
Preparation of TiO2-Decorated Boron Particles by Wet Ball Milling and their Photoelectrochemical Hydrogen and Oxygen Evolution Reactions



Abstract
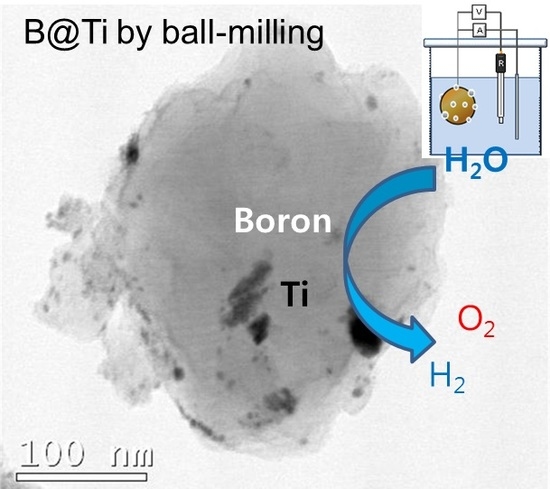
:
1. Introduction
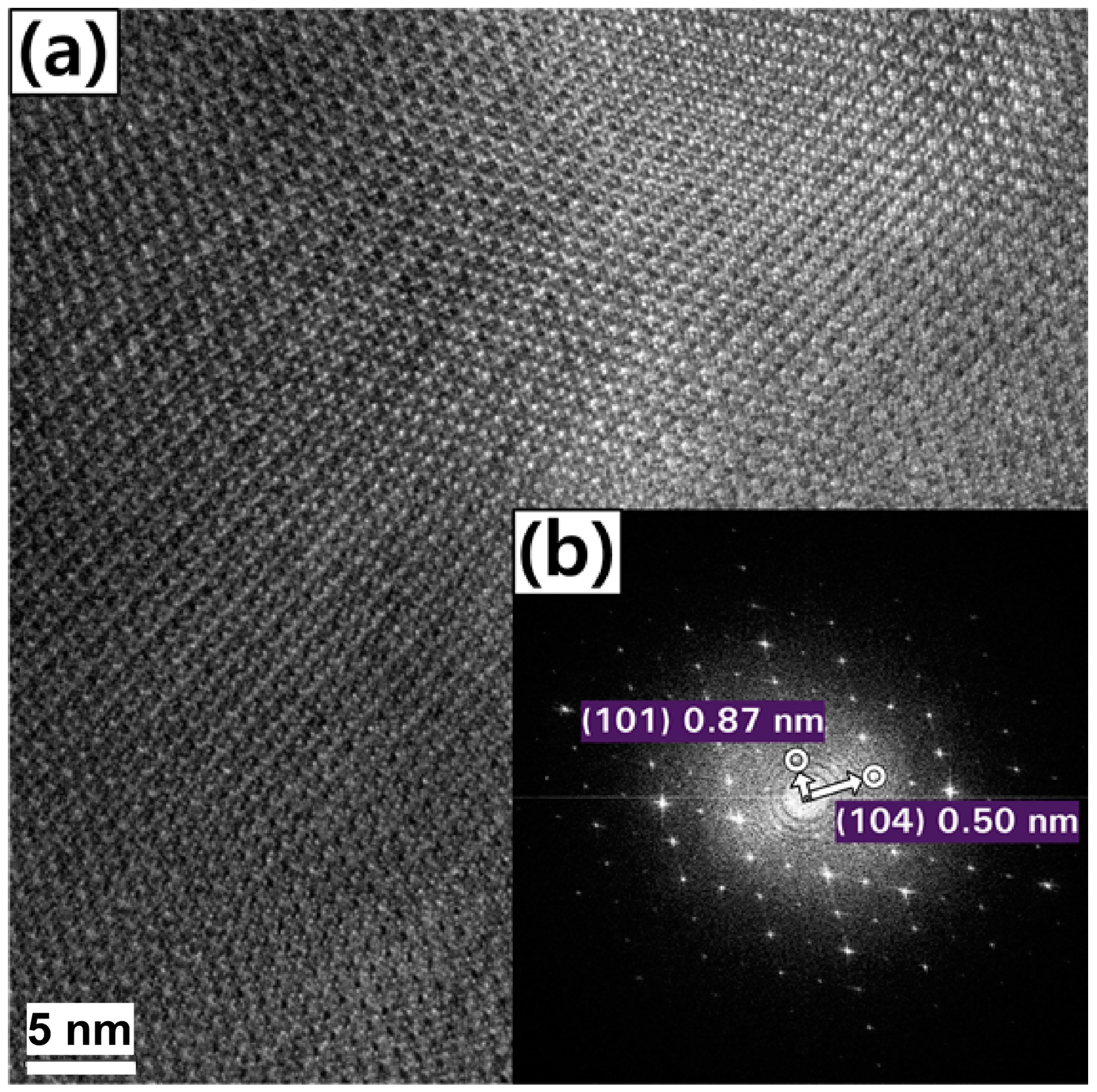
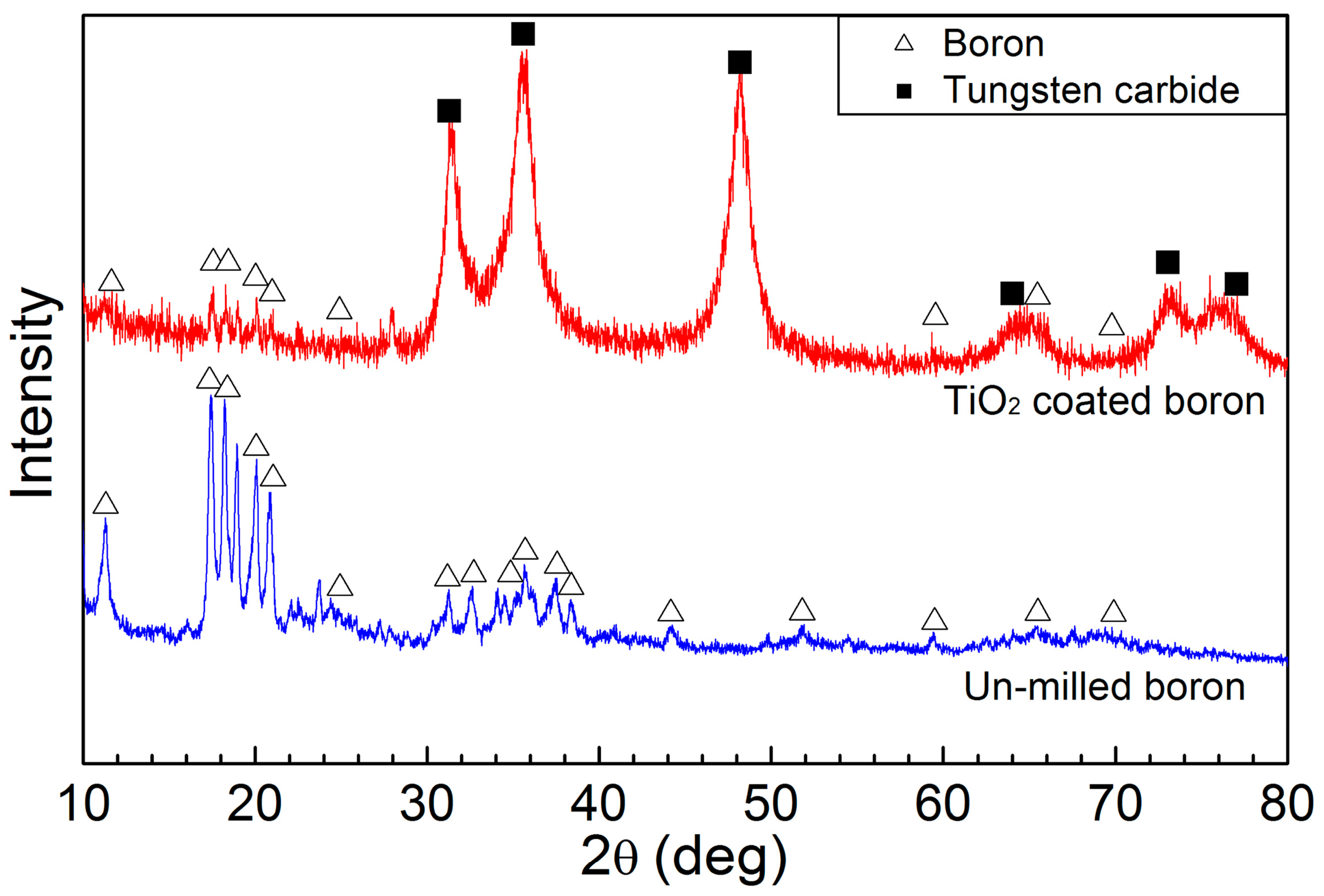
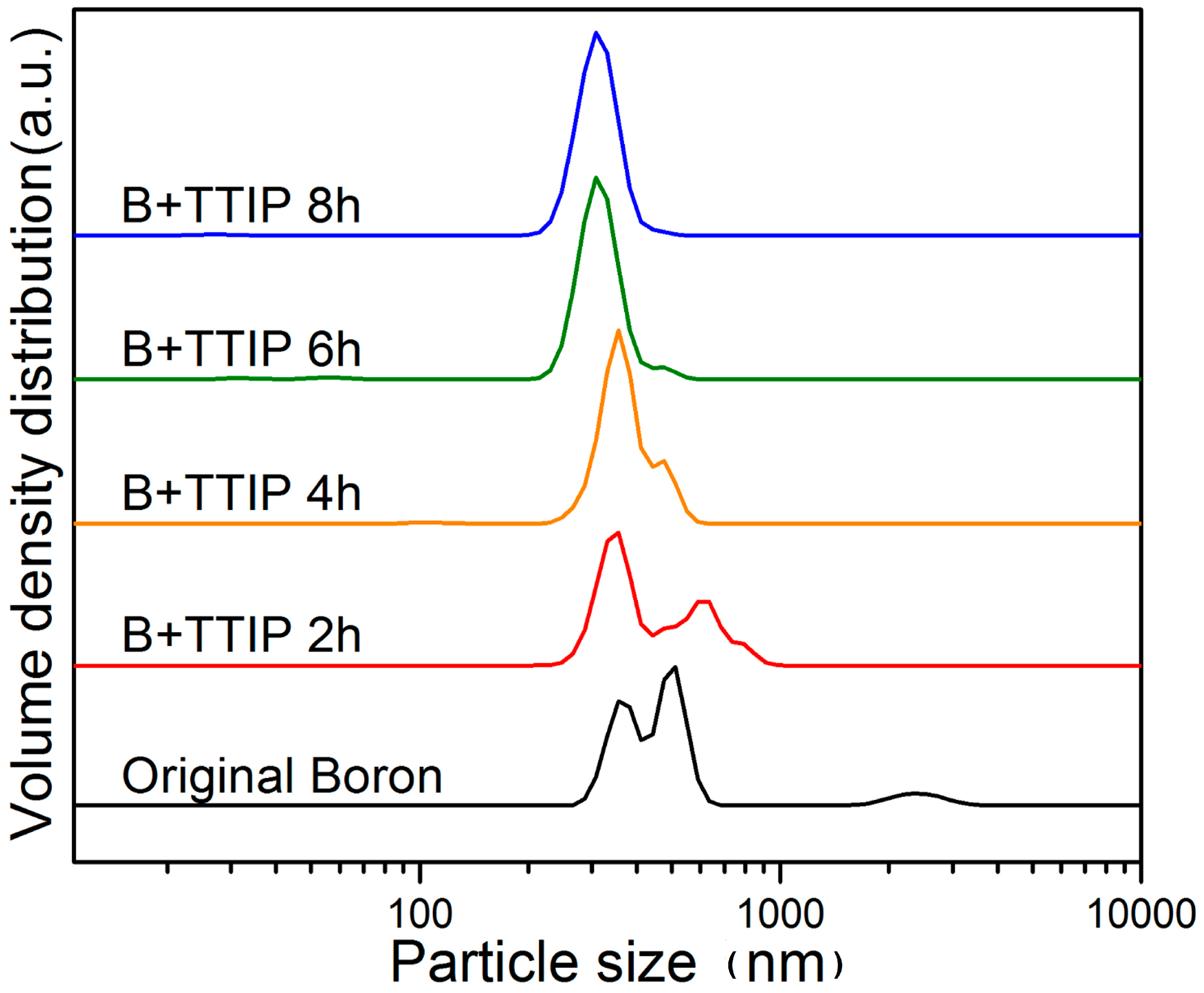
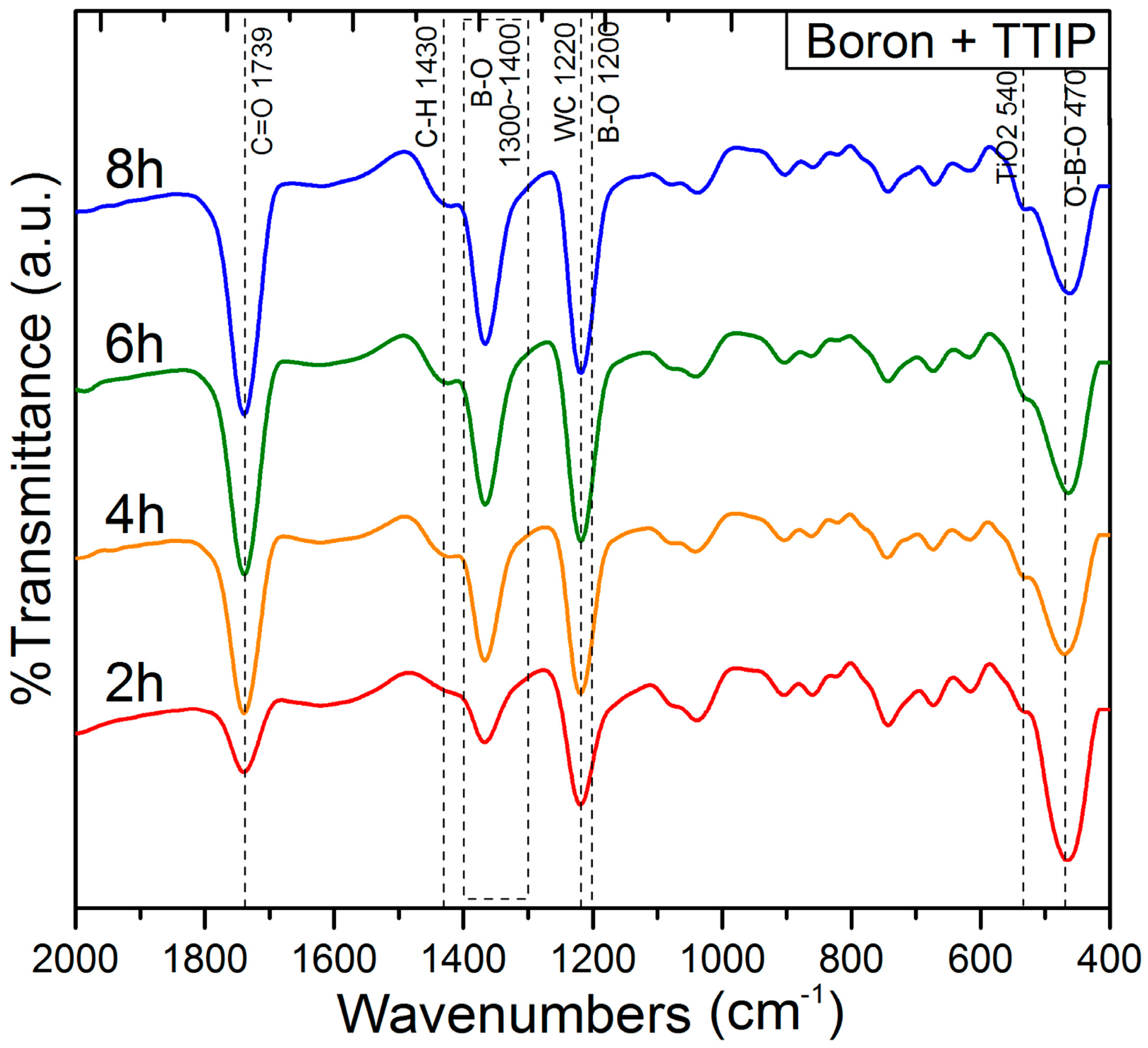
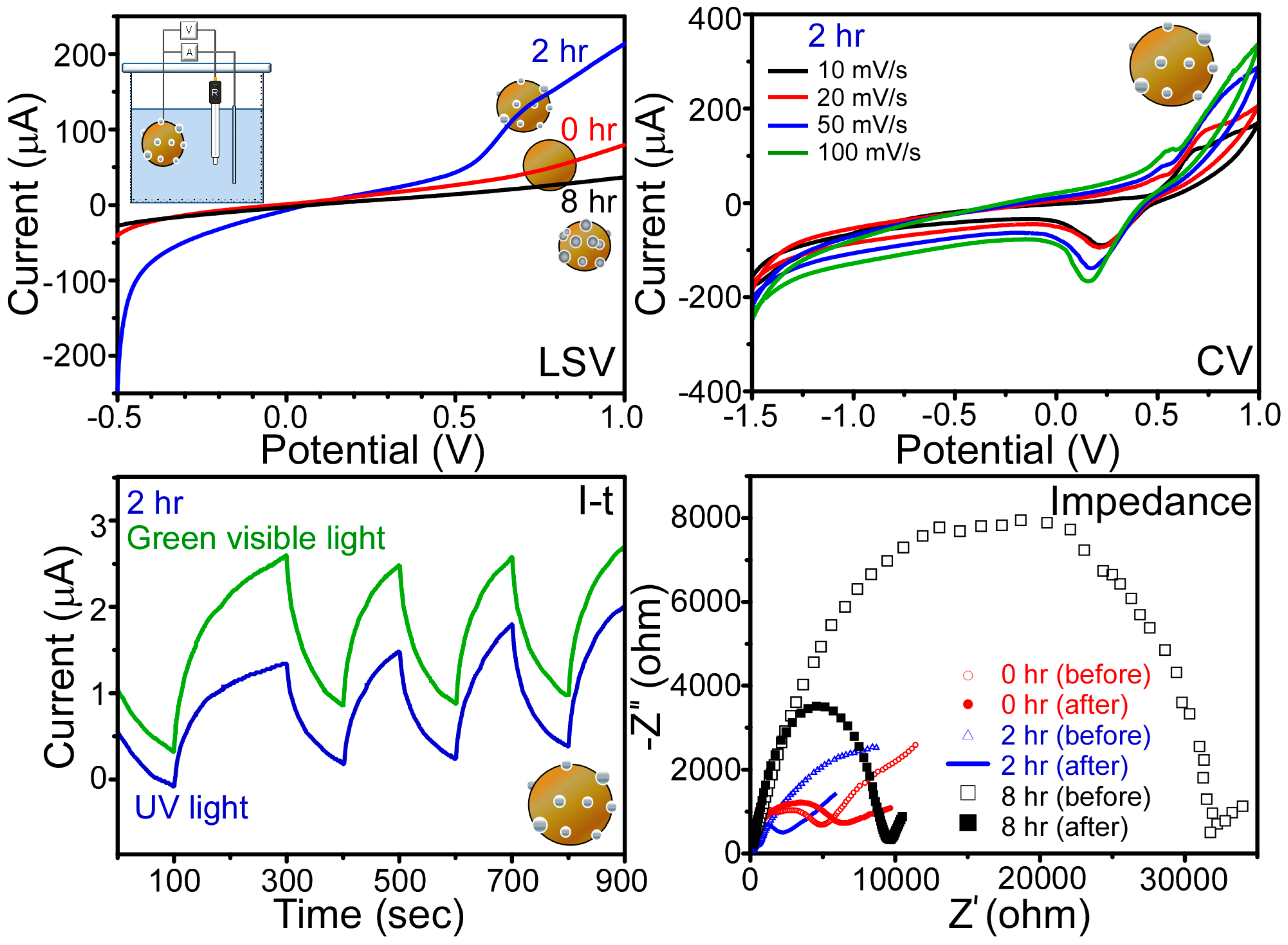
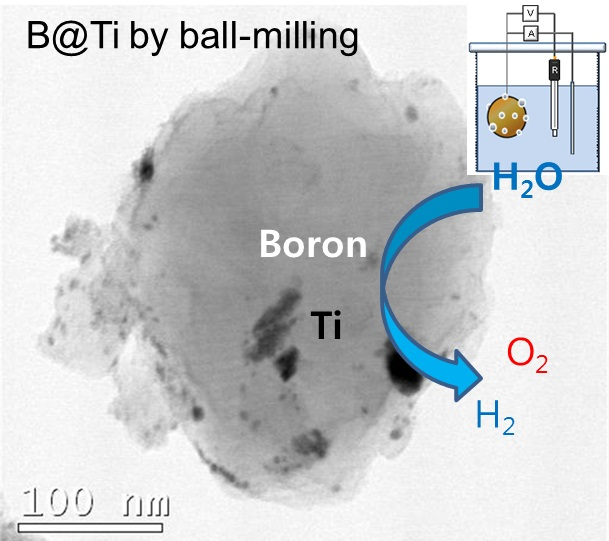
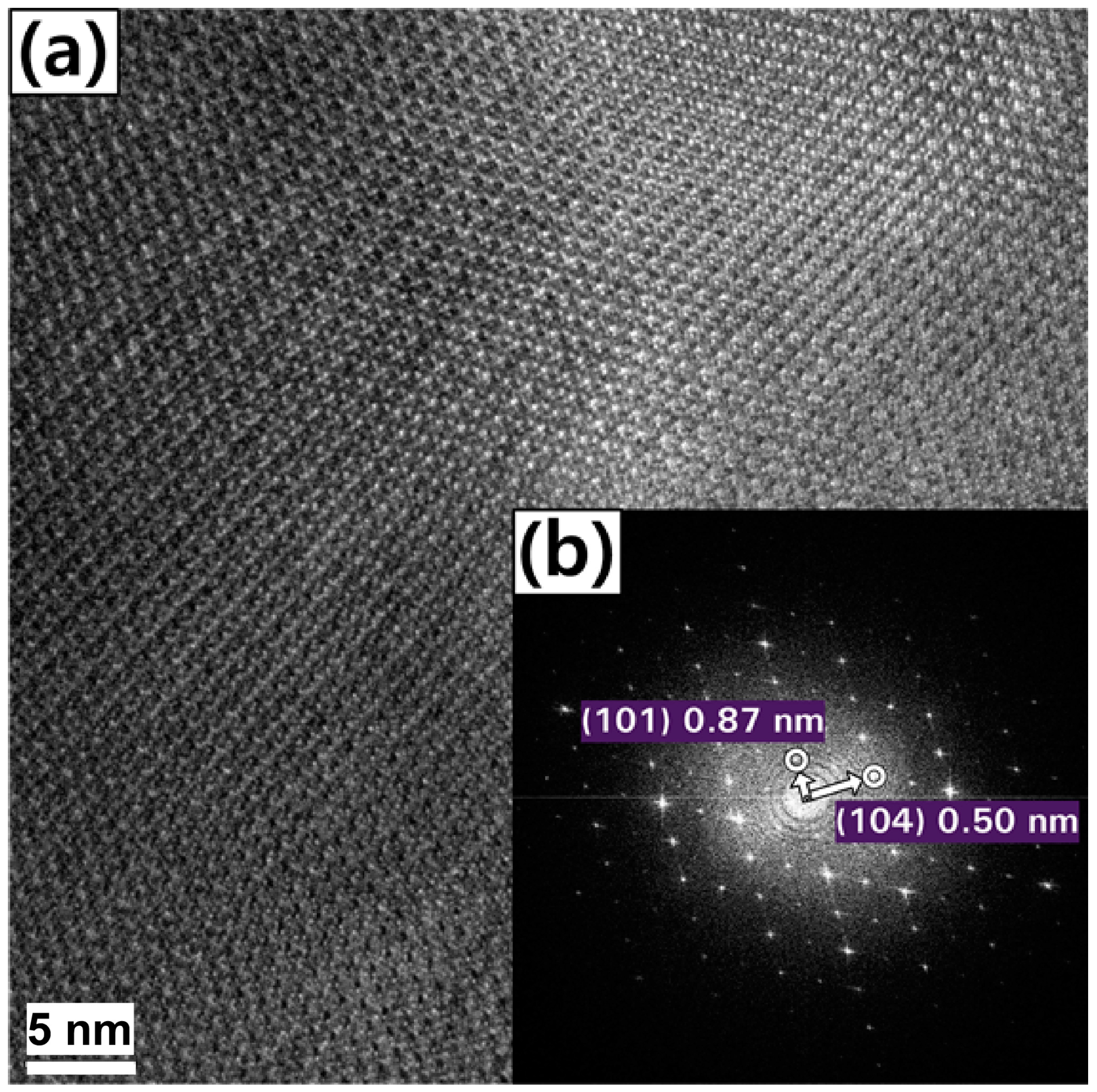
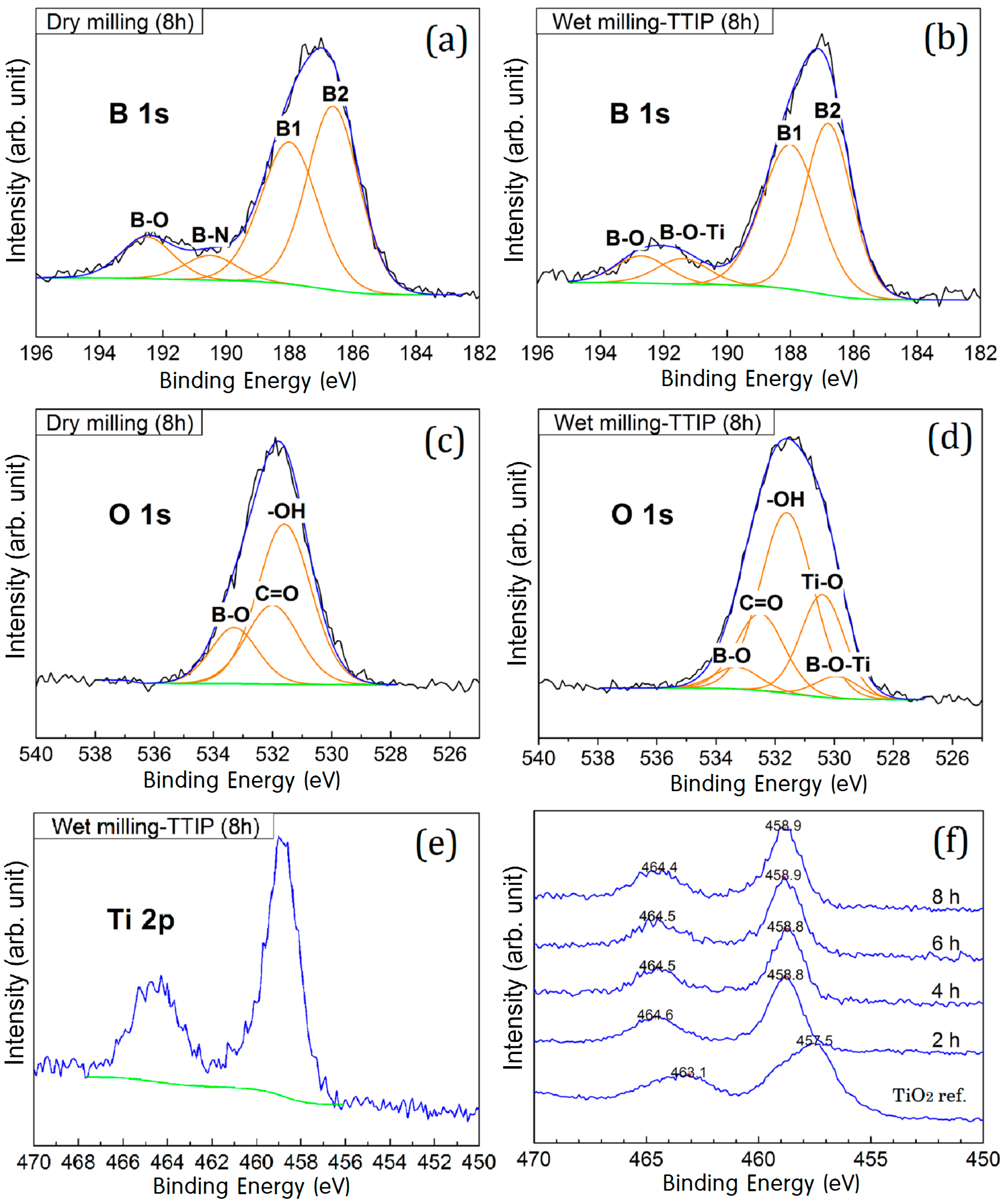
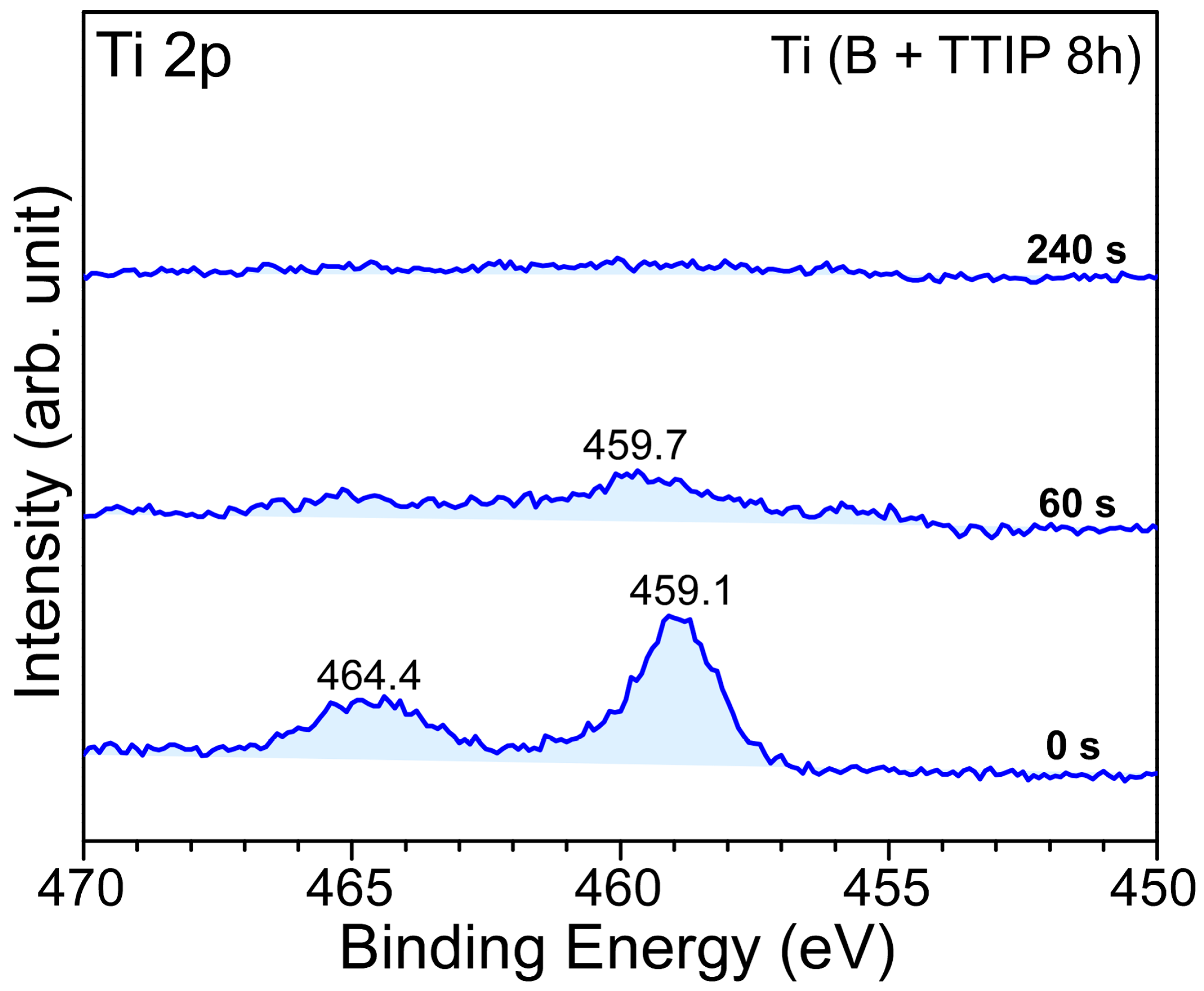
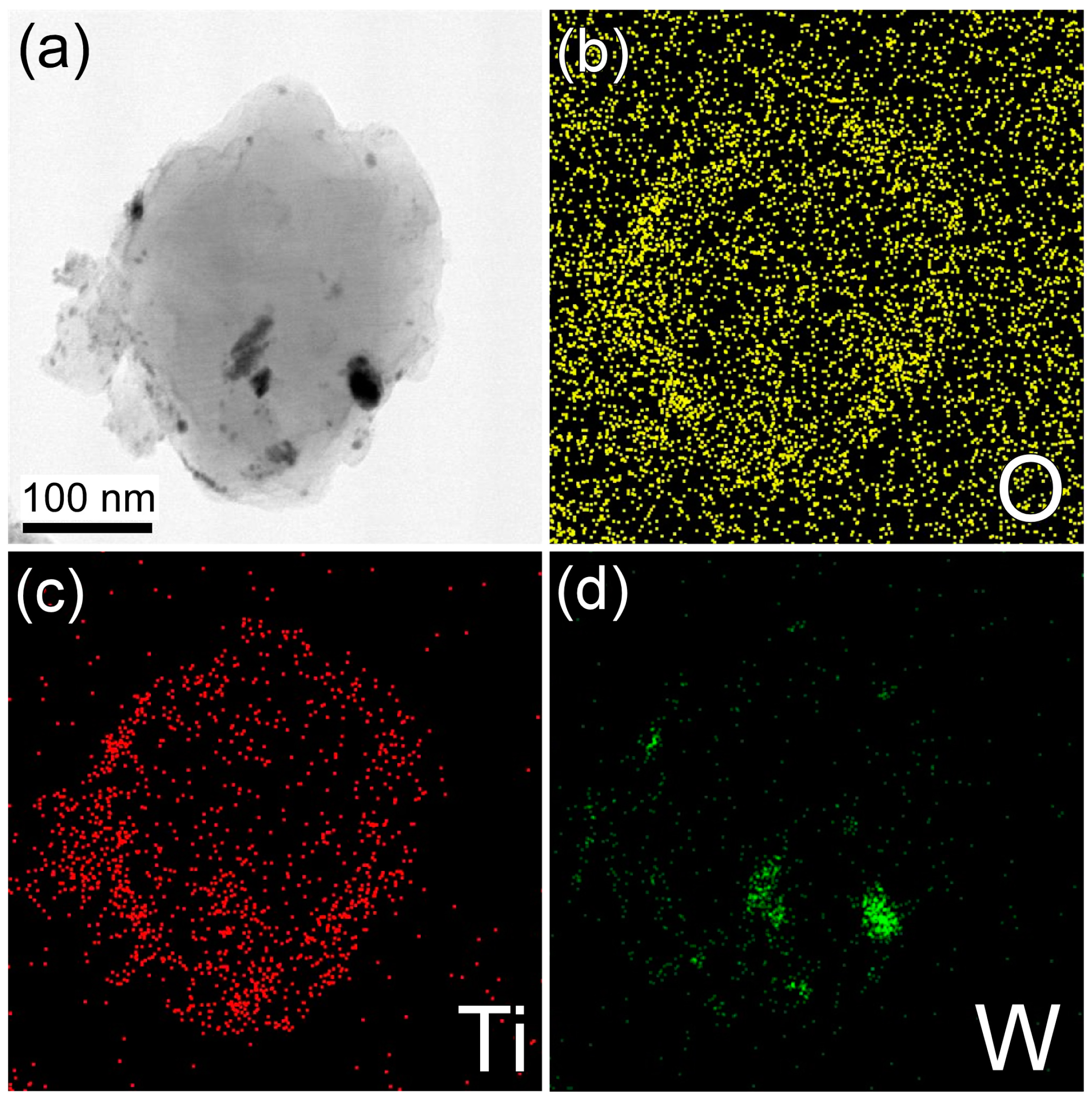
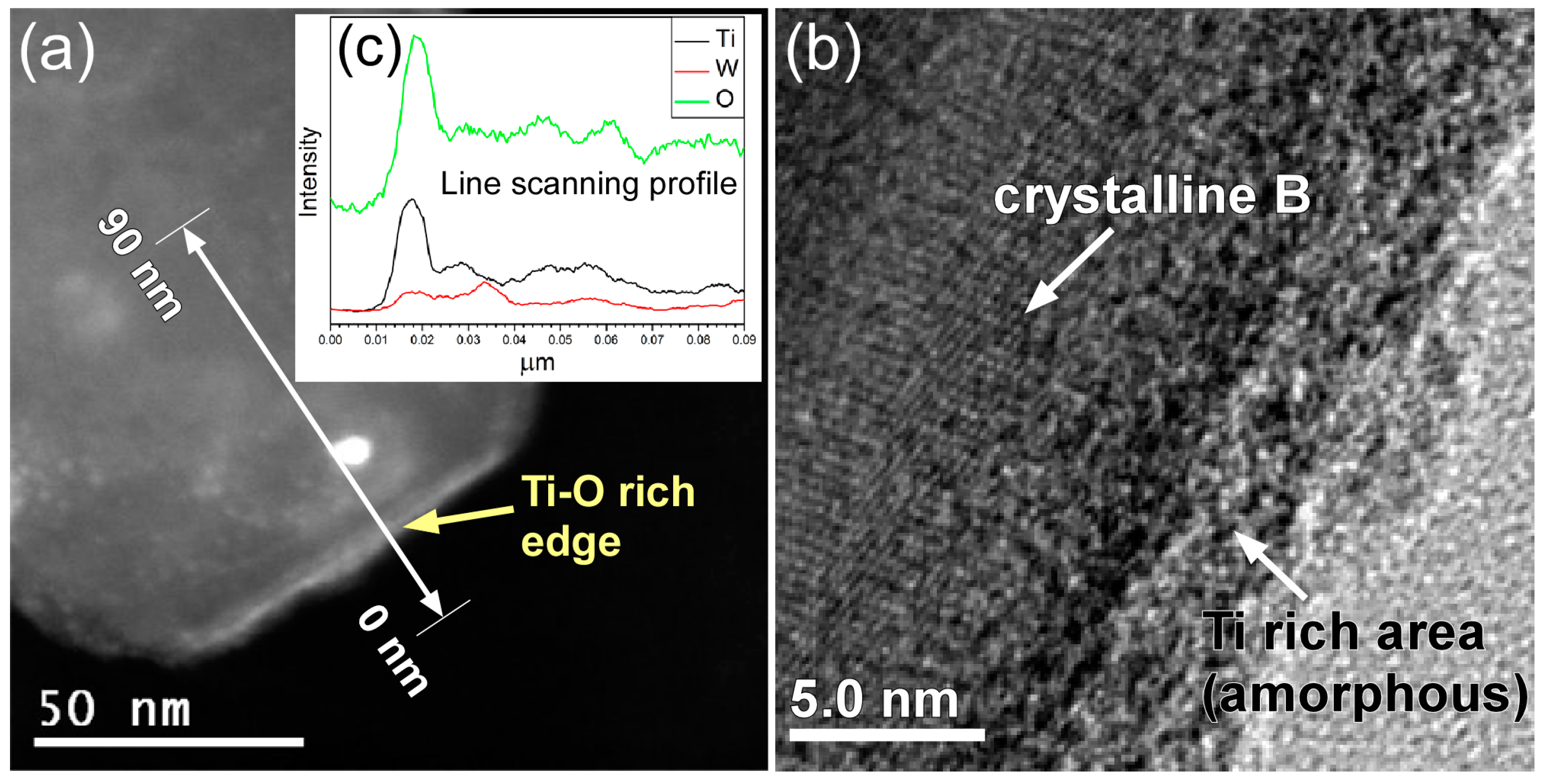
2. Results and Discussion
3. Materials and Methods
3.1. Metallic Oxide Coating on Boron by Wet Ball Milling
3.2. Characterization of the Coated Boron Particles
3.3. Photoelectrochemical Measurements
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Haddad, A.; Natan, B.; Arieli, R. The performance of a boron-loaded gel-fuel ramjet. Prog. Propuls. Phys. 2011, 2, 499–518. [Google Scholar]
- Pang, W.; Fan, X.; Zhang, W.; Xu, H.; Li, J.; Li, Y.; Shi, X.; Li, Y. Application of amorphous boron granulated with hydroxyl-terminated polybutadiene in fuel-rich solid propellant. Propellants Explos. Pyrotech. 2011, 36, 360–366. [Google Scholar] [CrossRef]
- Shin, W.G.; Han, D.; Park, Y.; Hyun, H.S.; Sung, H.-G.; Sohn, Y. Combustion of boron particles coated with an energetic polymer material. Korean J. Chem. Eng. 2016, 33, 3016–3020. [Google Scholar] [CrossRef]
- Zhu, S.; Gao, X.; Zhu, Y.; Zhu, Y.; Zheng, H.; Li, Y. Promoting effect of boron oxide on Cu/SiO2 catalyst for glycerol hydrogenolysis to 1, 2-propanediol. J. Catal. 2013, 303, 70–79. [Google Scholar] [CrossRef]
- Zheng, J.; Xia, Z.; Li, J.; Lai, W.; Yi, X.; Chen, B.; Fang, W.; Wan, H. Promoting effect of boron with high loading on Ni-based catalyst for hydrogenation of thiophene-containing ethylbenzene. Catal. Commun. 2012, 21, 18–21. [Google Scholar] [CrossRef]
- Aramendia, M.A.; Borau, V.; Jimenez, C.; Marinas, J.M.; Porras, A.; Urbano, F.J. Synthesis and characterization of MgO-B2O3 mixed oxides prepared by coprecipitation; selective dehydrogenation of propan-2-ol. J. Mater. Chem. 1999, 9, 819–825. [Google Scholar] [CrossRef]
- Colorio, G.; Vedrine, J.C.; Auroux, A.; Bonnetot, B. Partial oxidation of ethane over alumina-boria catalysts. Appl. Catal. A 1996, 137, 55–68. [Google Scholar] [CrossRef]
- Shin, W.G.; Jung, H.J.; Sung, H.G.; Hyun, H.S.; Sohn, Y. Synergic CO oxidation activities of boron-CeO2 hybrid materials prepared by dry and wet milling methods. Ceram. Int. 2014, 40, 11511–11517. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, G.; Bahnemann, D.W. Photoelectrocatalytic materials for environmental applications. J. Mater. Chem. 2009, 19, 5089–5121. [Google Scholar] [CrossRef]
- Ma, Y.; Wang, X.; Jia, Y.; Chen, X.; Han, H.; Li, C. Titanium dioxide-based nanomaterials for photocatalytic fuel generations. Chem. Rev. 2014, 114, 9987–10043. [Google Scholar] [CrossRef] [PubMed]
- Ge, M.; Cao, C.; Huang, J.; Li, S.; Chen, Z.; Zhang, K.-Q.; Al-Deyab, S.S.; Lai, Y. A review of one-dimensional TiO2 nanostructured materials for environmental and energy applications. J. Mater. Chem. A 2016, 4, 6772–6801. [Google Scholar] [CrossRef]
- Wang, X.; Li, Z.; Shi, J.; Yu, Y. One-dimensional titanium dioxide nanomaterials: Nanowires, nanorods, and nanobelts. Chem. Rev. 2014, 114, 9346–9384. [Google Scholar] [CrossRef] [PubMed]
- Ola, O.; Maroto-Valer, M.M. Review of material design and reactor engineering on TiO2 photocatalysis for CO2 reduction. J. Photochem. Photobiol. C 2015, 24, 16–42. [Google Scholar] [CrossRef]
- Roy, N.; Sohn, Y.; Pradhan, D. Synergy of low-energy {101} and high-energy {001} TiO2 crystal facets for enhanced photocatalysis. ACS Nano 2013, 7, 2532–2540. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.M.; Ansari, S.A.; Pradhan, D.; Ansari, M.O.; Lee, J.; Cho, M.H. Band gap engineered TiO2 nanoparticles for visible light induced photoelectrochemical and photocatalytic studies. J. Mater. Chem. A 2014, 2, 637–644. [Google Scholar] [CrossRef]
- Kalathil, S.; Khan, M.M.; Ansari, S.A.; Lee, J.; Cho, M.H. Band gap narrowing of titanium dioxide (TiO2) nanocrystals by electrochemically active biofilms and their visible light activity. Nanoscale 2013, 5, 6323–6326. [Google Scholar] [CrossRef] [PubMed]
- Roy, N.; Sohn, Y.; Leung, K.T.; Pradhan, D. Engineered electronic states of transition metal doped TiO2 nanocrystals for low overpotential oxygen evolution reaction. J. Phys. Chem. C 2014, 118, 29499–29506. [Google Scholar] [CrossRef]
- Kang, J.G.; Sohn, Y. Interfacial nature of Ag nanoparticles supported on TiO2 photocatalysts. J. Mater. Sci. 2012, 47, 824–832. [Google Scholar] [CrossRef]
- Daghrir, R.; Drogui, P.; Robert, D. Modified TiO2 for Environmental Photocatalytic Applications: A Review. Ind. Eng. Chem. Res. 2013, 52, 3581–3599. [Google Scholar] [CrossRef]
- Xiang, Q.; Yu, J.; Jaroniec, M. Synergetic Effect of MoS2 and Graphene as Cocatalysts for Enhanced Photocatalytic H2 Production Activity of TiO2 Nanoparticles. J. Am. Chem. Soc. 2012, 134, 6575–6578. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Shaner, M.R.; Beardslee, J.A.; Lichterman, M.; Brunschwig, B.S.; Lewis, N.S. Amorphous TiO2 coatings stabilize Si, GaAs, and GaP photoanodes for efficient water oxidation. Science 2014, 344, 1005–1009. [Google Scholar] [CrossRef] [PubMed]
- Hou, H.; Shang, M.; Gao, F.; Wang, L.; Liu, Q.; Zheng, J.; Yang, Z.; Yang, W. Highly Efficient Photocatalytic Hydrogen Evolution in Ternary Hybrid TiO2/CuO/Cu Thoroughly Mesoporous Nanofibers. ACS Appl. Mater. Interfaces 2016, 8, 20128–20137. [Google Scholar] [CrossRef] [PubMed]
- Terashima, C.; Hishinuma, R.; Roy, N.; Sugiyama, Y.; Latthe, S.S.; Nakata, K.; Kondo, T.; Yuasa, M.; Fujishima, A. Charge Separation in TiO2/BDD Heterojunction Thin Film for Enhanced Photoelectrochemical Performance. ACS Appl. Mater. Interfaces 2016, 8, 1583–1588. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhang, L.; Xing, W.; Lu, K. A Novel Method to Prepare B/N Codoped Anatase TiO2. J. Phys. Chem. C 2015, 119, 7732–7737. [Google Scholar] [CrossRef]
- Li, L.H.; Chen, Y.; Glushenkov, A.M. Boron nitride nanotube films grown from boron ink painting. J. Mater. Chem. 2010, 20, 9679–9683. [Google Scholar] [CrossRef]
- Li, L.; Li, L.H.; Chen, Y.; Dai, X.J.; Xing, T.; Petravic, M.; Liu, X. Mechanically activated catalyst mixing for high-yield boron nitride nanotube growth. Nanoscale Res. Lett. 2012, 7, 417. [Google Scholar] [CrossRef] [PubMed]
- Li, L.H.; Chen, Y.; Glushenkov, A.M. Synthesis of boron nitride nanotubes by boron ink annealing. Nanotechnology 2010, 21, 105601. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.J.; Sohn, Y.; Sung, H.G.; Hyun, H.S.; Shin, W.G. Physicochemical properties of ball milled boron particles: Dry vs. wet ball milling process. Powder Technol. 2015, 269, 548–553. [Google Scholar] [CrossRef]
- Choi, Y.I.; Jung, H.J.; Shin, W.G.; Sohn, Y. Band gap-engineered ZnO and Ag/ZnO by ball-milling and their photocatalytic and Fenton-like photocatalytic activities. Appl. Surf. Sci. 2015, 356, 615–625. [Google Scholar] [CrossRef]
- Devener, B.V.; Perez, J.P.L.; Jankovich, J.; Anderson, S.L. Oxide-free, catalyst-coated, fuel-soluble, air-stable boron nanopowder as combined combustion catalyst and high energy density fuel. Energy Fuel 2009, 23, 6111–6120. [Google Scholar] [CrossRef]
- Shin, W.G.; Calder, S.; Ugurlu, O.; Girshick, S.L. Production and characterization of boron nanoparticles synthesized with a thermal plasma system. J. Nanopart. Res. 2011, 13, 7187–7191. [Google Scholar] [CrossRef]
- Xie, Y.P.; Liu, G.; Lu, G.Q.M.; Cheng, H.M. Boron oxynitride nanoclusters on tungsten trioxide as a metal-free cocatalyst for photocatalytic oxygen evolution from water splitting. Nanoscale 2012, 4, 1267–1270. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Yang, B.; Chen, J. Effects of calcination temperature on preparation of boron-doped TiO2 by sol-Gel method. Int. J. Photoenergy 2012, 2012, 528637–528644. [Google Scholar] [CrossRef]
- Liu, S.X.; Kim, J.T. Characterization of surface modification of polyethersulfone membrane. J. Adhes. Sci. Techonol. 2011, 25, 193–212. [Google Scholar] [CrossRef]
- Zhang, X.W.; Lei, L.C. Development of supported boron-doping TiO2 catalysts by chemical vapor deposition. J. Zhejiang Univ. Sci. A 2008, 9, 109–112. [Google Scholar] [CrossRef]
- Liu, P.; Cai, W.; Fang, M.; Li, Z.; Zeng, H.; Hu, J.; Jing, W. Room temperature synthesized rutile TiO2 nanoparticles induced by laser ablation in liquid and their photocatalytic activity. Nanotechnology 2009, 20, 285707–285712. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Wang, L.; Tang, Y.; Luo, S.; Liu, Y.; Zhang, S.; Zeng, Y.; Xu, Y. Vertical single or few-layer MoS2 nanosheets rooting into TiO2 nanofibers for highly efficient photocatalytic hydrogen evolution. Appl. Catal. B 2015, 164, 1–9. [Google Scholar] [CrossRef]
- Sohn, Y.; Pradhan, D.; Leung, K.T. Electrochemical Pd nanodeposits on a Au Nanoisland template supported on Si (100):formation of Pd-Au alloy and interfacial electronic structures. ACS Nano 2010, 4, 5111–5120. [Google Scholar] [CrossRef] [PubMed]
- Andres, E.S.; Toledano-Luque, M.; Prado, A.D.; Navacerrada, M.A.; Mártil, I.; González-Díaz, G.; Bohne, W.; Röhrich, J.; Strub, E. Physical properties of high pressure reactively sputtered TiO2. J. Vac. Sci. Technol. A 2005, 23, 1523–1530. [Google Scholar] [CrossRef]
- Chen, D.; Yang, D.; Wang, Q.; Jiang, Z. Effects of boron doping on photocatalytic activity and microstructure of titanium dioxide nanoparticles. Ind. Eng. Chem. Res. 2006, 45, 4110–4116. [Google Scholar] [CrossRef]
- Hoffmann, P.; Galindo, H.; Zambrano, G.; Rincon, C.; Prieto, P. FTIR studies of tungsten carbide in bulk material and thin film samples. Mater. Charact. 2003, 50, 255–259. [Google Scholar] [CrossRef]
- Lee, S.; Kang, J.S.; Leung, K.T.; Kim, S.K.; Sohn, Y. Magnetic Ni-Co alloys induced by water gas shift reaction, Ni-Co oxides by CO oxidation and their supercapacitor applications. Appl. Surf. Sci. 2016, 386, 393–404. [Google Scholar] [CrossRef]
- Choi, Y.I.; Yoon, H.J.; Kim, S.K.; Sohn, Y. Crystal-facet dependent CO oxidation, preferential oxidation of CO in H2-rich, water-gas shift reactions, and supercapacitor application over Co3O4 nanostructures. Appl. Catal. A 2016, 519, 56–67. [Google Scholar] [CrossRef]
- Lee, S.; Kang, J.-S.; Leung, K.T.; Lee, W.; Kim, D.; Han, S.; Yoo, W.; Yoon, H.J.; Nam, K.; Sohn, Y. Unique multi-phase Co/Fe/CoFe2O4 by water-gas shift reaction, CO oxidation and enhanced supercapacitor performances. J. Ind. Eng. Chem. 2016, 43, 69–77. [Google Scholar] [CrossRef]

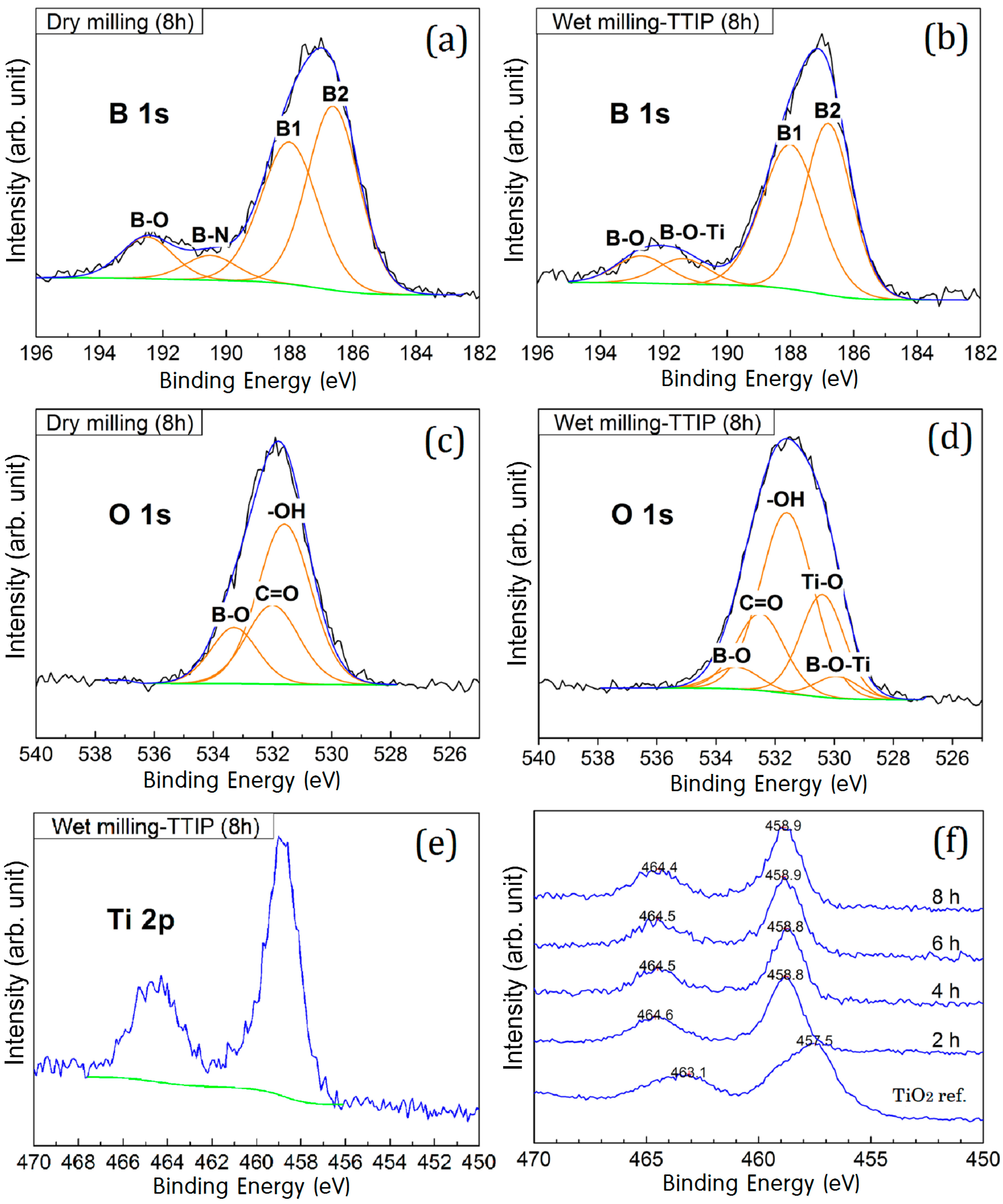
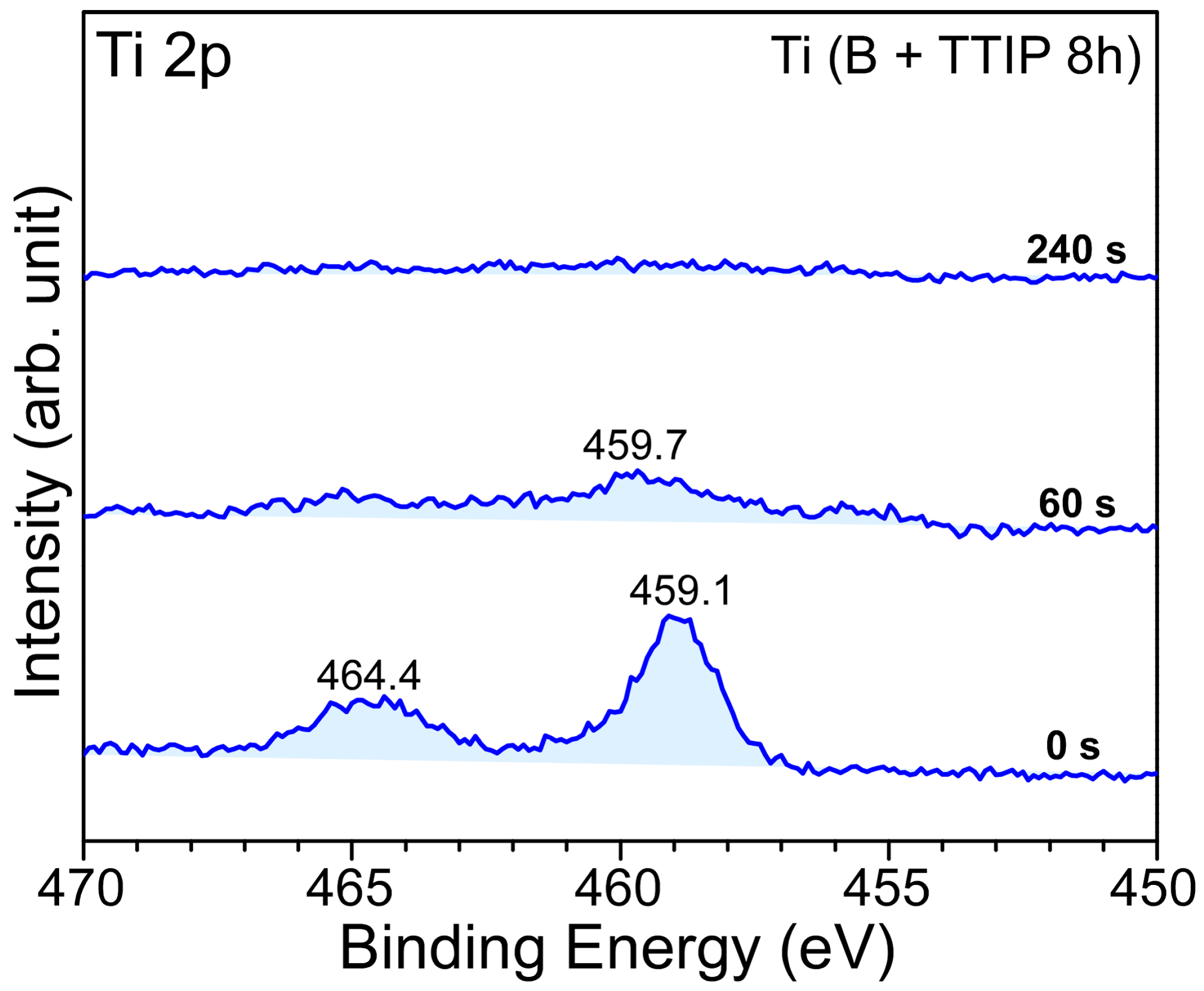
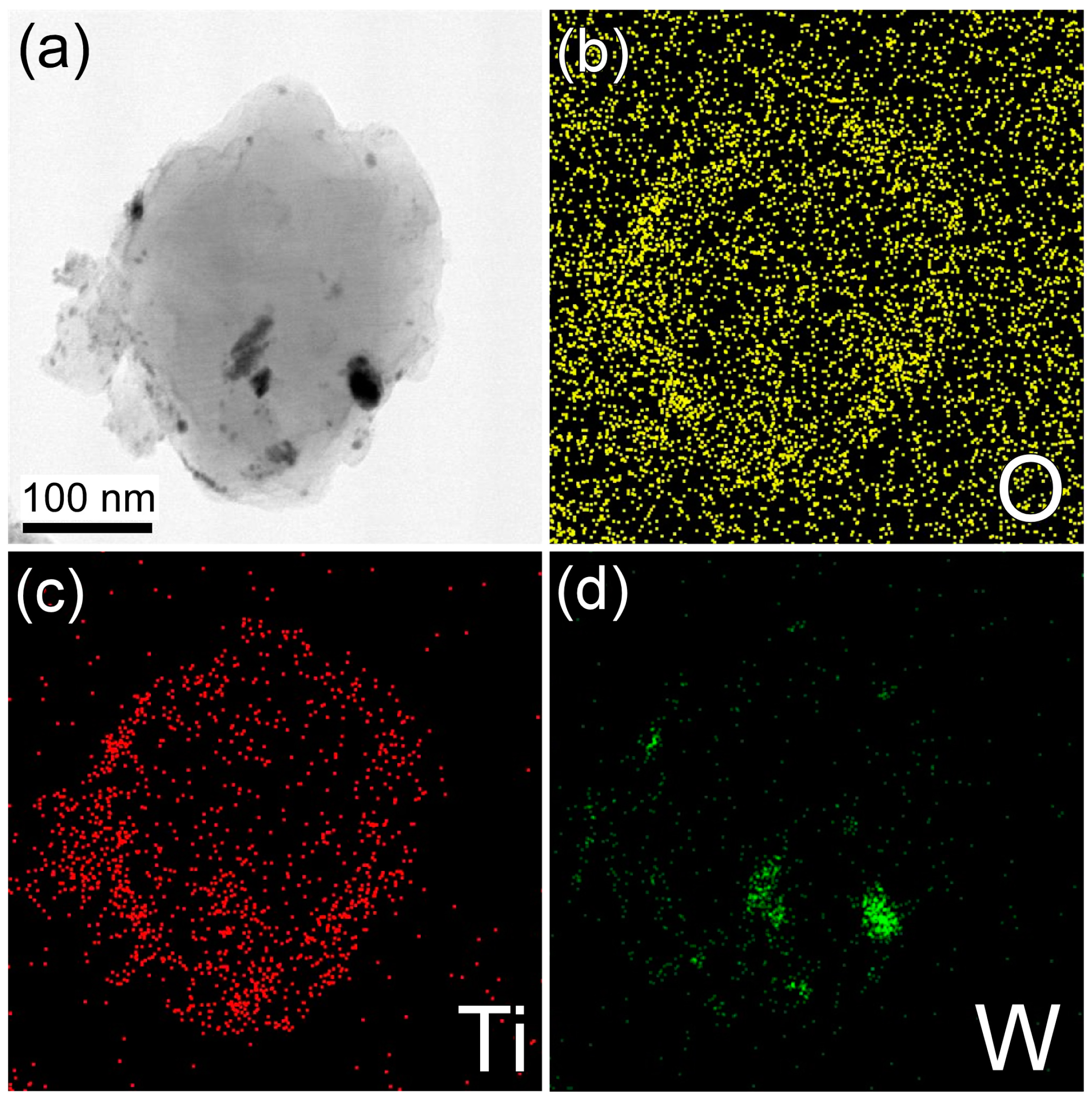
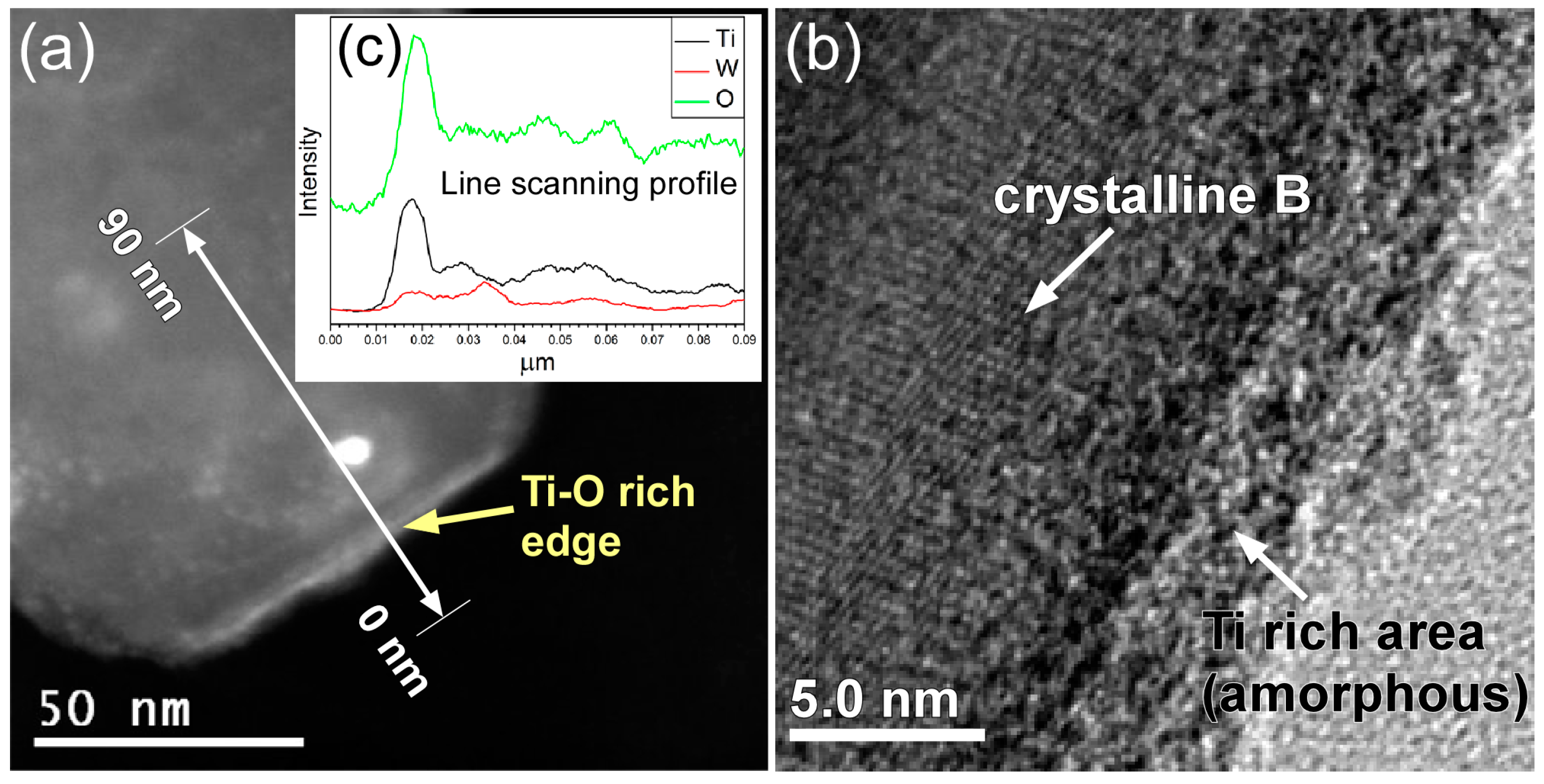


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
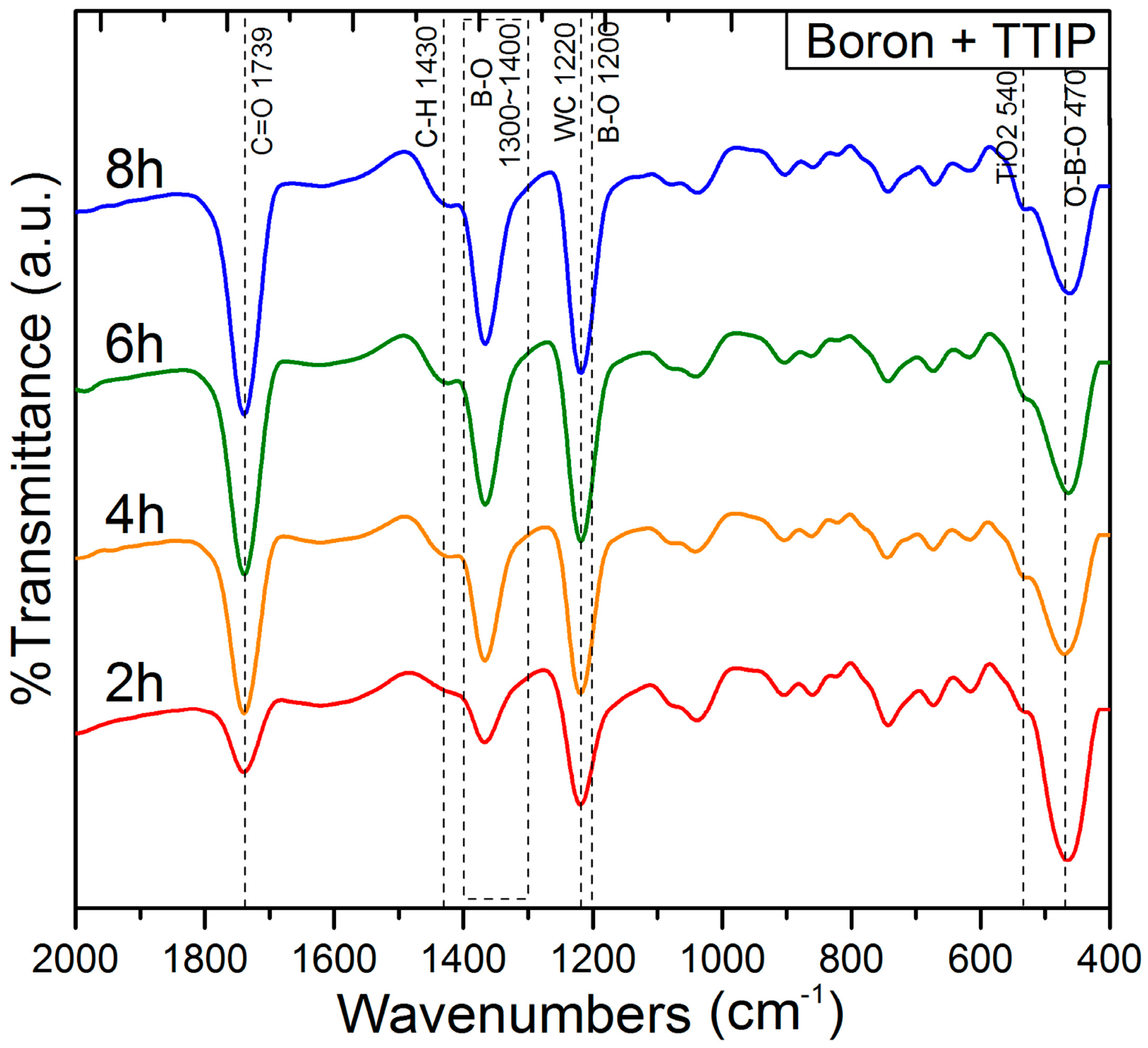{kind=link}
{kind=link}
{kind=link}
| Atom | 1 h | 2 h | 4 h | 6 h | 8 h |
|---|---|---|---|---|---|
| B | 95.5 | 96.29 | 92.86 | 93.27 | 92.75 |
| O | 4.56 | 3.12 | 6.03 | 4.56 | 4.85 |
| Ti | 0.0 | 0.0 | 0.15 | 0.21 | 0.35 |
| W | 0.40 | 0.59 | 0.96 | 1.97 | 2.05 |
| Total | 100 | 100 | 100 | 100 | 100 |
| Un-Milled Boron Sputter Time (s) | 4 h Wet Milled Boron Sputter Time (s) | 8 h Wet Milled Boron Sputter Time (s) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Peaks | 0 | 60 | 240 | 0 | 60 | 240 | 0 | 60 | 240 |
| B 1s | 53.92 | 71.22 | 79.45 | 47.42 | 61.21 | 73.02 | 34.16 | 44.74 | 68.49 |
| C 1s | 19.27 | 12.38 | 6.25 | 21.16 | 12.32 | 6.1 | 22.23 | 19.42 | 9.56 |
| O 1s | 23.5 | 13.35 | 13.08 | 25.1 | 20.84 | 16.15 | 33.37 | 26.18 | 16.16 |
| Ti 2p | - | - | - | 1.42 | 1.01 | 0.99 | 3.59 | 2.17 | 1.04 |
| W 4f | - | - | - | 1.47 | 1.88 | 2.34 | 2.17 | 3.58 | 3.26 |
| F 1s | 3.31 | 3.05 | 1.22 | 1.98 | 1.69 | 0.65 | 1.85 | 1.48 | 0.57 |
| Co 2p | - | - | - | 1.44 | 1.04 | 0.75 | 2.63 | 2.45 | 0.91 |
| Total | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jung, H.J.; Nam, K.; Sung, H.-G.; Hyun, H.S.; Sohn, Y.; Shin, W.G. Preparation of TiO2-Decorated Boron Particles by Wet Ball Milling and their Photoelectrochemical Hydrogen and Oxygen Evolution Reactions. Materials 2016, 9, 1012. https://doi.org/10.3390/ma9121012
Jung HJ, Nam K, Sung H-G, Hyun HS, Sohn Y, Shin WG. Preparation of TiO2-Decorated Boron Particles by Wet Ball Milling and their Photoelectrochemical Hydrogen and Oxygen Evolution Reactions. Materials. 2016; 9(12):1012. https://doi.org/10.3390/ma9121012
Chicago/Turabian StyleJung, Hye Jin, Kyusuk Nam, Hong-Gye Sung, Hyung Soo Hyun, Youngku Sohn, and Weon Gyu Shin. 2016. "Preparation of TiO2-Decorated Boron Particles by Wet Ball Milling and their Photoelectrochemical Hydrogen and Oxygen Evolution Reactions" Materials 9, no. 12: 1012. https://doi.org/10.3390/ma9121012
APA StyleJung, H. J., Nam, K., Sung, H.-G., Hyun, H. S., Sohn, Y., & Shin, W. G. (2016). Preparation of TiO2-Decorated Boron Particles by Wet Ball Milling and their Photoelectrochemical Hydrogen and Oxygen Evolution Reactions. Materials, 9(12), 1012. https://doi.org/10.3390/ma9121012