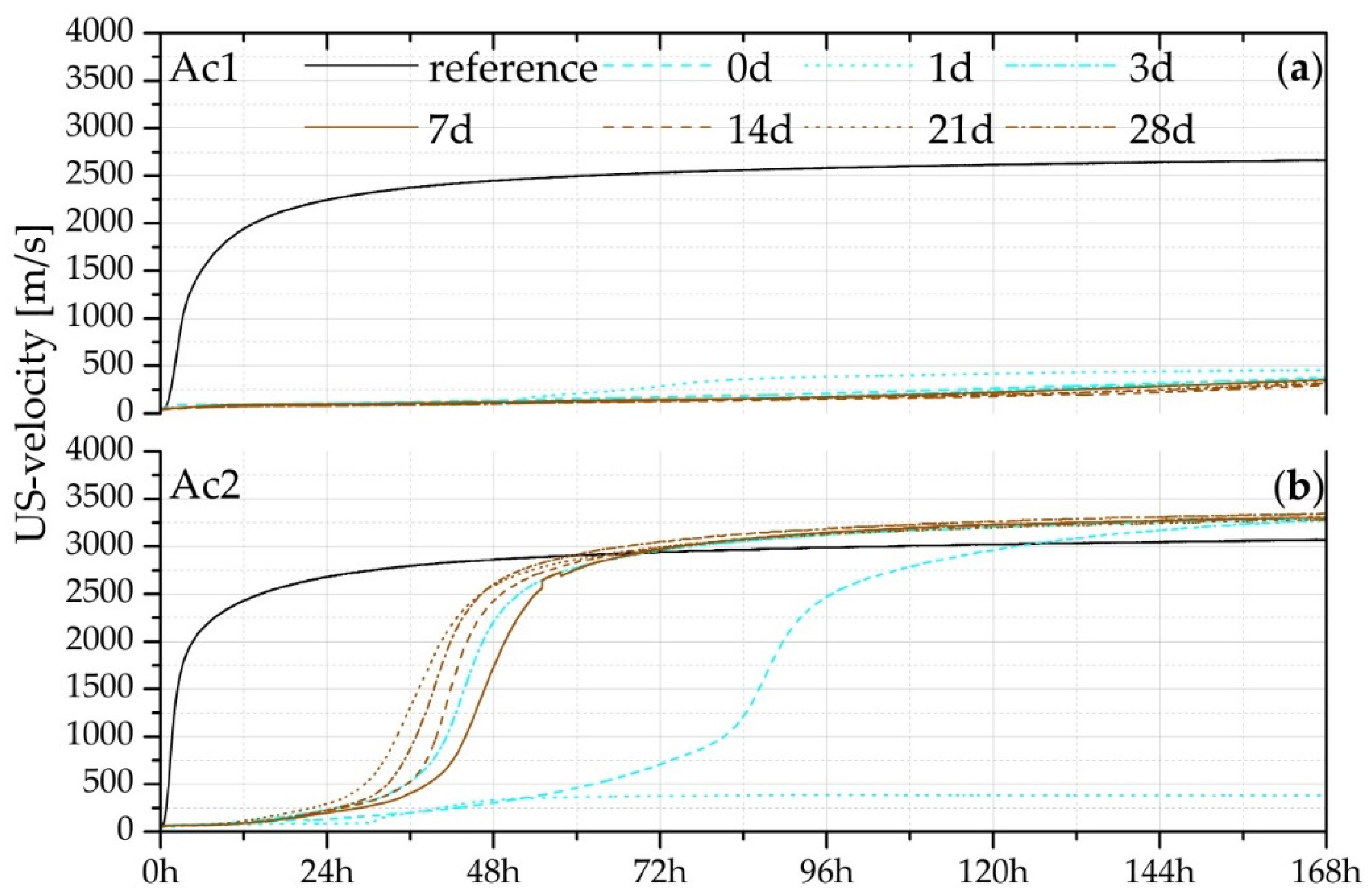
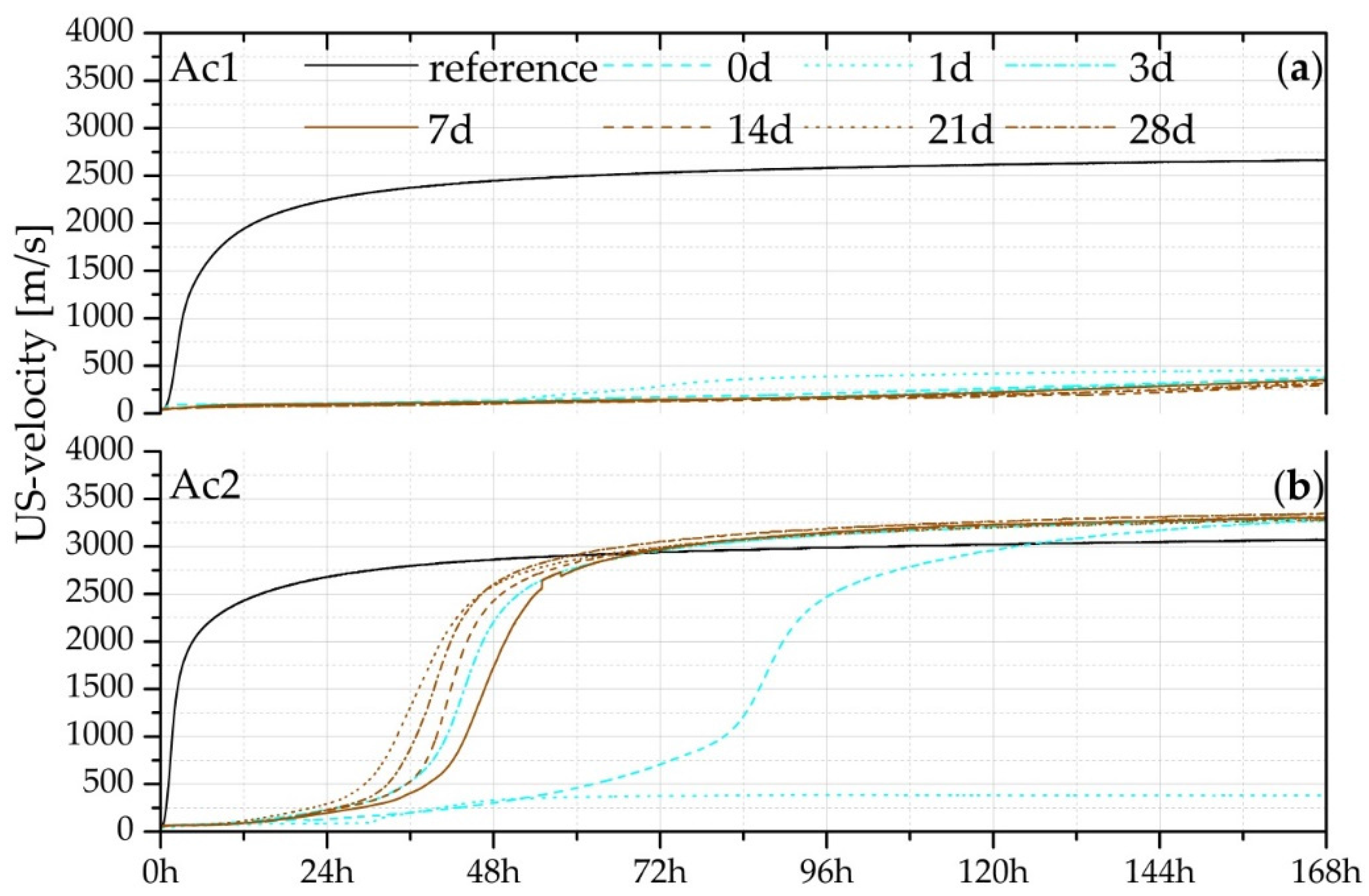
3.2. Measurement of US-Velocity
3.3. Compressive Strength and Gross Density
3.4. TG
3.5. SEM
4. Conclusions
- Setting within 48 h after the addition of the activator (from a retardation of three days)
- Compressive strength after seven days already exceeds the reference
- Formation of more hydration products than the reference, especially C-S-H-phases
- Formation of a denser structure
- The beginning of setting is extended, due to the overcoming of the retarded state.
- Higher activator concentration causes a higher absolute compressive strength (also correct for non-retarded systems) and a quicker setting.
- Activator concentration has no effect on the relative increase of the compressive strength from the 28 days of the hydration (also correct for non-retarded systems).
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tenoutasse, N.; Singh, N.B. Effect of glucose and calcium gluconate on the hydration of Portland cement. Indian J. Technol. 1978, 16, 184–189. [Google Scholar]
- Langenfeld, M.; Stark, J. The Influence of Retarding Admixtures on the Early Hydration of Portland Cement Phases, Shown in ESEM-FEG; Wissenschaftliche Zeitschrift der Bauhaus-Universität Weimar: Weimar, Germany, 1998; pp. 82–90. [Google Scholar]
- Stephan, D.; Plank, J. Einfluss von Verzögerern auf Alit und Zemente mit unterschiedlichem Gehalt an Klinkerphasen. In GDCh-Monographie; Gesellschaft Dt. Chemiker: Frankfurt am Main, Germany, 2003; pp. 31–38. [Google Scholar]
- Richartz, W. Einfluß von Zusätzen auf das Erstarrungsverhalten von Zement. Beton 1983, 33, 425–429, 465–471. [Google Scholar]
- Rickert, J. Influence of Retarders on the Hydration of Clinker and Cement; Bau und Technik: Düsseldorf, Germany, 2004. [Google Scholar]
- Pollard, S.J.T.; Mongomery, D.M.; Sollars, C.J.; Perry, R. Organic compounds in the cement-based stabilisation/solidification of hazardous mixed wastes—Mechanistic and process considerations. J. Hazard. Mater. 1991, 28, 313–327. [Google Scholar] [CrossRef]
- Pang, X.; Boontheung, P.; Boul, P.J. Dynamic retarder exchange as a trigger for Portland cement hydration. Cem. Concr. Res. 2014, 63, 20–28. [Google Scholar] [CrossRef]
- Thomas, N.L.; Birchall, J.D. The retarding action of sugars on cement hydration. Cem. Concr. Res. 1983, 13, 830–842. [Google Scholar] [CrossRef]
- Ludwig, U.; Urrutia, C. The mechanism of the action of saccharose on the setting and hardening of cements. ZKG Int. 1989, 42, 431–436. [Google Scholar]
- Ma, S.; Li, W.; Zhang, S.; Ge, D.; Yu, J.; Shen, X. Influence of sodium gluconate on the performance and hydration of Portland cement. Constr. Build. Mater. 2015, 91, 138–144. [Google Scholar] [CrossRef]
- Benedix, R. Bauchemie: Einführung in Die Chemie für Bauingenieure und Architekten, 5th ed.; Vieweg+Teubner Verlag/Springer Fachmedien Wiesbaden GmbH: Wiesbaden, Germany, 2011. [Google Scholar]
- Rixom, M.R.; Mailvaganam, N.P. Chemical Admixtures for Concrete, 3rd ed.; Routledge: New York, NY, USA, 1999. [Google Scholar]
- Tatarin, R.; Erfurt, W.; Stark, J. Continuous ultrasonic investigations during the hydration of cement paste, mortar and concrete. ZKG Int. 2004, 57, 69–78. [Google Scholar]
- Herb, A.T.; Grosse, C.U.; Reinhardt, H.-W. Ultrasonic testing device for mortar. Otto Graf J. 1999, 10, 144–155. [Google Scholar]
- Reinhardt, H.-W.; Große, C.U.; Herb, A.T. Ultrasonic monitoring of setting and hardening of cement mortar—A new device. Mater. Struct. 2000, 33, 580–583. [Google Scholar] [CrossRef]
- Grosse, C.U.; Reinhardt, H.-W. Fresh concrete monitored by ultrasound methods. Otto Graf J. 2001, 12, 157–168. [Google Scholar]
- De Belie, N.; Grosse, C.U.; Kurz, J.; Reinhardt, H.-W. Ultrasound monitoring of the influence of different accelerating admixtures and cement types for shotcrete on setting and hardening behaviour. Cem. Concr. Res. 2005, 35, 2087–2094. [Google Scholar] [CrossRef]
- Robeyst, N.; Gruyaert, E.; Grosse, C.U.; De Belie, N. Monitoring the setting of concrete containing blast-furnace slag by measuring the ultrasonic p-wave velocity. Cem. Concr. Res. 2008, 38, 1169–1176. [Google Scholar] [CrossRef]
- Reinhardt, H.W.; Grosse, C.U. Continuous monitoring of setting and hardening of mortar and concrete. Constr. Build. Mater. 2004, 18, 145–154. [Google Scholar] [CrossRef]
- Pal, S.; Mukherjee, A.; Pathak, S. Investigation of hydraulic activity of ground granulated blast furnace slag in concrete. Cem. Concr. Res. 2003, 33, 1481–1486. [Google Scholar] [CrossRef]
- Ehrenberg, A.; Israel, D.; Kühn, A.; Ludwig, H.-M.; Tigges, V.; Wassing, W. Granulated blastfurnace slag: Reaction potential and production of optimized cements, part 1. Cem. Int. 2008, 6, 90–96. [Google Scholar]
- Stark, J.; Wicht, B. Dauerhaftigkeit von Beton, 2nd ed.; Springer Vieweg: Heidelberg, Germany, 2013. [Google Scholar]
- Kühl, H. Zement-Chemie: Die Erhärtung und die Verarbeitung der hydraulischen Bindemittel; Verlag Technik: Berlin, Germany, 1961. [Google Scholar]
- Luke, G.; Luke, K. Effect of sucrose on retardation of Portland cement. Adv. Cem. Res. 2000, 12, 9–18. [Google Scholar] [CrossRef]
- Lieber, W.; Richartz, W. Effect of triethanolamine, sugar and boric acids on the setting and hardening of cements. ZKG Int. 1972, 25, 403–409. [Google Scholar]
- Han, M.-C.; Han, C.-G. Use of maturity methods to estimate the setting time of concrete containing super retarding agents. Cem. Concr. Compos. 2010, 32, 164–172. [Google Scholar] [CrossRef]
- Živica, V. Effects of type and dosage of alkaline activator and temperature on the properties of alkali-activated slag mixtures. Constr. Build. Mater. 2007, 21, 1463–1469. [Google Scholar] [CrossRef]
- Fernández-Jiménez, A.; Puertas, F.; Sobrados, I.; Sanz, J. Structure of calcium silicate hydrates formed in alkaline-activated slag: Influence of the type of alkaline activator. J. Am. Ceram. Soc. 2003, 86, 1389–1394. [Google Scholar] [CrossRef]
- Fernández-Jiménez, A.; Puertas, F. Effect of activator mix on the hydration and strength behaviour of alkali-activated slag cements. Adv. Cem. Res. 2003, 15, 129–136. [Google Scholar] [CrossRef]
- Pacheco-Torgal, F.; Castro-Gomes, J.; Jalali, S. Alkali-activated binders: A review. Part 1. Historical background, terminology, reaction mechanisms and hydration products. Constr. Build. Mater. 2008, 22, 1305–1314. [Google Scholar] [CrossRef]
- Pacheco-Torgal, F.; Castro-Gomes, J.; Jalali, S. Alkali-activated binders: A review. Part 2. About materials and binders manufacture. Constr. Build. Mater. 2008, 22, 1315–1322. [Google Scholar] [CrossRef]
- Fernández-Jiménez, A.; Palomo, J.G.; Puertas, F. Alkali-activated slag mortars mechanical strength behaviour. Cem. Concr. Res. 1999, 29, 1313–1321. [Google Scholar] [CrossRef]
- Ravikumar, D.; Peethamparan, S.; Neithalath, N. Structure and strength of NaOH activated concretes containing fly ash or GGBFS as the sole binder. Cem. Concr. Compos. 2010, 32, 399–410. [Google Scholar] [CrossRef]
- Tänzer, R.; Buchwald, A.; Stephan, D. Effect of slag chemistry on the hydration of alkali-activated blast-furnace slag. Mater. Struct. 2015, 48, 629–641. [Google Scholar] [CrossRef]
- Escalante-Garcia, J.I.; Fuentes, A.F.; Gorokhovsky, A.; Fraire-Luna, P.E.; Mendoza-Suarez, G. Hydration products and reactivity of blast-furnace slag activated by various alkalis. J. Am. Ceram. Soc. 2003, 86, 2148–2153. [Google Scholar] [CrossRef]
- Stephan, D.; Tänzer, R.; Braun, T.; Schmidt, M. Alkali activation—An alternative to binders that contain clinker; Part 1. Cem. Int. 2010, 8, 72–85. [Google Scholar]
- Stephan, D.; Tänzer, R.; Braun, T.; Schmidt, M. Alkali activation—An alternative to binders that contain clinker; Part 2. Cem. Int. 2010, 8, 74–81. [Google Scholar]
- Stark, J.; Möser, B.; Eckart, A. New approaches to cement hydration, Part 1. ZKG Int. 2001, 54, 52–60. [Google Scholar]
- Stark, J.; Möser, B.; Eckart, A. New approaches to cement hydration, Part 2. ZKG Int. 2001, 54, 114–119. [Google Scholar]
- Gruskovnjak, A.; Lothenbach, B.; Holzer, L.; Figi, R.; Winnefeld, F. Hydration of alkali-activated slag: Comparison with ordinary Portland cement. Adv. Cem. Res. 2006, 18, 119–128. [Google Scholar] [CrossRef]
- Burciaga-Díaz, O.; Escalante-García, J.I.; Rigaud, M. Structure, mechanisms of reaction, and strength of an alkali-activated blast-furnace slag. J. Am. Ceram. Soc. 2013, 96, 3939–3948. [Google Scholar] [CrossRef]
- Provis, J.L. Geopolymers and other alkali activated materials: Why, how, and what? Mater. Struct. 2014, 47, 11–25. [Google Scholar] [CrossRef]
- Ben Haha, M.; Le Saout, G.; Winnefeld, F.; Lothenbach, B. Influence of activator type on hydration kinetics, hydrate assemblage and microstructural development of alkali activated blast-furnace slags. Cem. Concr. Res. 2011, 41, 301–310. [Google Scholar] [CrossRef]
- Oh, J.E.; Monteiro, P.J.; Jun, S.S.; Choi, S.; Clark, S.M. The evolution of strength and crystalline phases for alkali-activated ground blast furnace slag and fly ash-based geopolymers. Cem. Concr. Res. 2010, 40, 189–196. [Google Scholar] [CrossRef]
- Brothers, L.E.; Pisklak, T.J. Set-Delayed Cement Compositions Comprising Pumice and Associated Methods. U.S. Patent 8,851,173, 9 March 2012. [Google Scholar]
- Ehrenberg, A.; Israel, D.; Kühn, A.; Ludwig, H.-M.; Tigges, V.; Wassing, W. Granulated blastfurnace slag: Reaction potential and production of optimized cements, part 2. Cem. Int. 2008, 6, 82–92. [Google Scholar]
- DIN Deutsches Institut für Normung e.V. Methods of Test for Mortar for Masonry—Part 3: Determination of Consistence of Fresh Mortar (by Flow Table); German Version EN 1015-3:1999+A1:2004+A2:2006; Beuth: Berlin, Germany, 2007. [Google Scholar]
- DIN Deutsches Institut für Normung e.V. Methods of Testing Cement—Part 1: Determination of Strength; German Version EN 196-1:2005; Beuth: Berlin, Germany, 2005. [Google Scholar]
- Walz, K.; Mathieu, H. Einfluß der Zusatzmenge von Betonverflüssigern auf die Festigkeitsentwicklung. Beton 1961, 11, 619–625, 691–696. [Google Scholar]
- Richartz, W. Über die Gefüge- und Festigkeitsentwicklung des Zementsteins. Beton 1969, 19, 203–206, 503–506. [Google Scholar]
- Roy, A.; Schilling, P.J.; Eaton, H.C.; Malone, P.G.; Brabston, W.N.; Wakeley, L.D. Activation of ground blast-furnace slag by alkali-metal and alkaline-earth hydroxides. J. Am. Ceram. Soc. 1992, 75, 3233–3240. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical Composition | GGBFS |
|---|---|
| SiO2 | 35.6 wt.% |
| Al2O3 | 10.6 wt.% |
| Fe2O3 | 0.7 wt.% |
| MgO | 7.4 wt.% |
| CaO | 43.2 wt.% |
| Na2O | 0.2 wt.% |
| K2O | 0.4 wt.% |
| TiO2 | 0.7 wt.% |
| MnO | 0.2 wt.% |
| Physical Properties | |
| Density | 2.92 kg/dm3 |
| Specific surface area (Blaine) | 3218 cm2/g |
| Water demand (Puntke) | 19.1 wt.% |
| RRSB-Distribution x0/n | 20.7/1.7 |
| Samples of US-Velocity | Reference | 0 Day | 3 Days | 7 Days | 14 Days | 21 Days | 28 Days |
|---|---|---|---|---|---|---|---|
| US-velocity at 168 h (m/s) | 3071 | 3278 | 3292 | 3299 | 3313 | 3279 | 3340 |
| 10% of max. at time (h) | 0.90 | 50.18 | 30.45 | 33.58 | 30.78 | 25.53 | 29.20 |
| 50% of max. at time (h) | 2.50 | 87.16 | 44.48 | 47.50 | 42.48 | 38.08 | 40.15 |
| 90% of max. at time (h) | 26.65 | 119.12 | 72.52 | 71.76 | 71.28 | 67.12 | 67.45 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schneider, N.; Stephan, D. Reactivation of a Retarded Suspension of Ground Granulated Blast-Furnace Slag. Materials 2016, 9, 174. https://doi.org/10.3390/ma9030174
Schneider N, Stephan D. Reactivation of a Retarded Suspension of Ground Granulated Blast-Furnace Slag. Materials. 2016; 9(3):174. https://doi.org/10.3390/ma9030174
Chicago/Turabian StyleSchneider, Nick, and Dietmar Stephan. 2016. "Reactivation of a Retarded Suspension of Ground Granulated Blast-Furnace Slag" Materials 9, no. 3: 174. https://doi.org/10.3390/ma9030174
APA StyleSchneider, N., & Stephan, D. (2016). Reactivation of a Retarded Suspension of Ground Granulated Blast-Furnace Slag. Materials, 9(3), 174. https://doi.org/10.3390/ma9030174