
Use of Gypsum as a Preventive Measure for Strength Deterioration during Curing in Class F Fly Ash Geopolymer System



Abstract
:1. Introduction
2. Experimental Section
2.1. Materials

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SiO2 | Al2O3 | CaO | Na2O | K2O | MgO | MnO | TiO2 | SO3 | P2O5 | Fe2O3 | Others |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 51.8 | 20.0 | 10.1 | 0.6 | 1.0 | 2.0 | 0.1 | 1.2 | 0.9 | 1.4 | 10.3 | 0.6 |
2.2. Experimental Design and Sample Preparation
| Sample ID | Binder | Activator | Activator/binder (wt./wt.) | Curing temp. (°C) |
|---|---|---|---|---|
| Fly ash(F):gypsum(G) (wt.:wt.) | 10M NaOH(N)/Na-silicate solution(W) (wt./wt.) | |||
| F-NW | 100:0 | 1.0 | 0.4 | 60 |
| FG2-NW | 98:2 | |||
| FG4-NW | 96:4 | |||
| FG6-NW | 94:6 |
3. Results and Discussions
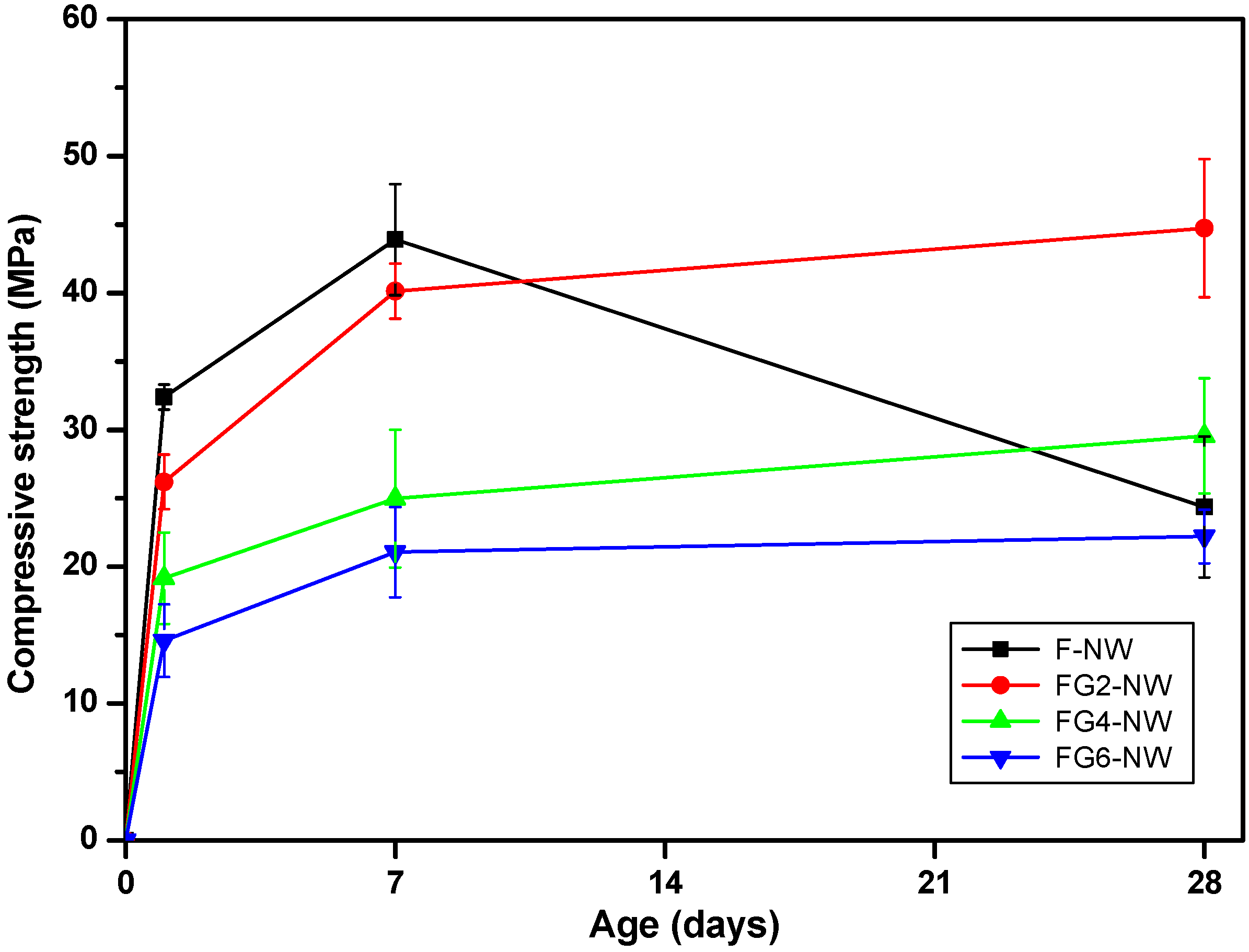
| Sample ID | Compressive strength (standard deviation), MPa | ||
|---|---|---|---|
| 1-day | 7-day | 28-day | |
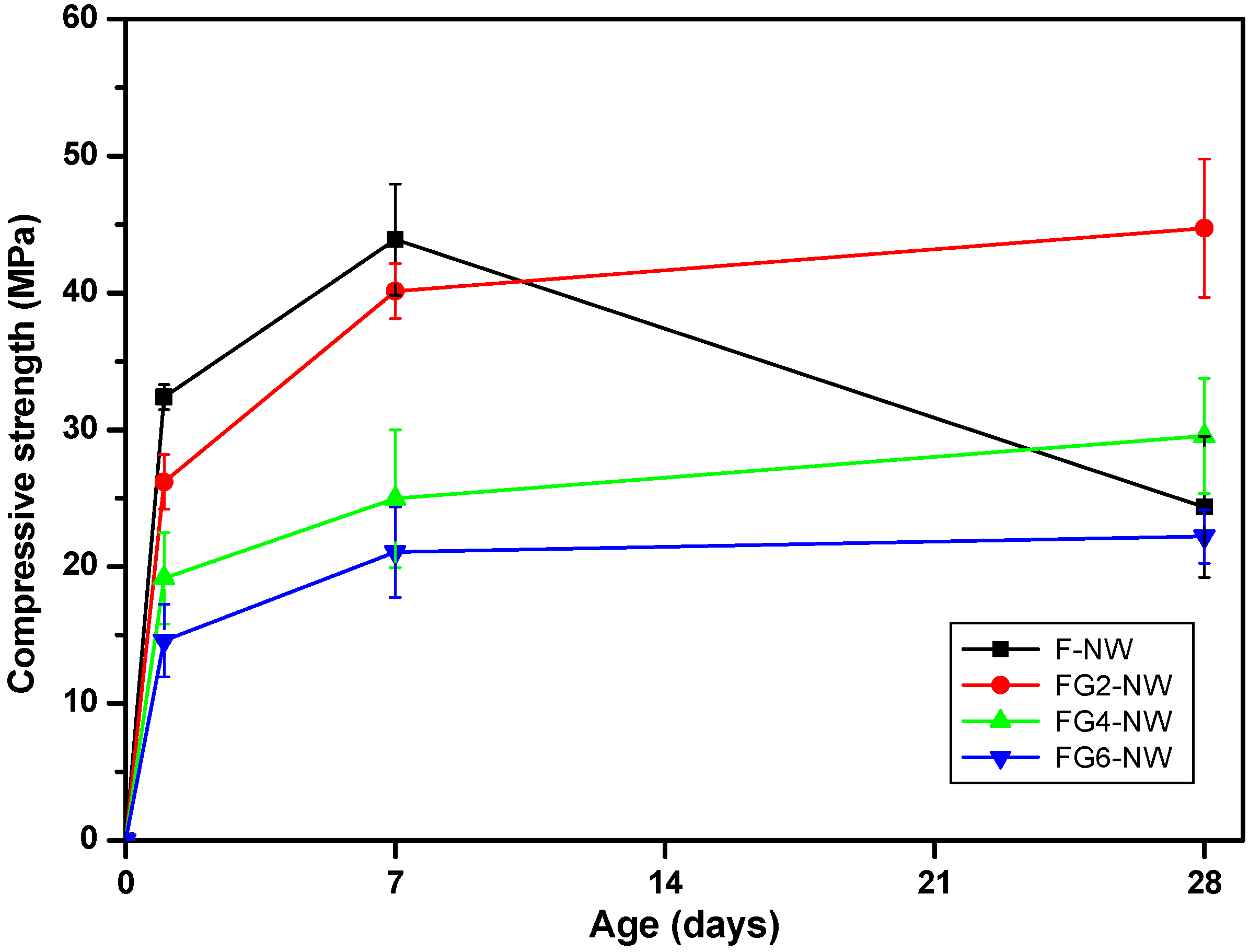
| F-NW | 32 (1) | 44 (4) | 24 (5) |
| FG2-NW | 26 (2) | 40 (2) | 45 (5) |
| FG4-NW | 19 (3) | 25 (5) | 30 (4) |
| FG6-NW | 15 (3) | 21 (3) | 22 (2) |
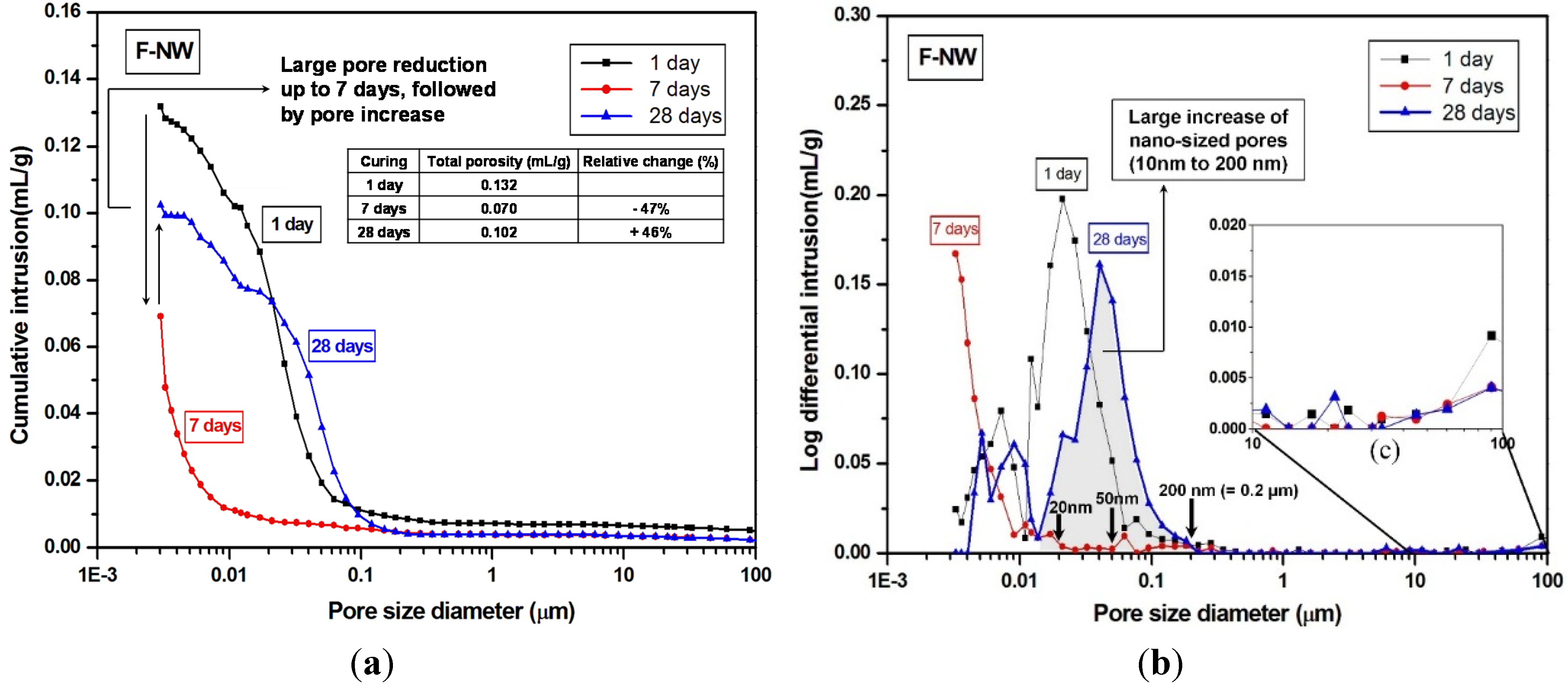
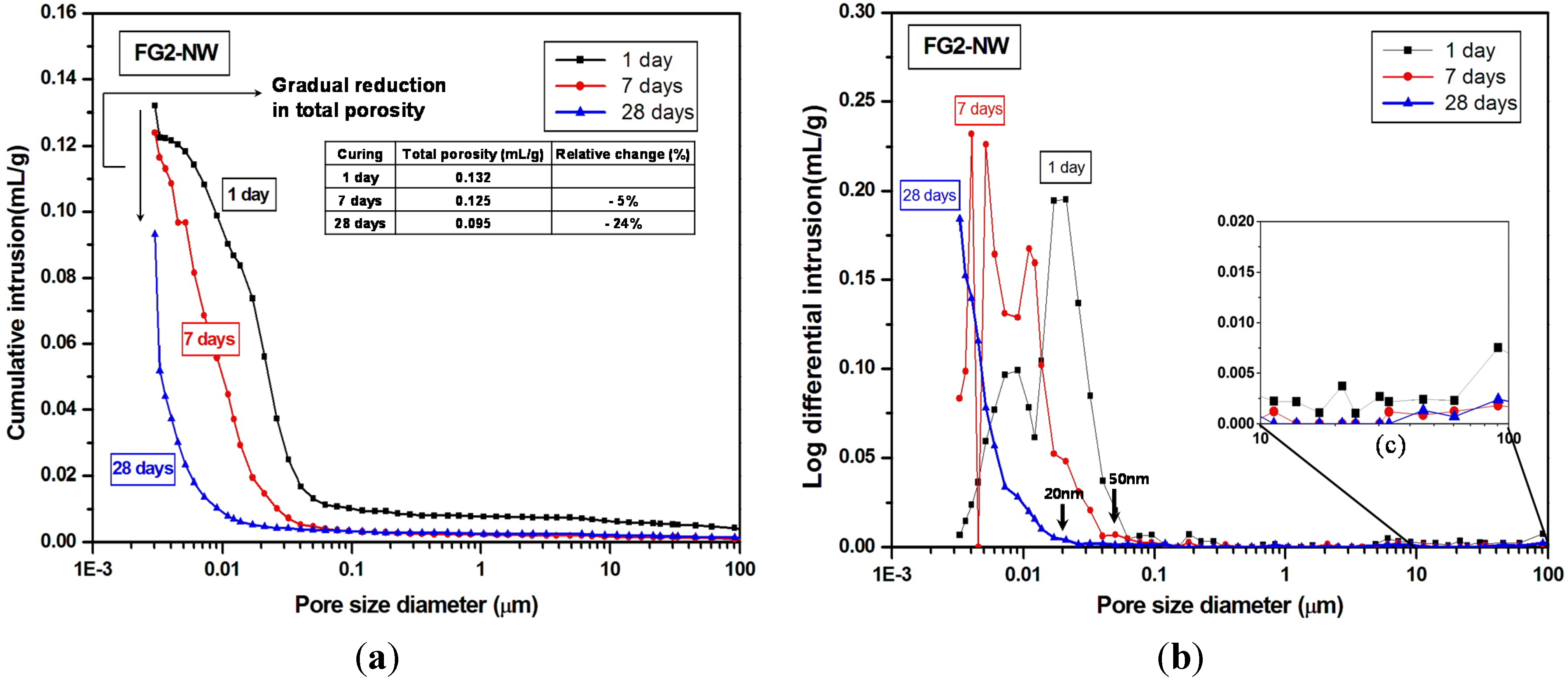
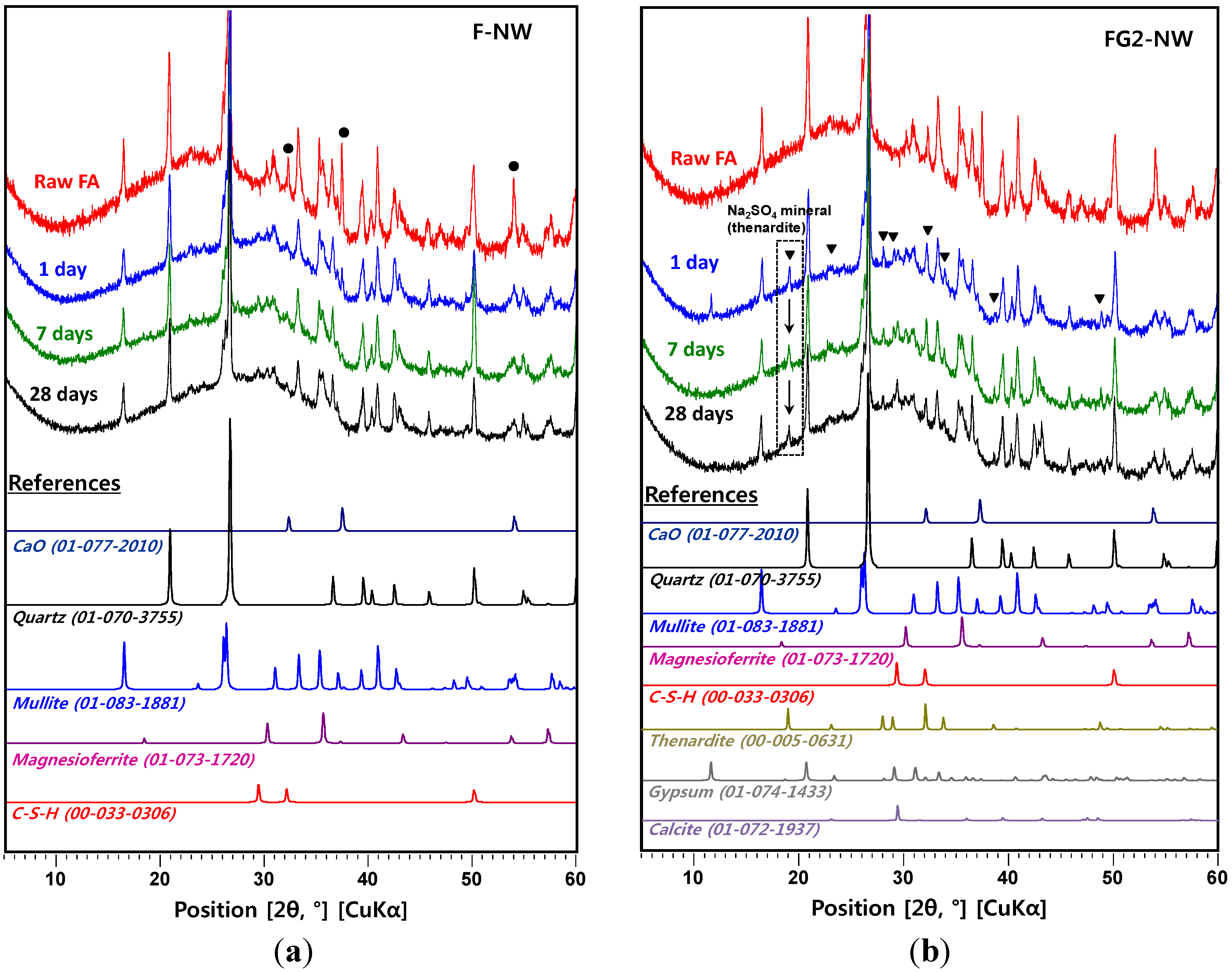
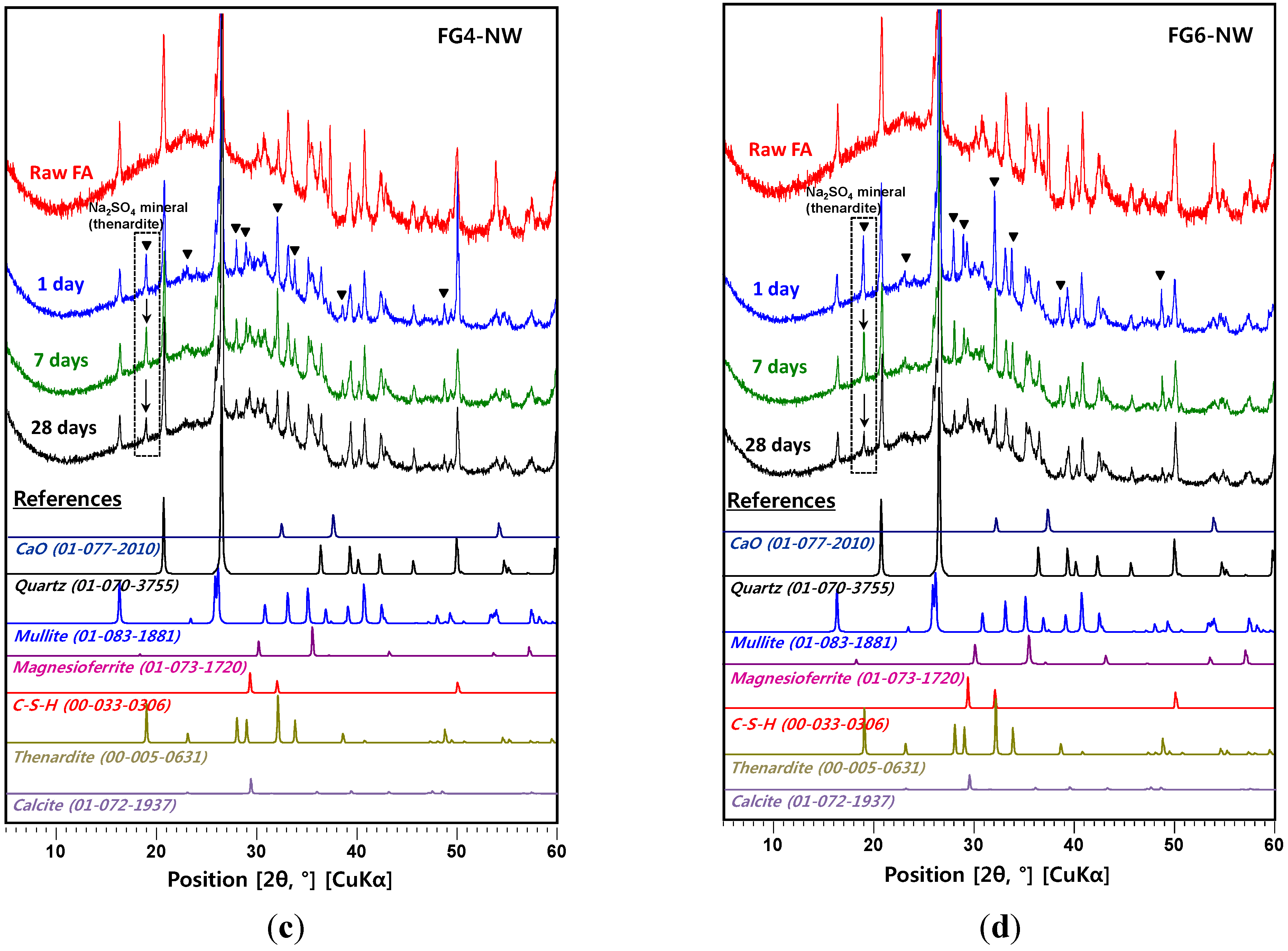
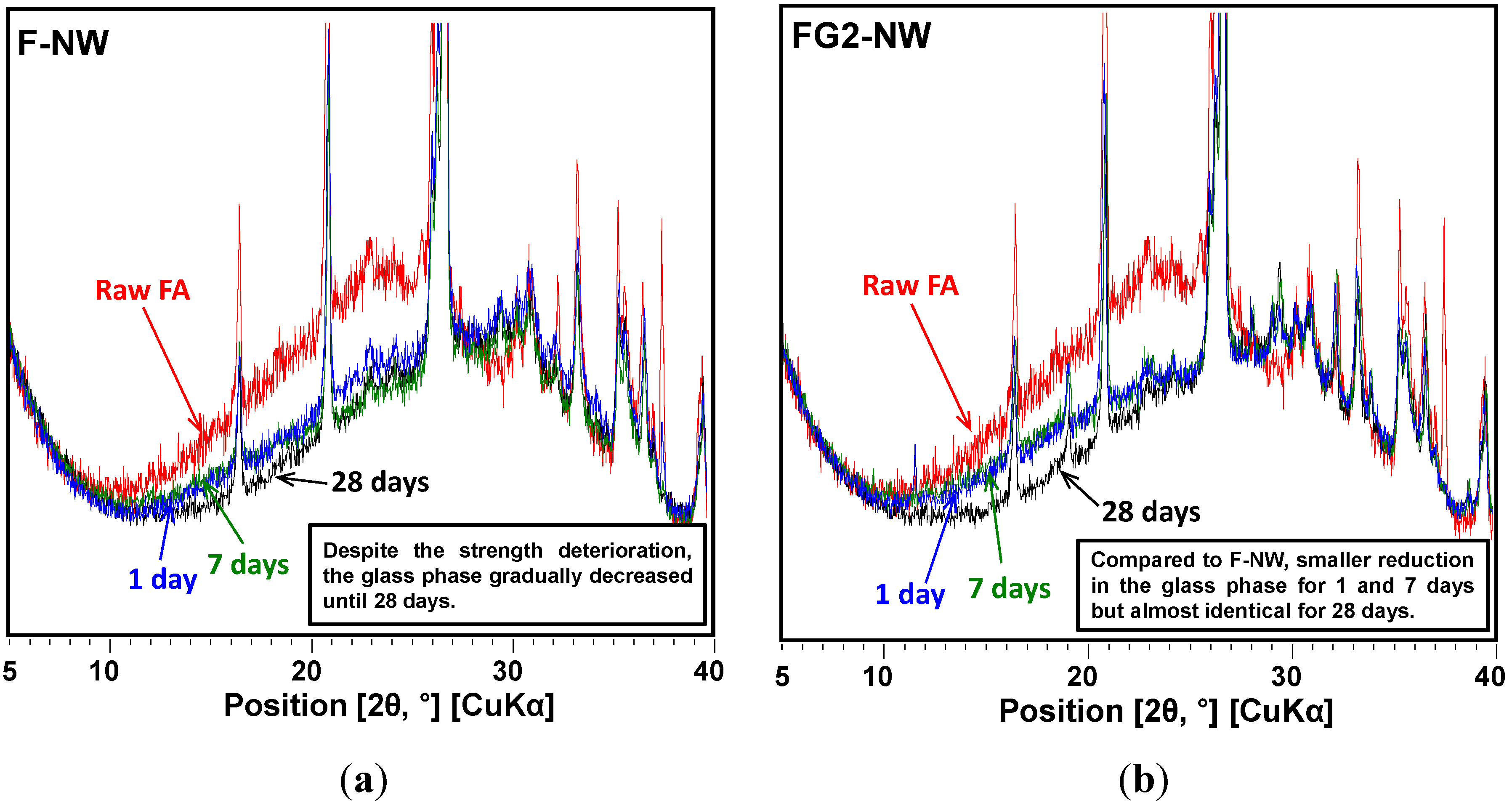
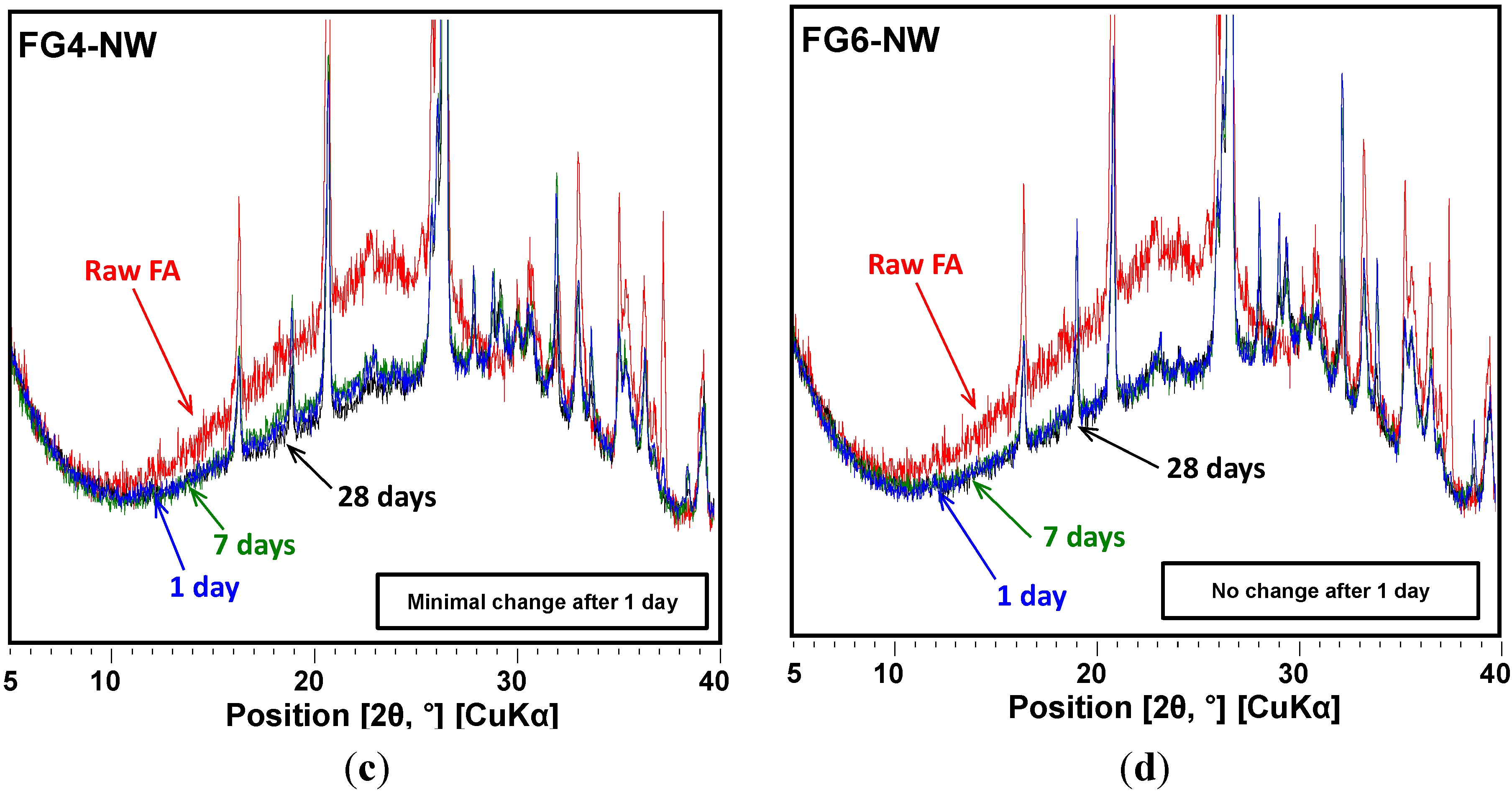

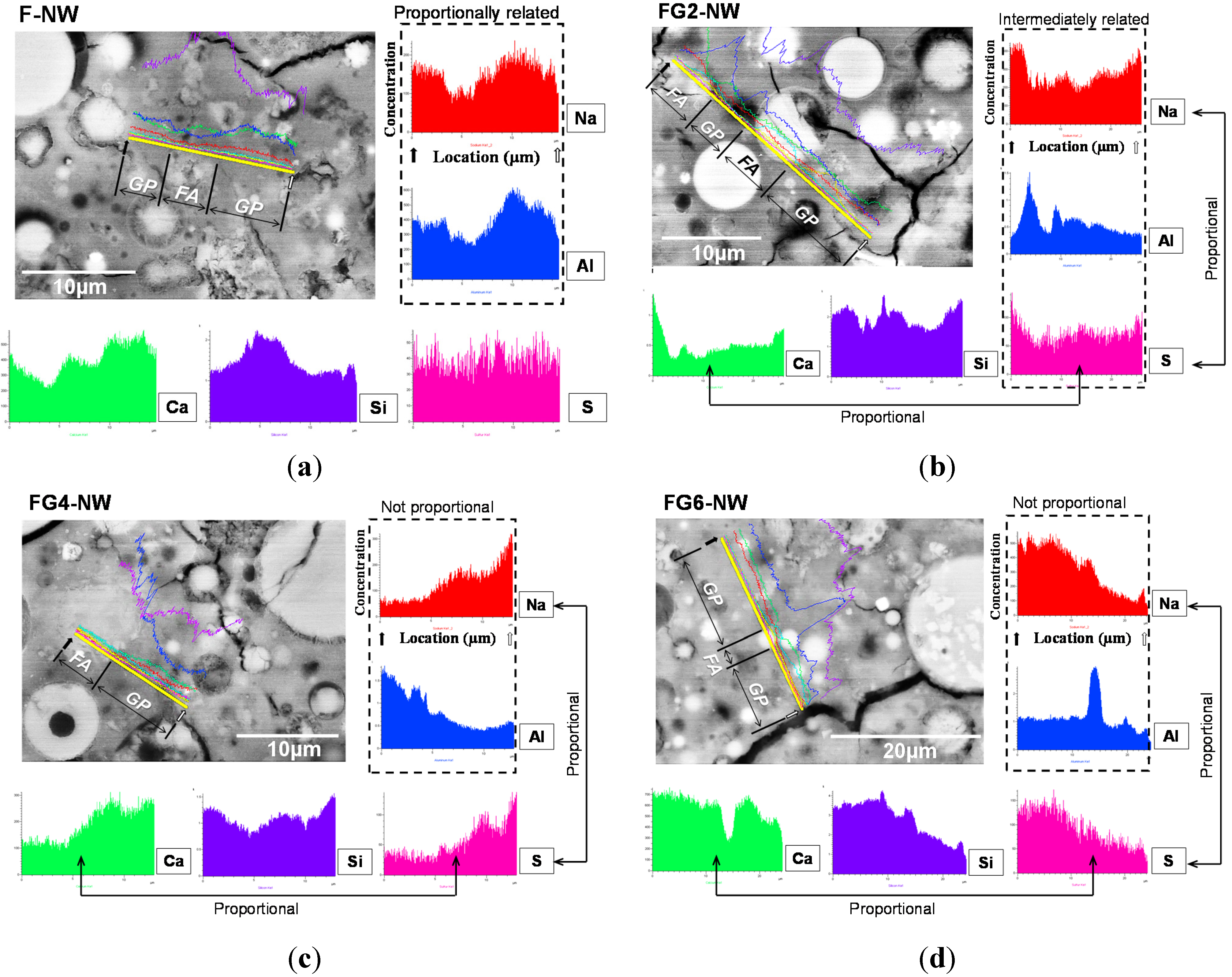
4. Conclusions
- When Class F fly ash is activated with a highly alkaline silicate solution under an elevated temperature (i.e., F-NW), the hardened matrix has a chance to show significant strength deterioration with increased curing time.
- The strength deterioration is not solely associated with the high Na concentration or the composition of the fly ash but is more likely related to a high simultaneous concentration of Na and Si ions at an early stage of geopolymer formation.
- The replacement of fly ash with 2% to 6% gypsum eliminated the strength deterioration; the 2% replacement resulted in the best strength value at 28 days; however, at higher replacement ratios, greater than 4%, a markedly lowered overall strength was observed.
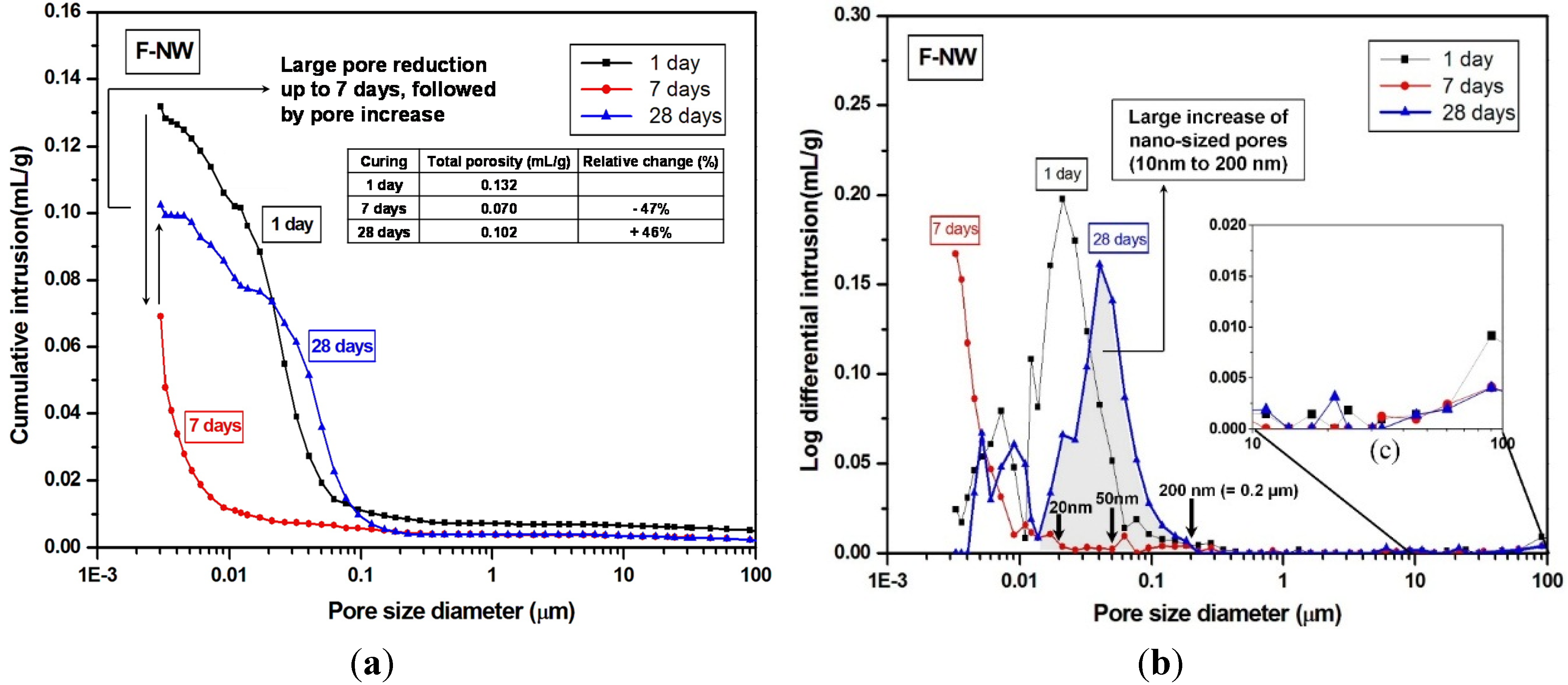
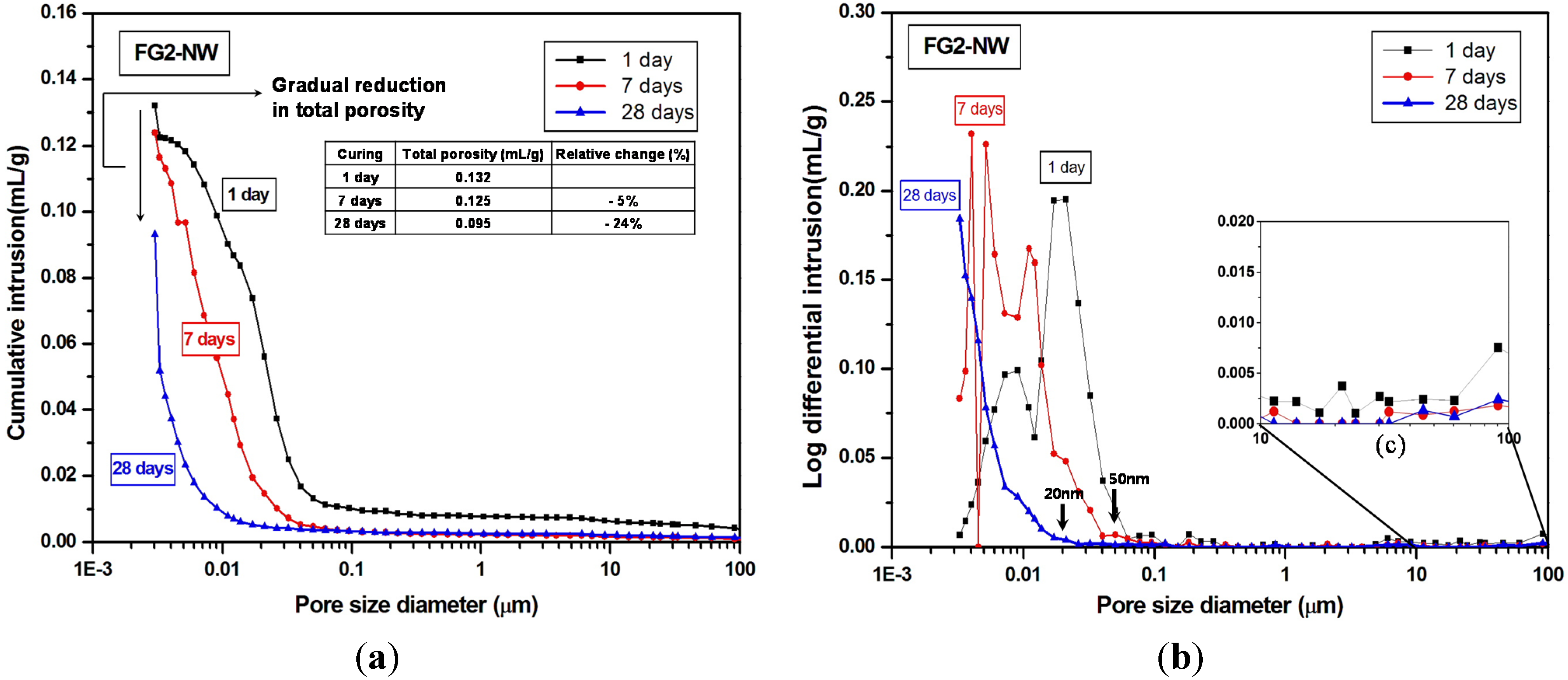
- The MIP result identified a large volume of increased porosity in F-NW after seven days, indicating extensive nano-sized crack formation (~10 to 200 nm), which is related to strength deterioration; moreover, the sample with 2% gypsum did not show any evidence of similar crack formation.
- The use of gypsum obviously delayed the evolution of pore structures over time, indirectly suggesting that the rate of the geopolymeric reaction was also slowed.
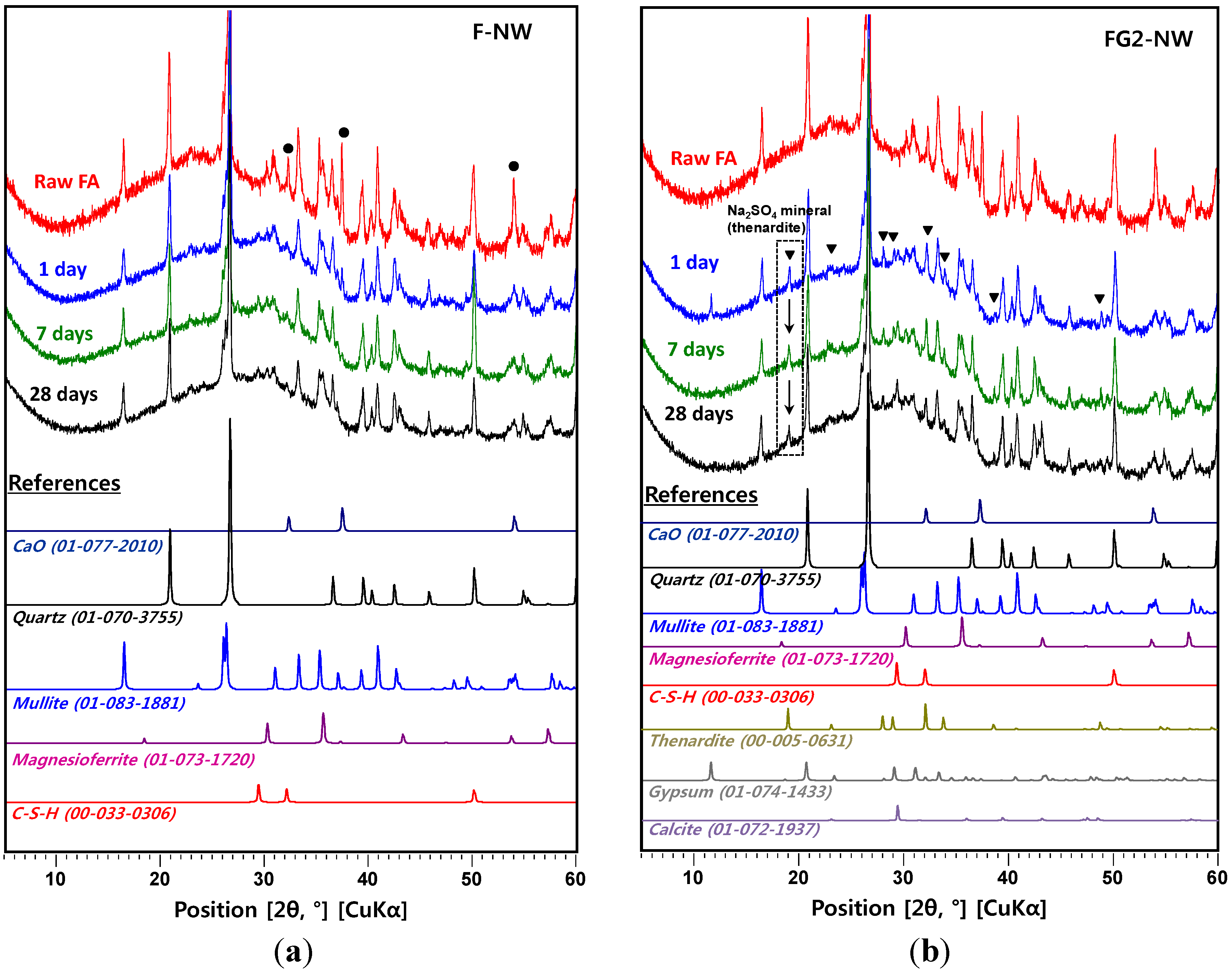
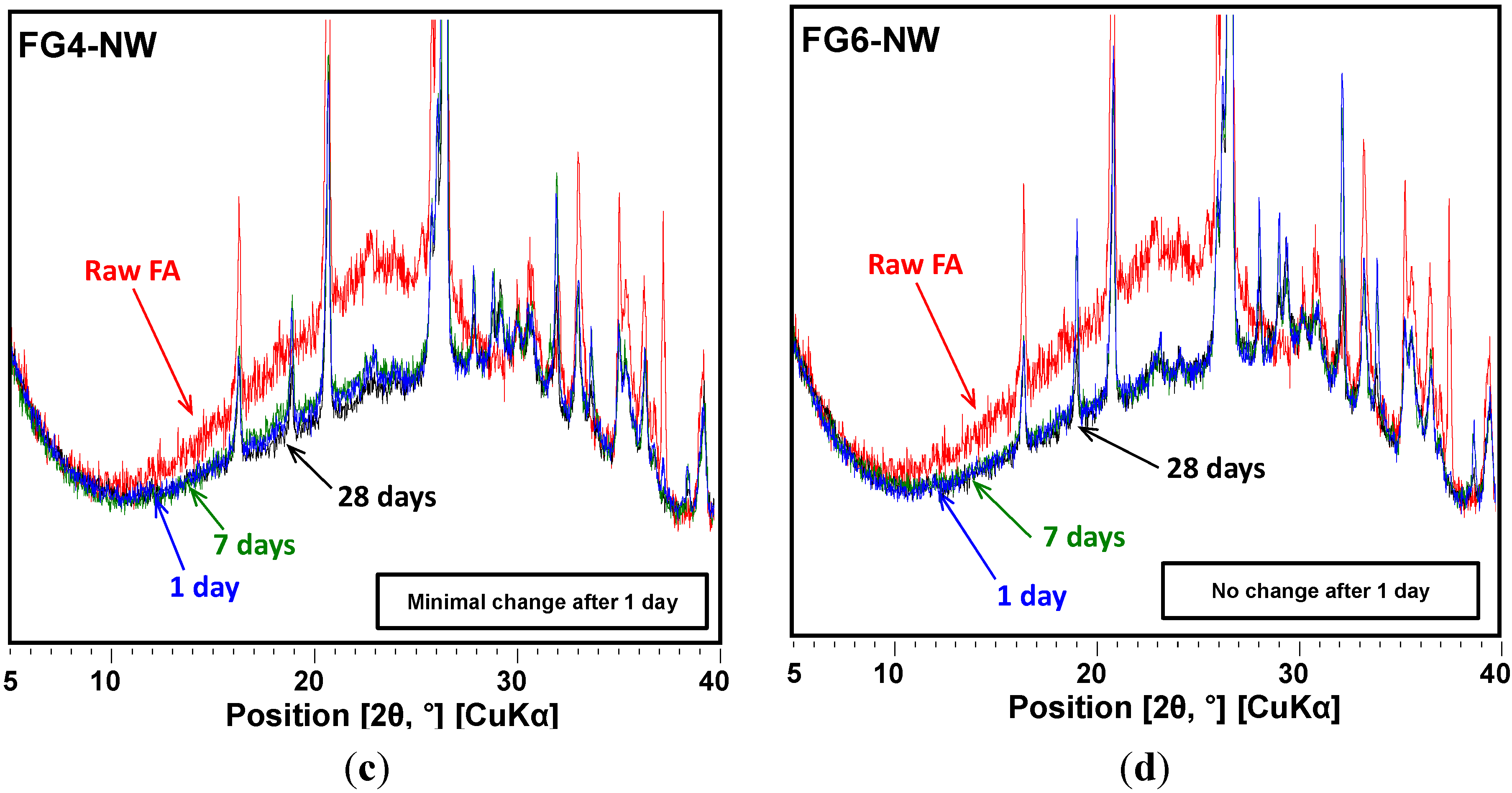
- In the XRD results, no observable crystalline phase was directly related to the strength deterioration and nano-sized cracking. The cracking was likely due to the fast growth of the nano-crystalline zeolitic phase, exerting a destructive pressure on the hardened matrix. The presence of soluble silicate largely promoted the growth rate of the zeolitic crystals; however, the sizes of the crystals formed over the 28-day curing interval were too small to be detected by XRD.
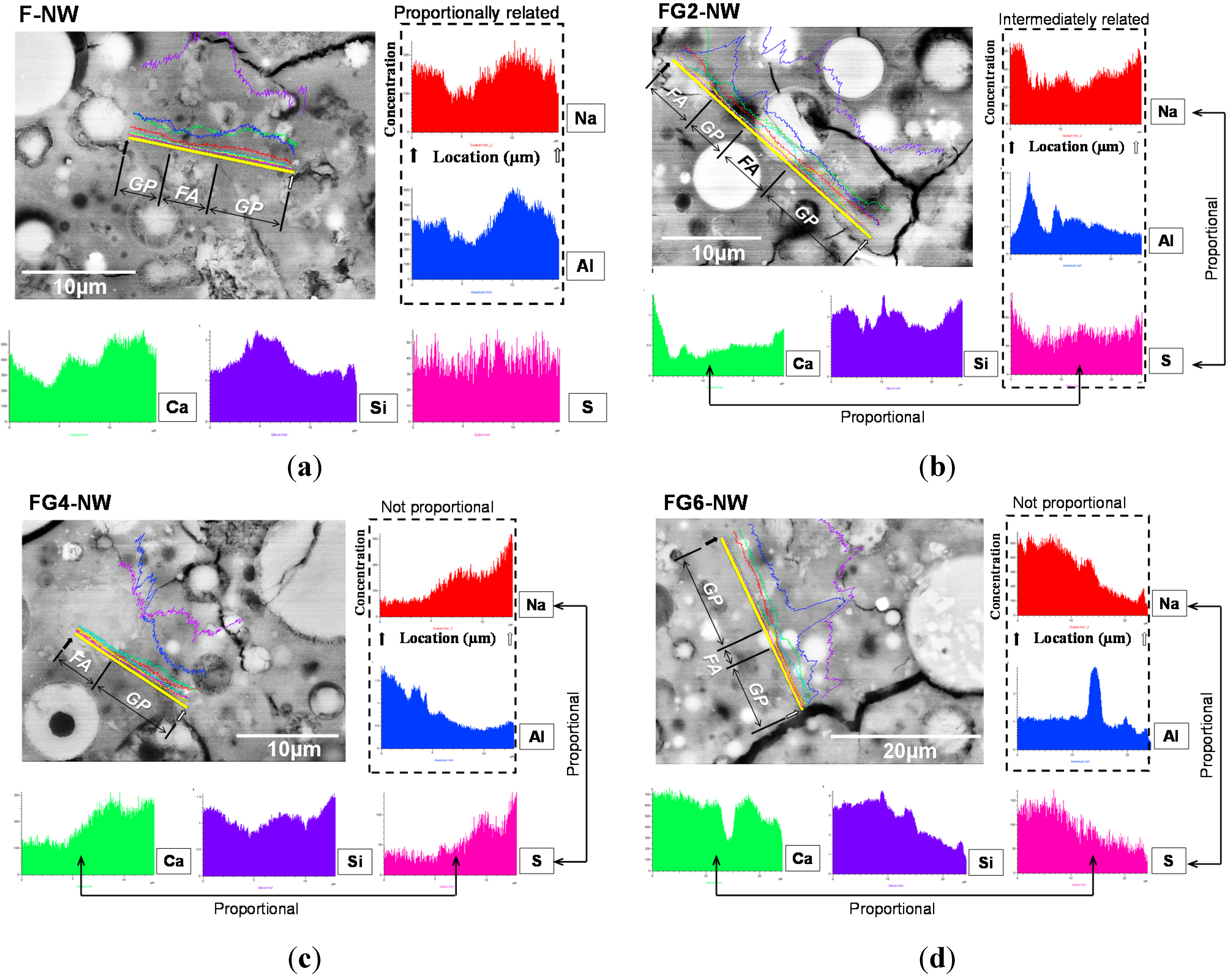
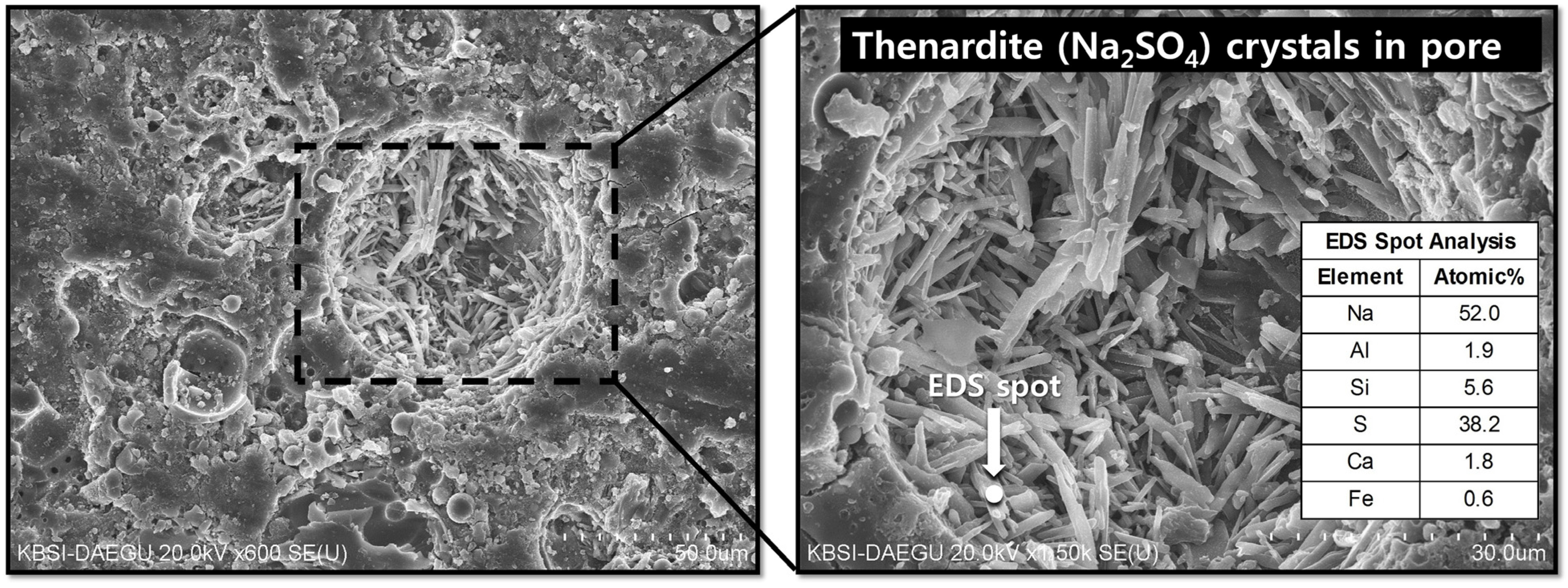
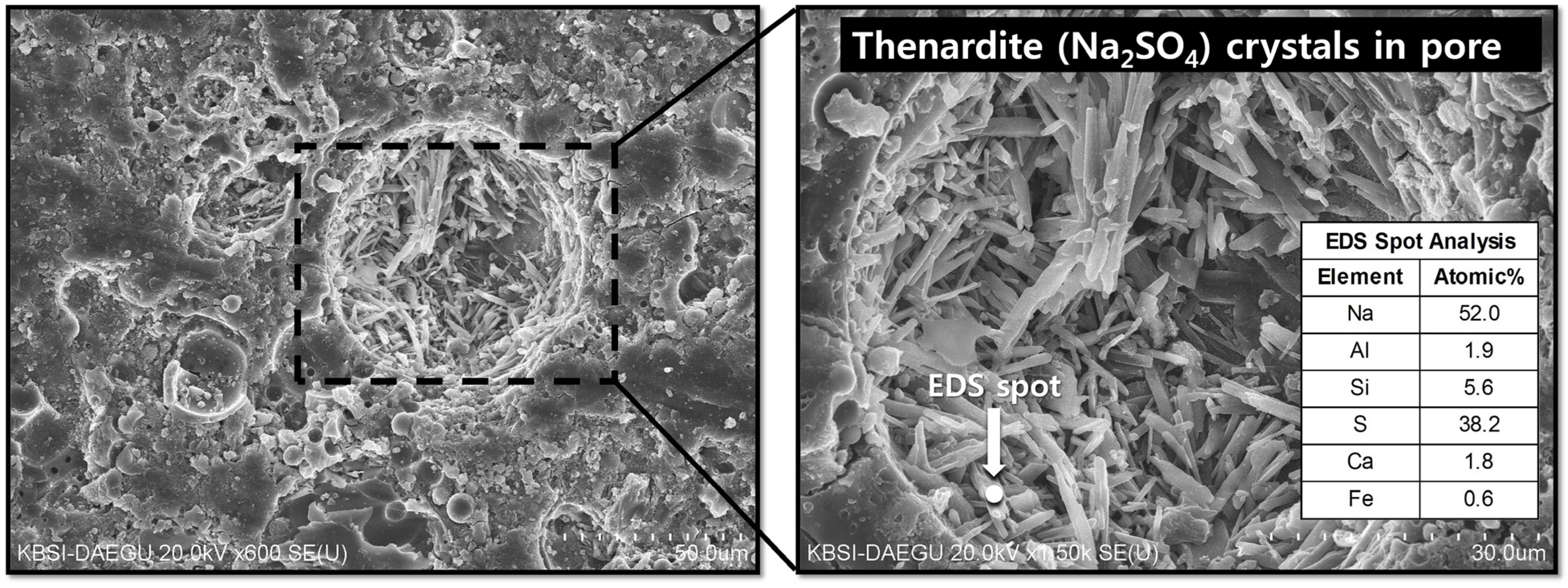
- In the samples with gypsum, a crystalline phase of Na2SO4 (thenardite) formed, suggesting that a considerable portion of Na ions were reserved by SO42−, widely forming thenardite in nanometer-sized pores at the early stages of the reaction, and were continuously released by dissolution processes as curing progresses. Thus, the addition of 2% gypsum can temporarily reduce the availability of Na+, up to ~11%, in the matrix. The small quantity of Na+ reduction appears to assist in preventing the strength deterioration, possibly due to a significant reduction in the growth rate of zeolitic crystals.
- The gypsum addition not only affected the geopolymeric reaction by reserving Na ions, but also delayed the glass dissolution process, which is a Si provider. When a proper amount of gypsum was used, the strength deterioration might have been prevented by temporarily reducing excessive Si concentration during geopolymer formation.
- The elemental line scanning results of the SEM work illustrated that the synthesized Na2SO4 phase was widely dispersed in the geopolymer, implying extensive reservation of Na+ by SO42−; when gypsum addition was excessive, a substantial amount of Na ions were eliminated without participating in the geopolymeric reaction.
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Part, W.K.; Ramli, M.; Cheah, C.B. An overview on the influence of various factors on the properties of geopolymer concrete derived from industrial by-products. Constr. Build. Mater. 2015, 77, 370–395. [Google Scholar] [CrossRef]
- Singh, B.; Ishwarya, G.; Gupta, M.; Bhattacharyya, S.K. Geopolymer concrete: A review of some recent developments. Constr. Build. Mater. 2015, 85, 78–90. [Google Scholar] [CrossRef]
- Rattanasak, U.; Chindaprasirt, P. Influence of NaOH solution on the synthesis of fly ash geopolymer. Miner. Eng. 2009, 22, 1073–1078. [Google Scholar] [CrossRef]
- Guo, X.; Shi, H.; Dick, W.A. Compressive strength and microstructural characteristics of class c fly ash geopolymer. Cem. Concr. Comp. 2010, 32, 142–147. [Google Scholar] [CrossRef]
- Somna, K.; Jaturapitakkul, C.; Kajitvichyanukul, P.; Chindaprasirt, P. NaOH-activated ground fly ash geopolymer cured at ambient temperature. Fuel. 2011, 90, 2118–2124. [Google Scholar] [CrossRef]
- Palomo, A.; Grutzeck, M.W.; Blanco, M.T. Alkali-activated fly ashes: a cement for the future. Cem. Concr. Res. 1999, 29, 1323–1329. [Google Scholar] [CrossRef]
- Duxson, P.; Fernández-Jiménez, A.; Provis, J.L.; Lukey, G.C.; Palomo, A.; van Deventer, J.S.J. Geopolymer technology: The current state of the art. J. Mater. Sci. 2007, 42, 2917–2933. [Google Scholar] [CrossRef]
- Boonserm, K.; Sata, V.; Pimraksa, K.; Chindaprasirt, P. Microstructure and strength of blended FBC-PCC fly ash geopolymer containing gypsum as an additive. ScienceAsia 2012, 38, 175–181. [Google Scholar] [CrossRef]
- Bakharev, T. Geopolymeric materials prepared using class f fly ash and elevated temperature curing. Cem. Concr. Res. 2005, 35, 1224–1232. [Google Scholar] [CrossRef]
- Zhang, Z.; Yao, X.; Zhu, H.; Chen, Y. Role of water in the synthesis of calcined kaolin-based geopolymer. Appl. Clay Sci. 2009, 43, 218–223. [Google Scholar]
- Wongpa, J.; Kiattikomol, K.; Jaturapitakkul, C.; Chindaprasirt, P. Compressive strength, modulus of elasticity, and water permeability of inorganic polymer concrete. Mater. Des. 2010, 31, 4748–4754. [Google Scholar] [CrossRef]
- De Silva, P.; Sagoe-Crenstil, K. Medium-term phase stability of Na2O–Al2O3–SiO2–H2O geopolymer systems. Cem. Concr. Res. 2008, 38, 870–876. [Google Scholar] [CrossRef]
- Boonserm, K.; Sata, V.; Pimraksa, K.; Chindaprasirt, P. Improved geopolymerization of bottom ash by incorporating fly ash and using waste gypsum as additive. Cem. Concr. Comp. 2012, 34, 819–824. [Google Scholar] [CrossRef]
- Rattanasak, U.; Pankhet, K.; Chindaprasirt, P. Effect of chemical admixtures on properties of high-calcium fly ash geopolymer. Int. J. Miner. Metall. Mater. 2011, 18, 364–369. [Google Scholar] [CrossRef]
- Zhang, J.; Scherer, G.W. Comparison of methods for arresting hydration of cement. Cem. Concr. Res. 2011, 41, 1024–1036. [Google Scholar] [CrossRef]
- X'Pert HighScore Plus; PANalytical: Almelo, The Netherlands, 2012.
- PDF-2 Database; International Center for Diffraction Data (ICDD): Newton Square, PA, USA, 2000.
- Oh, J.E.; Monteiro, P.J.M.; Jun, S.S.; Choi, S.; Clark, S.M. The evolution of strength and crystalline phases for alkali-activated ground blast furnace slag and fly ash-based geopolymers. Cem. Concr. Res. 2010, 40, 189–196. [Google Scholar] [CrossRef]
- Oh, J.E.; Jun, Y.; Jeong, Y. Characterization of geopolymers from compositionally and physically different class f fly ashes. Cem. Concr. Comp. 2014, 50, 16–26. [Google Scholar] [CrossRef]
- Mehta, P.K.; Monteiro, P.J.M. Concrete: Microstructure, Properties, and Materials; McGraw-Hill: New York, NY, USA, 2006. [Google Scholar]
- Provis, J.L.; Lukey, G.C.; van Deventer, J.S.J. Do geopolymers actually contain nanocrystalline zeolites? A reexamination of existing results. Chem. Mater. 2005, 17, 3075–3085. [Google Scholar] [CrossRef]
- Criado, M.; Fernández-Jiménez, A.; Palomo, A. Alkali Activation of fly ash: Effect of the SiO2/Na2O ratio part I: FTIR study. Micropor. Mesopor. Mater. 2007, 106, 180–191. [Google Scholar] [CrossRef]
- Yu, S.; Oguchi, C.T. Is Sheer thenardite attack impotent compared with cyclic conversion of thenardite–mirabilite mechanism in laboratory simulation tests? Eng. Geol. 2013, 152, 148–154. [Google Scholar] [CrossRef]
- Popović, D.; Stupar, G.; Miladinović, J.; Todorović, M.; Zrilić, M. Solubility in the ternary system CaSO4+ Na2SO4+ H2O at 298.15 K1. Russ. J. Phys. Chem. A 2011, 85, 2349–2353. [Google Scholar] [CrossRef]
- Babushkin, V.I.; Matveev, G.M.; Mchedlov-Petrosian, O.P. Thermodynamics of Silicates; Springer-Verlag: Berlin, Germany, 1985. [Google Scholar]
- Alexander, G.B.; Heston, W.; Iler, R.K. The solubility of amorphous silica in water. J. Phys. Chem. 1954, 58, 453–455. [Google Scholar] [CrossRef]
- Nath, P.; Sarker, P.K. Effect of GGBFS on Setting, Workability and early strength properties of fy aash geopolymer concrete cured in ambient condition. Constr. Build. Mater. 2014, 66, 163–171. [Google Scholar] [CrossRef]
- Nath, P.; Sarker, P.K. Use of OPC to improve setting and early strength properties of low calcium fly ash geopolymer concrete cured at room temperature. Cem. Concr. Comp. 2015, 55, 205–214. [Google Scholar] [CrossRef]
- Davidovits, J. Geopolymer Chemistry and Properties; Geopolymer: Compiegne, France, 1988; pp. 25–48. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jun, Y.; Oh, J.E. Use of Gypsum as a Preventive Measure for Strength Deterioration during Curing in Class F Fly Ash Geopolymer System. Materials 2015, 8, 3053-3067. https://doi.org/10.3390/ma8063053
Jun Y, Oh JE. Use of Gypsum as a Preventive Measure for Strength Deterioration during Curing in Class F Fly Ash Geopolymer System. Materials. 2015; 8(6):3053-3067. https://doi.org/10.3390/ma8063053
Chicago/Turabian StyleJun, Yubin, and Jae Eun Oh. 2015. "Use of Gypsum as a Preventive Measure for Strength Deterioration during Curing in Class F Fly Ash Geopolymer System" Materials 8, no. 6: 3053-3067. https://doi.org/10.3390/ma8063053
APA StyleJun, Y., & Oh, J. E. (2015). Use of Gypsum as a Preventive Measure for Strength Deterioration during Curing in Class F Fly Ash Geopolymer System. Materials, 8(6), 3053-3067. https://doi.org/10.3390/ma8063053