
Synthesis of Copper Nanoparticles in Ethylene Glycol by Chemical Reduction with Vanadium (+2) Salts




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
- -
- vanadium (+2) sulphate, different from other routinely adopted inorganic reductants with basic properties, has an acid behavior, and this proved to be beneficial in hindering the formation of a surface layer of mixed oxides on Cu-NPs [23];
- -
- vanadium (+2) sulphate reduces copper salts at room temperature with fast kinetics comparable to those typical of NaBH4, thus allowing much lower temperatures with respect to those typical of processes where conventional reductants operating in non-aqueous solvents are used;
- -
- vanadium (+2) cations interfere with copper cations in the process only in some cases, namely when non-ionic stabilizing agents are used to prevent nanoparticles aggregation in a non-aqueous solvent embedding the precursor. This side effect will be discussed in the following section.
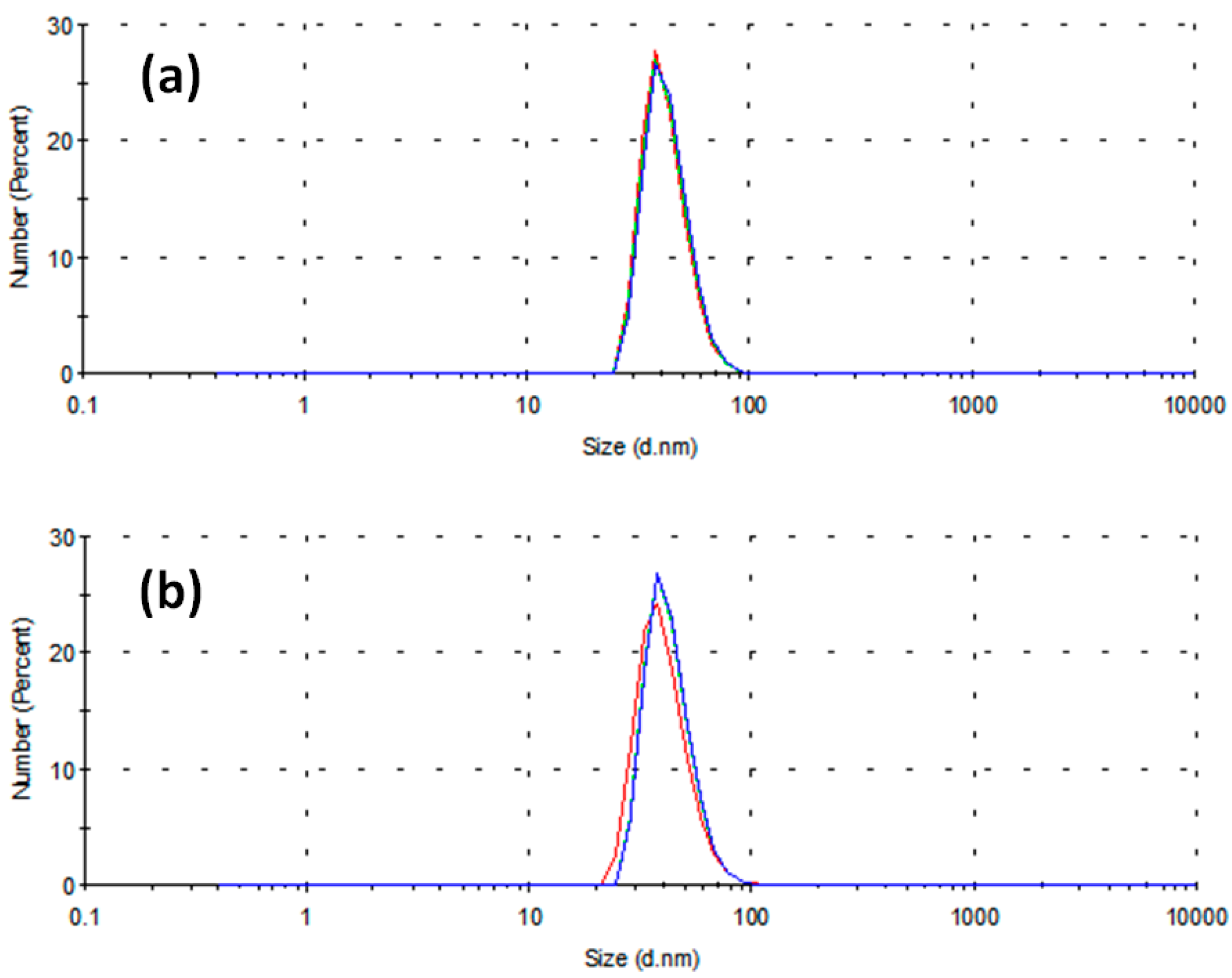
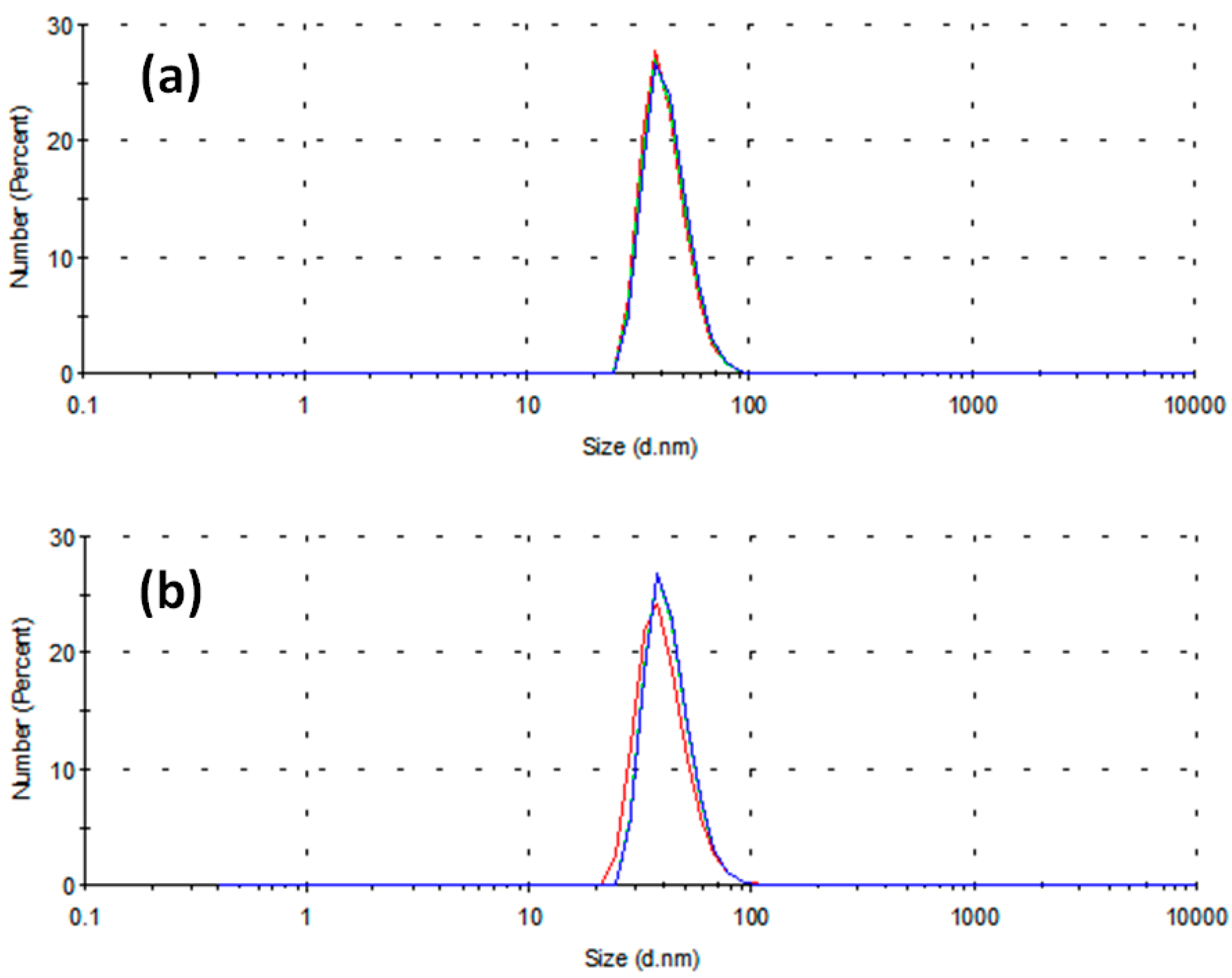
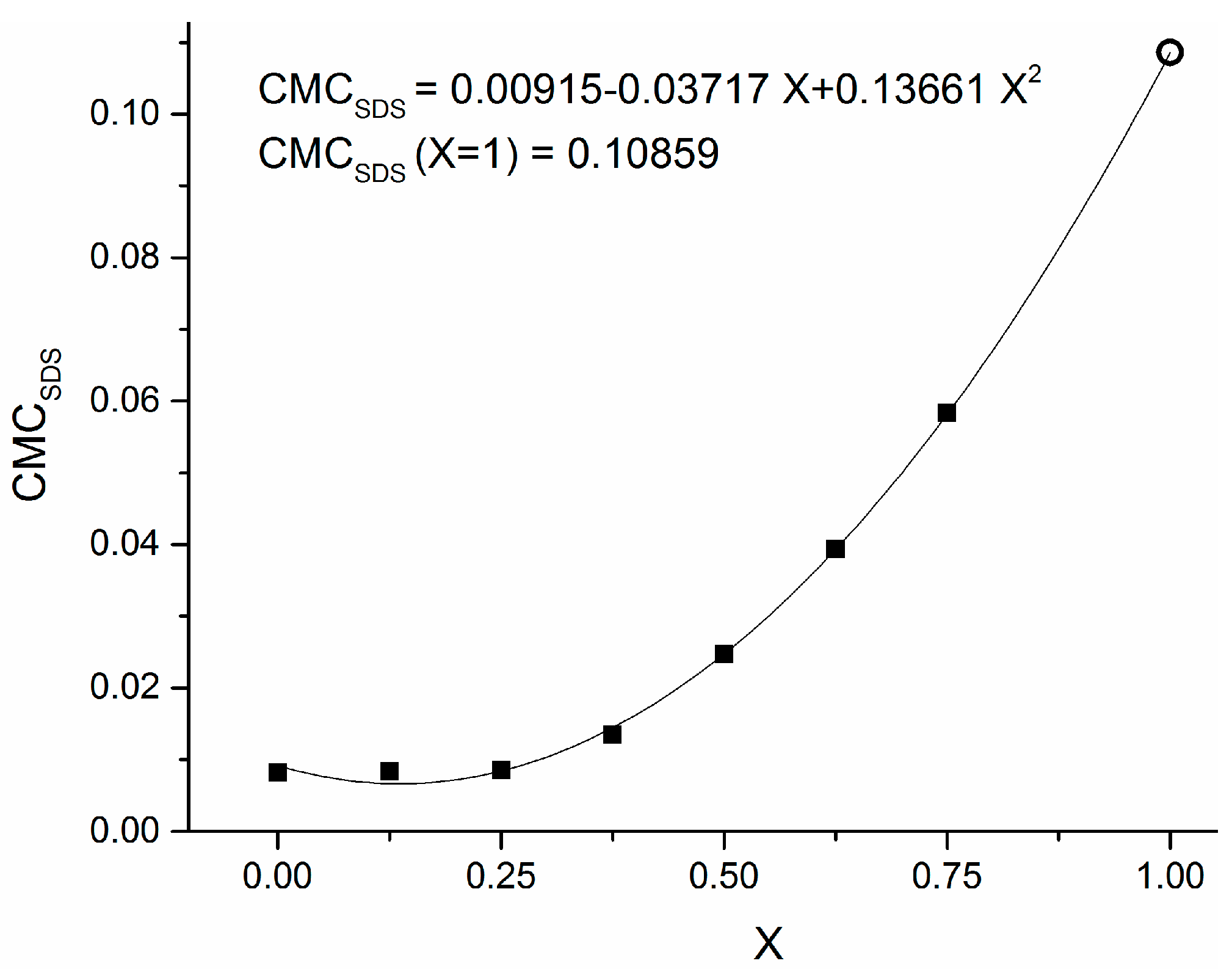
2. Results and Discussion
- -
- The capping activity of a non-ionic stabilizing agent toward Cu2+ ions is due to the presence of active sites on its molecule as electron-pair donors to the Cu2+ ion itself. The oxygen atom in the ketonic group of PVP, with its double pair of free electrons, is one of the best candidates for this role.
- -
- Vanadium, being a transition element as copper, may compete against copper in forming coordination bonds with the aforementioned electron-pair donors, possibly deactivating the PVP in its capping activity. Therefore, this latter effect might be damped at lower concentrations of reductant, and this fact might explain how smaller diameters have been obtained in this condition. To the best of our knowledge, this is the first case when a reductant containing a transition element is adopted in the synthesis of metal nanoparticles.
3. Materials and Methods
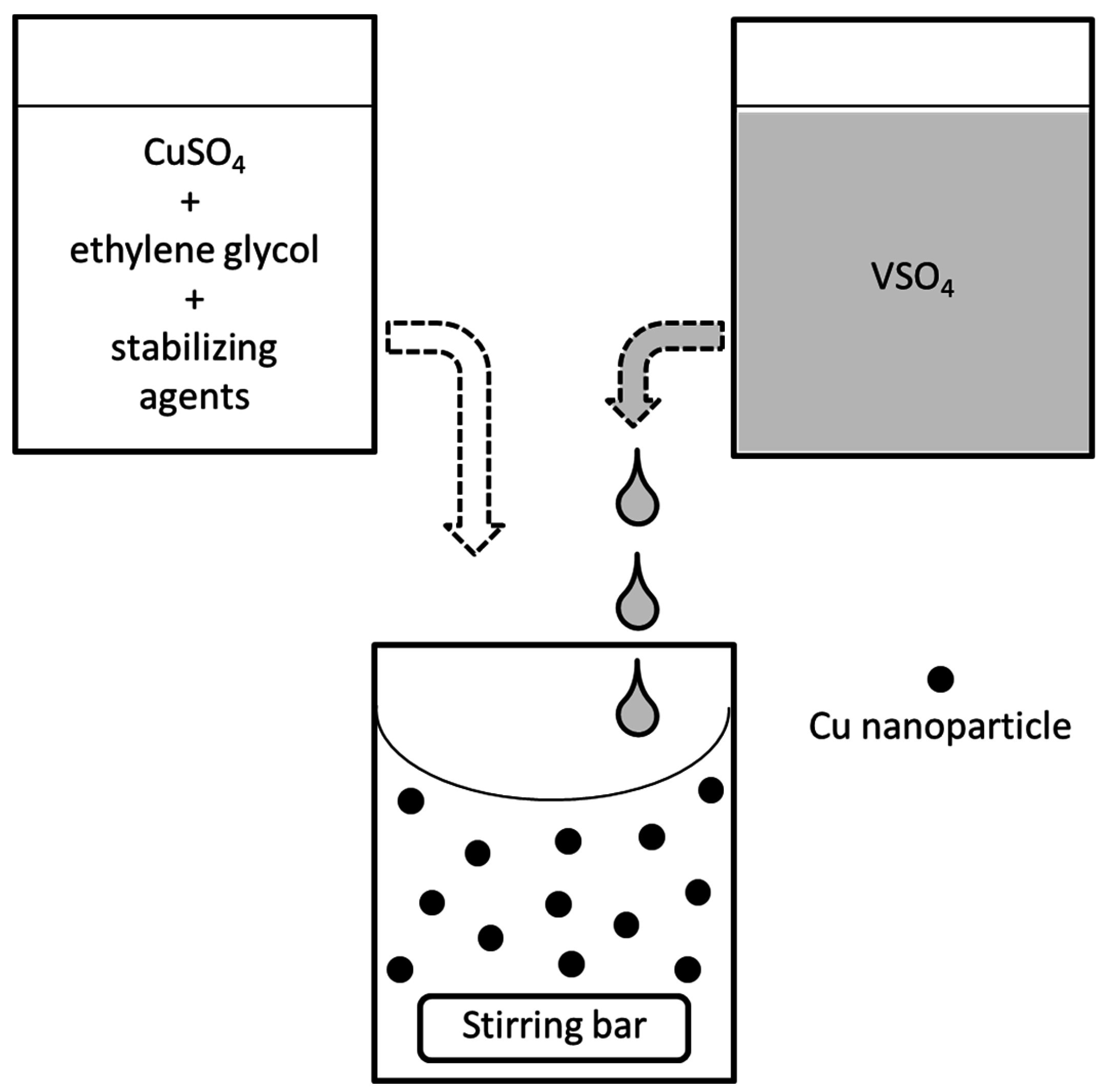
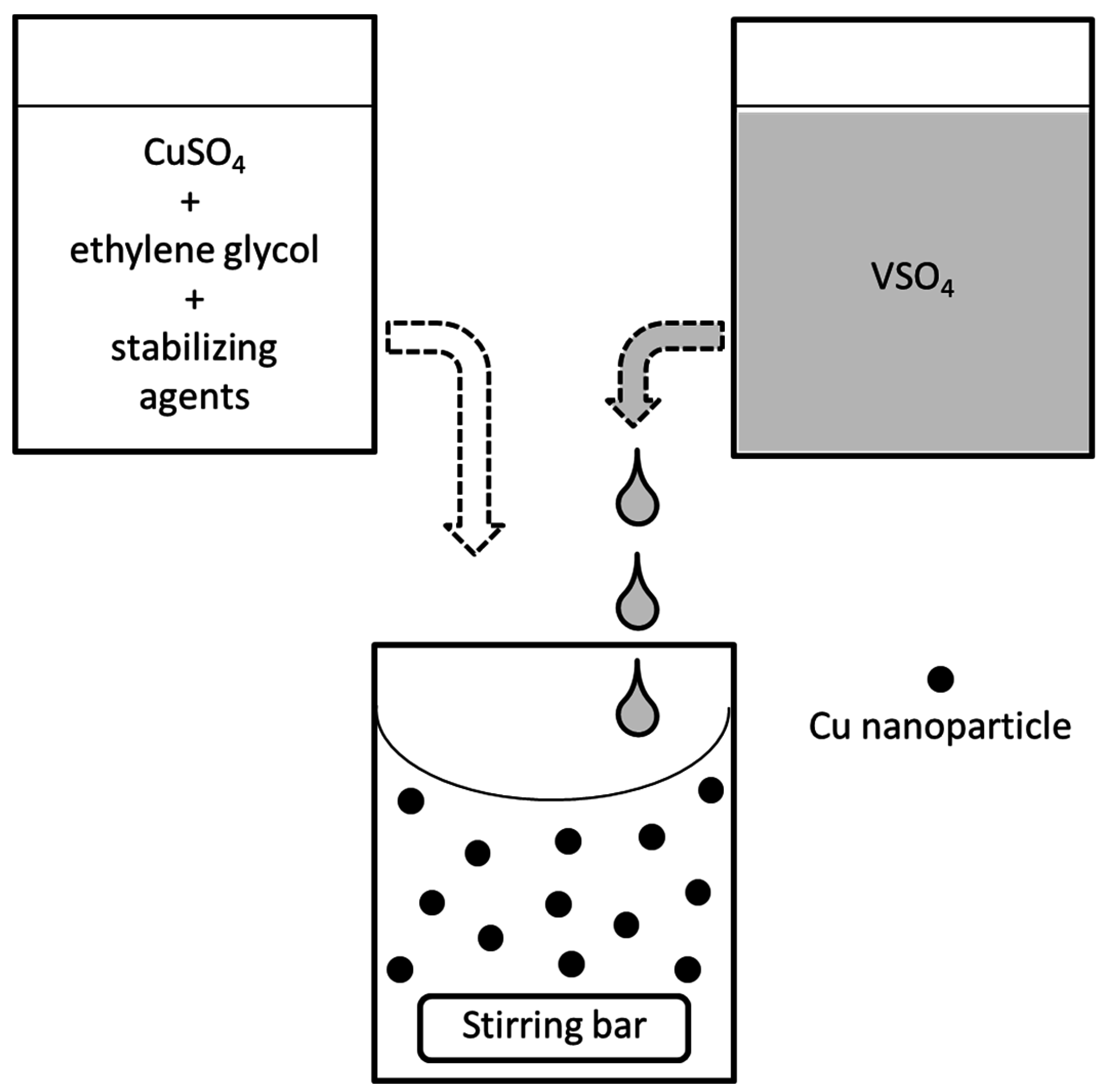
3.1. Reduction Process
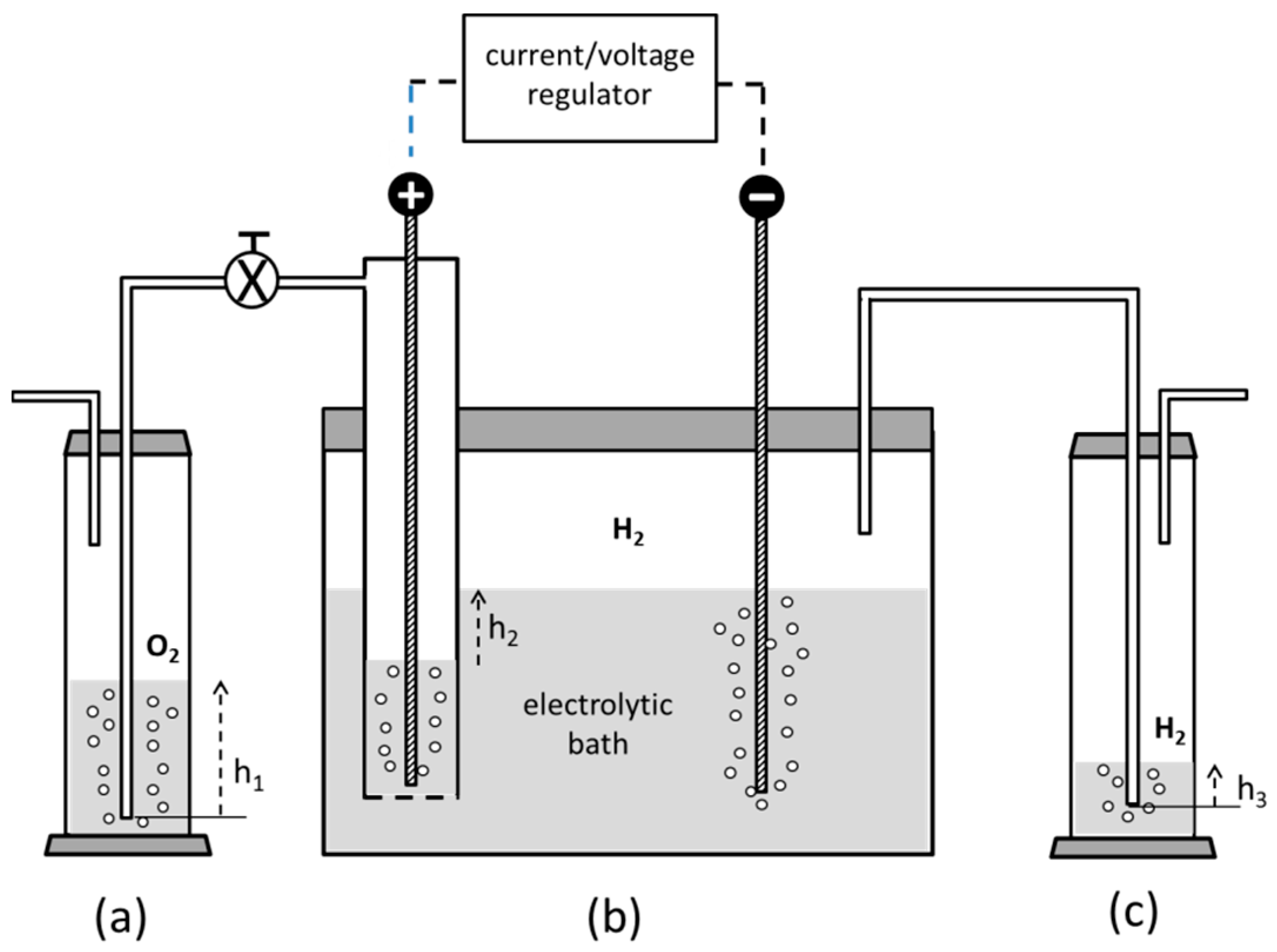
3.2. Preparation of the Reductant
- -
- in all situations, an inert atmosphere preserving from oxidation is generally needed;
- -
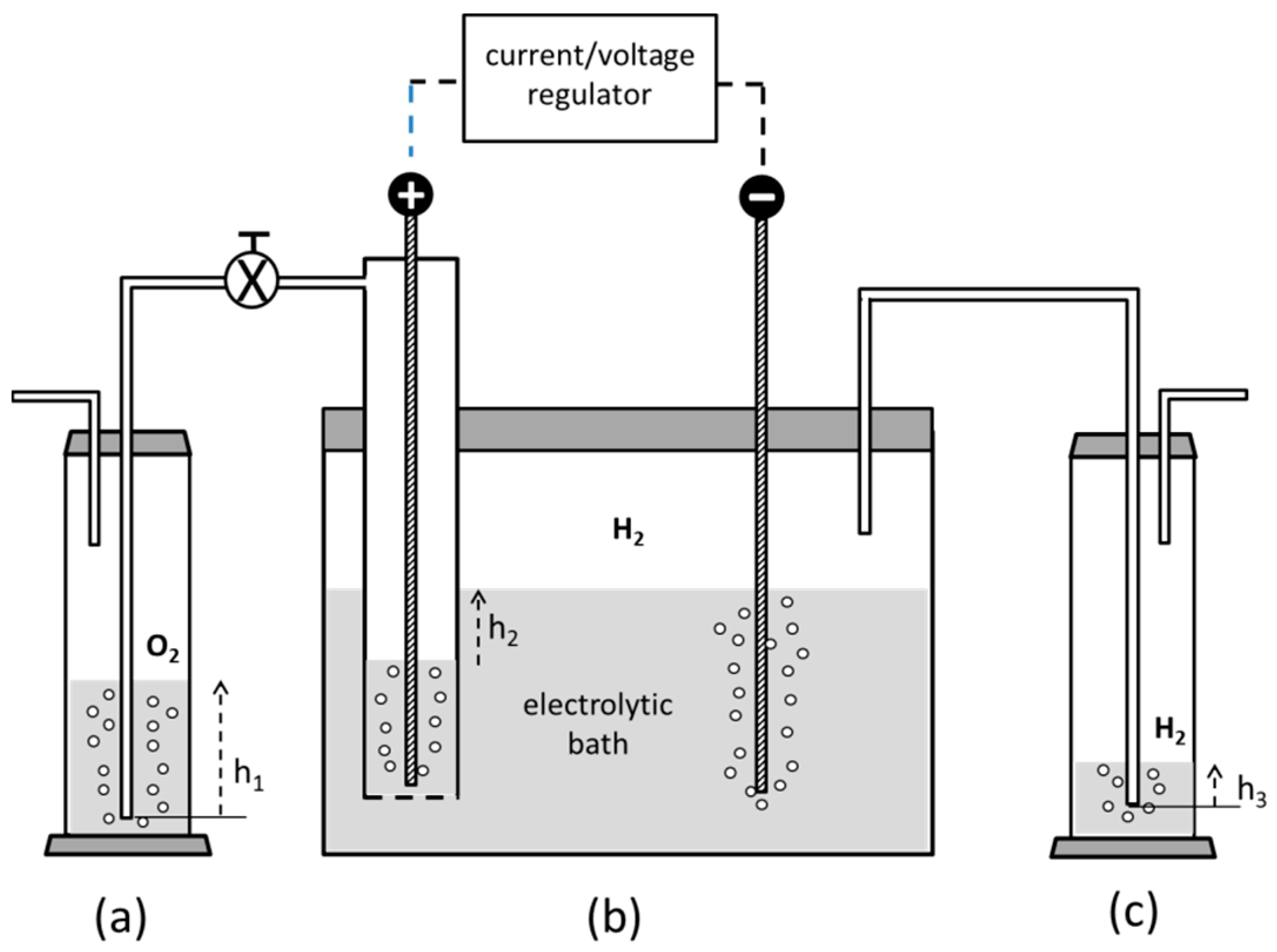
- if an electrochemical process is adopted in aqueous solvent, a cathode having the highest overvoltage to the hydrogen discharge has to be adopted in order to maximize the current yield toward the vanadium reduction. As a consequence, a mercury, an amalgamated-metal cathode or a surface-pretreated lead cathode represent a basic choice to fulfil this stringent constraint [33].
- -
- V2O5 in crystalline form is only sparingly soluble in water and a considerable amount of zinc is attacked by H2SO4, giving off hydrogen instead of taking part in Reaction (2).
- -
- The presence of ZnSO4, which is dissolved together with VSO4, may trigger the formation of insoluble double zinc–vanadium sulphates. Regretfully, this insoluble phase may contaminate the Cu-NPs produced by Reaction (1).
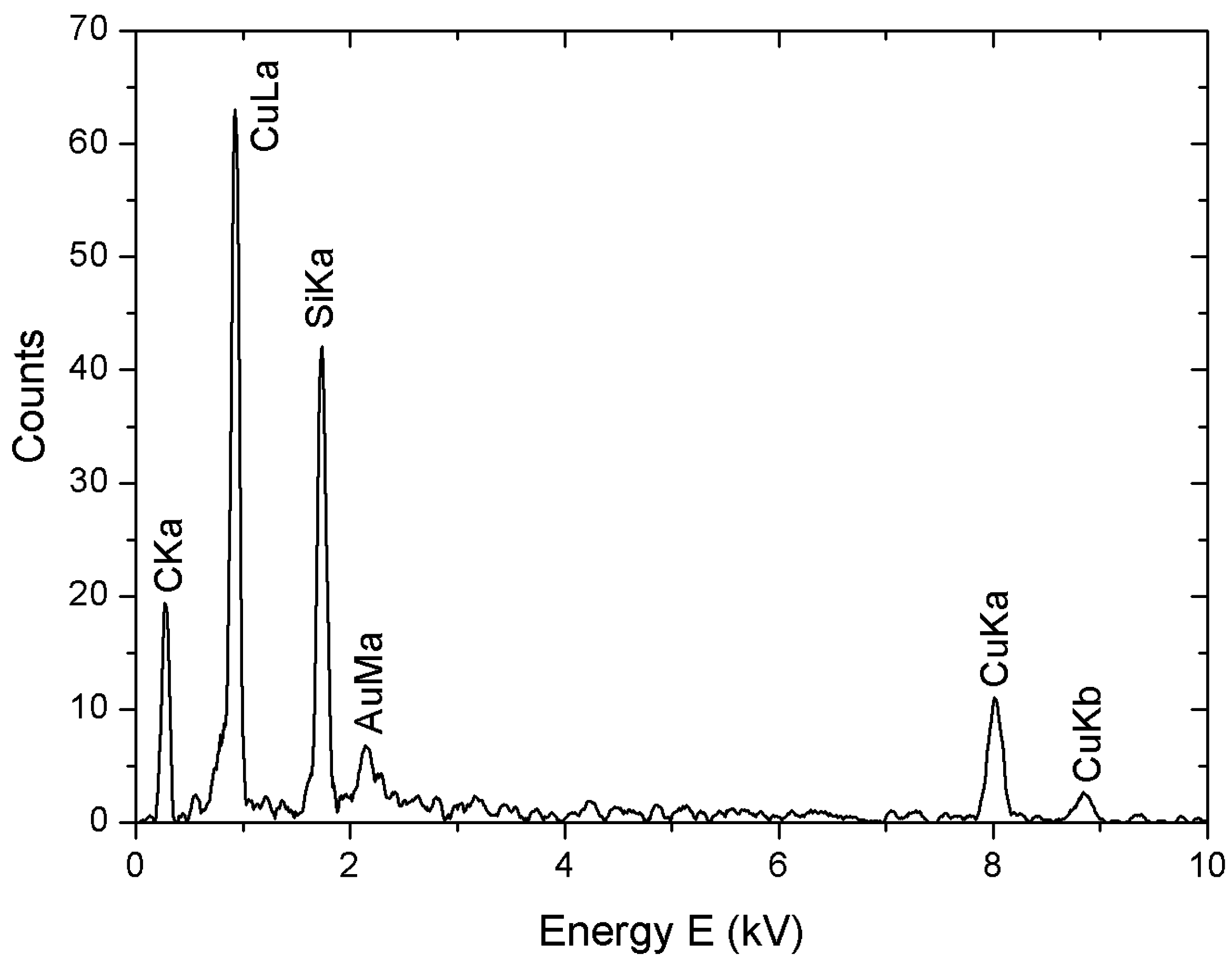
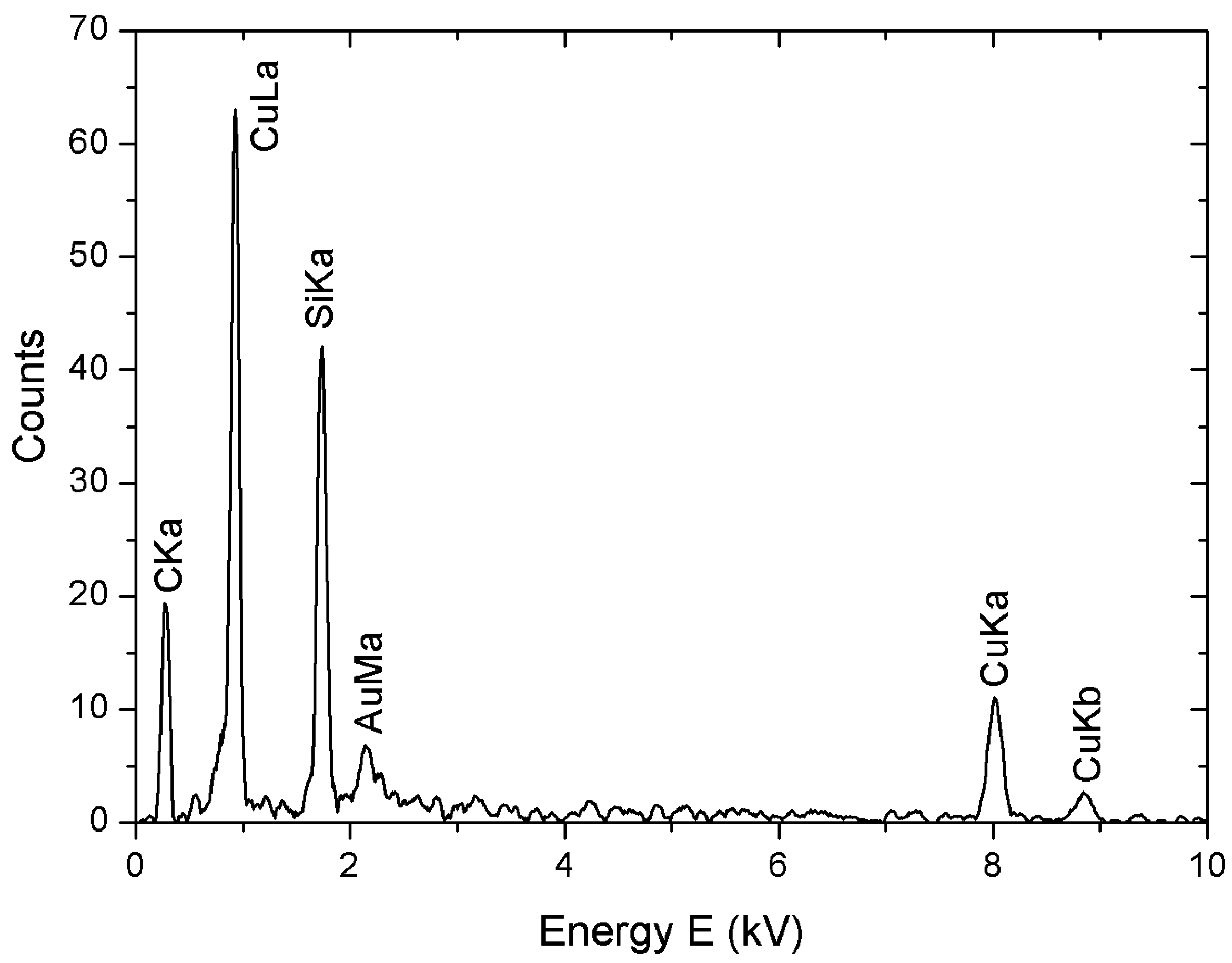
3.3. Characterization Techniques
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| NPs | nanoparticles |
| DLS | Dynamic Light Scattering |
| SEM | Scanning Electron Microscopy |
| EDS | Energy Dispersive X-ray Spectroscopy |
| SPR | Surface Plasmon Resonance |
| TEM | Transmission Electron Microscopy |
| PVP | Polyvynilpyrrolidone |
| SDS | Sodium dodecyl sulphate |
| CTAB | cetyl-trimethylammonium bromide |
| MTAB | myristyl-trimethylammonium bromide |
| TBAB | tetrabutylammonium bromide |
| EG | ethylene glycol |
| CMC | critical micellar concentration |
References
- Zazo, H.; Colino, C.I.; Lanao, J.M. Current applications of nanoparticles in infectious diseases. J. Control. Release 2016, 224, 86–102. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, R.V.; Wojcieszak, R.; Wender, H.; Sato, B.; Dias, C.; Vono, L.L.R.; Eberhardt, D.; Teixeira, S.R.; Rossi, L.M. Easy access to metallic copper nanoparticles with high activity and stability for CO oxidation. ACS Appl. Mater. Interfaces 2015, 7, 7987–7994. [Google Scholar] [CrossRef] [PubMed]
- Pascariu, V.; Avadanei, O.; Gasner, P.; Stoica, I.; Reverberi, A.P.; Mitoseriu, L. Preparation and characterization of PbTiO3-epoxy resin compositionally graded thick films. Phase Transit. 2013, 86, 715–725. [Google Scholar] [CrossRef]
- Baghbanzadeh, M.; Rana, D.; Lan, C.Q.; Matsuura, T. Effects of inorganic nano-additives on properties and performance of polymeric membranes in water treatment. Sep. Purif. Rev. 2016, 45, 141–167. [Google Scholar] [CrossRef]
- Palazzi, E.; Perego, P.; Fabiano, B. Mathematical modelling and optimization of hydrogen continuous production in a fixed bed bioreactor. Chem. Eng. Sci. 2002, 57, 3819–3830. [Google Scholar] [CrossRef]
- Reverberi, A.P.; Klemeš, J.J.; Varbanov, P.S.; Fabiano, B. A review on hydrogen production from hydrogen sulphide by chemical and photochemical methods. J. Clean. Prod. 2016. [Google Scholar] [CrossRef]
- Reverberi, A.P.; Kuznetsov, N.T.; Meshalkin, V.P.; Salerno, M.; Fabiano, B. Systematical analysis of chemical methods in metal nanoparticle synthesis. Theor. Found. Chem. Eng. 2016, 50, 59–66. [Google Scholar] [CrossRef]
- Vautrin-Ul, C. Overview of the field. In Nanosciences and Nanotechnology: Evolution or Revolution? Lourtioz, J.-M., Lahmani, M., Dupas-Haeberlin, C., Hesto, P., Eds.; Springer: Basel, Switzerland, 2016; pp. 113–174. [Google Scholar]
- Tamilvanan, A.; Balamurugan, K.; Ponappa, K.; Kumar, B.M. Copper nanoparticles: Synthetic strategies, properties and multifunctional application. Int. J. Nanosci. 2014, 13, 1430001. [Google Scholar] [CrossRef]
- De Rademaeker, E.; Suter, G.; Pasman, H.J.; Fabiano, B. A review of the past, present and future of the European Loss Prevention and Safety Promotion in the Process Industries. Process Saf. Environ. 2014, 92, 280–291. [Google Scholar] [CrossRef]
- Parveen, F.; Sannakki, B.; Mandke, M.V.; Pathan, H.M. Copper nanoparticles: Synthesis methods and its light harvesting performance. Sol. Energy Mater. Sol. Cells 2016, 144, 371–382. [Google Scholar] [CrossRef]
- Toudert, J.; Serna, R.; de Castro, M.J. Exploring the optical potential of nano-bismuth: Tunable surface plasmon resonances in the near ultraviolet-to-near infrared range. J. Phys. Chem. C 2012, 116, 20530–20539. [Google Scholar] [CrossRef]
- Khan, B.A.A.; Megarajan, S.; Suresh, K.P.; Anbazhagan, V. Highly selective colorimetric cysteine sensor based on the formation of cysteine layer on copper nanoparticles. Sens. Actuator B 2016, 233, 431–437. [Google Scholar]
- Chaudhari, M.B.; Mohammed, W.S.; Hornyak, G.L.; Dutta, J. Chromatic tuning of plasmon resonance of tri-layered composites: Silver, gold and copper nanoparticles for optical thin film colour filter. Micro Nano Lett. 2012, 7, 146–148. [Google Scholar] [CrossRef]
- Luechinger, N.A.; Loher, S.; Athanassiou, E.K.; Grass, R.N.; Stark, W.J. Highly sensitive optical detection of humidity on polymer/metal nanoparticle hybrid films. Langmuir 2007, 23, 3473–3477. [Google Scholar] [CrossRef] [PubMed]
- Marras, C.; Loche, D.; Carta, D.; Casula, M.F.; Schirru, M.; Cutrufello, M.G.; Corrias, A. Copper-based catalysts supported on highly porous silica for the Water Gas Shift reaction. ChemPlusChem 2016, 81, 421–432. [Google Scholar] [CrossRef]
- Gawande, M.B.; Goswami, A.; Felpin, F.-X.; Asefa, T.; Huang, X.; Silva, R.; Zou, X.; Zboril, R.; Varma, R.S. Cu and Cu-Based Nanoparticles: Synthesis and Applications in Catalysis. Chem. Rev. 2016, 116, 3722–3811. [Google Scholar] [CrossRef] [PubMed]
- Umer, A.; Naveed, S.; Ramzan, N.; Rafique, M.S. Selection of a suitable method for the synthesis of copper nanoparticles. Nano 2012, 7, 1230005. [Google Scholar] [CrossRef]
- Primerano, P.; Milazzo, M.F.; Risitano, F.; Matarazzo, A. Sustainable improvement of the tetrabromoethylcyclohexane synthesis using Amino ILs as Catalysts in Water. A facile and environmentally-friendly procedure. J. Chem. Technol. Biotechnol. 2016, 91, 1274–1279. [Google Scholar] [CrossRef]
- Li, M.; Xiang, K.; Luo, G.; Gong, D.; Shen, Q.; Zhang, L. Preparation of monodispersed copper nanoparticles by an environmentally friendly chemical reduction. Chin. J. Chem. 2013, 31, 1285–1289. [Google Scholar] [CrossRef]
- Tan, K.S.; Cheong, K.Y. Advances of Ag, Cu and Ag-Cu alloy nanoparticles synthesized via chemical reduction route. J. Nanopart. Res. 2013, 15, 1–29. [Google Scholar] [CrossRef]
- Magdassi, S.; Grouchko, M.; Kamyshny, A. Copper nanoparticles for printed electronics: Routes towards achieving oxidation stability. Materials 2010, 3, 4626–4638. [Google Scholar] [CrossRef]
- Bashir, O.; Hussain, S.; Al-Thabaiti, S.A.; Khan, Z. Synthesis, optical properties stability and encapsulation of Cu-nanoparticles. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2015, 140, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.S.; Ragupathy, R.; Mandal, A.B. Self-association, mixed micellization, and thermodynamic studies of sodium dodecyl sulfate (SDS) and hexanediyl-1,6-bis(dimethylcetylammonium bromide) (16-6-16). J. Disper. Sci. Technol. 2016, 37, 1760–1766. [Google Scholar] [CrossRef]
- Cheng, X.; Zhang, X.; Yin, H.; Wang, A.; Xu, Y. Modifier effects on chemical reduction synthesis of nanostructured copper. Appl. Surf. Sci. 2006, 253, 2727–2732. [Google Scholar] [CrossRef]
- Ali, A.; Ansari, N.H. Studies on the effect of amino acids/peptide on micellization of SDS at different temperatures. J. Surfactants Deterg. 2010, 13, 441–449. [Google Scholar] [CrossRef]
- Tanhaei, B.; Saghatoleslami, N.; Chenar, M.P.; Ayati, A.; Hesampour, M.; Mänttäri, M. Experimental study of CMC evaluation in single and mixed surfactant systems, using the UV-Vis spectroscopic method. J. Surfactants Deterg. 2013, 16, 357–362. [Google Scholar] [CrossRef]
- Yuan, H.Z.; Luo, L.; Zhang, L.; Zhao, S.; Mao, S.Z.; Yu, J.Y.; Shen, L.F.; Du, Y.R. Aggregation of sodium dodecyl sulfate in poly(ethylene glycol) aqueous solution studied by 1H NMR spectroscopy. Colloid Polym. Sci. 2002, 280, 479–484. [Google Scholar] [CrossRef]
- Bakshi, M.S. Micelle formation by anionic and cationic surfactants in binary aqueous solvents. J. Chem. Soc. Faraday Trans. 1993, 89, 4323–4326. [Google Scholar] [CrossRef]
- Qazi, U.Y.; Kajimoto, S.; Fukumura, H. Effect of sodium dodecyl sulfate on the formation of silver nanoparticles by biphotonic reduction of silver nitrate in water. Chem. Lett. 2014, 43, 1693–1695. [Google Scholar] [CrossRef]
- De, S.; Mandal, S. Surfactant-assisted shape control of copper nanostructures. Colloids Surf. A 2013, 421, 72–83. [Google Scholar] [CrossRef]
- Cotton, F.A.; Falvello, L.R.; Llusar, R.; Libby, E.; Murillo, C.A.; Schwotzer, W. Synthesis and characterization of four vanadium (II) compounds, including vanadium (II) sulfate hexahydrate and vanadium (II) saccharinates. Inorg. Chem. 1986, 25, 3423–3428. [Google Scholar] [CrossRef]
- Brauer, G. Handbook of Preparative Inorganic Chemistry, 2nd ed.; Academic Press: New York, NY, USA, 1965. [Google Scholar]
- Leigh, G.J.; De Souza, J.S. New chemistry of vanadium. Coord. Chem. Rev. 1996, 154, 71–81. [Google Scholar] [CrossRef]
- Nagy, Z. Electrochemical Synthesis of Inorganic Compounds. A Bibliography; Springer: New York, NY, USA, 1985. [Google Scholar]
- Fabiano, B.; Reverberi, A.P.; Del Borghi, A.; Dovì, V.G. Biodiesel production via transesterification: Process safety insights from kinetic modeling. Theor. Found. Chem. Eng. 2012, 46, 673–680. [Google Scholar] [CrossRef]
- Mittal, R.K.; Mehrotra, R.C. Estimation of vanadium in different oxidation states. Fresenius’ Z. Anal. Chem. 1965, 209, 405–409. [Google Scholar] [CrossRef]

© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reverberi, A.P.; Salerno, M.; Lauciello, S.; Fabiano, B. Synthesis of Copper Nanoparticles in Ethylene Glycol by Chemical Reduction with Vanadium (+2) Salts. Materials 2016, 9, 809. https://doi.org/10.3390/ma9100809
Reverberi AP, Salerno M, Lauciello S, Fabiano B. Synthesis of Copper Nanoparticles in Ethylene Glycol by Chemical Reduction with Vanadium (+2) Salts. Materials. 2016; 9(10):809. https://doi.org/10.3390/ma9100809
Chicago/Turabian StyleReverberi, Andrea Pietro, Marco Salerno, Simone Lauciello, and Bruno Fabiano. 2016. "Synthesis of Copper Nanoparticles in Ethylene Glycol by Chemical Reduction with Vanadium (+2) Salts" Materials 9, no. 10: 809. https://doi.org/10.3390/ma9100809
APA StyleReverberi, A. P., Salerno, M., Lauciello, S., & Fabiano, B. (2016). Synthesis of Copper Nanoparticles in Ethylene Glycol by Chemical Reduction with Vanadium (+2) Salts. Materials, 9(10), 809. https://doi.org/10.3390/ma9100809