
Evaluation of Lapatinib Powder-Entrapped Biodegradable Polymeric Microstructures Fabricated by X-Ray Lithography for a Targeted and Sustained Drug Delivery System



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Fabrication Results of Lapatinib Powder-Entrapped Microstructures
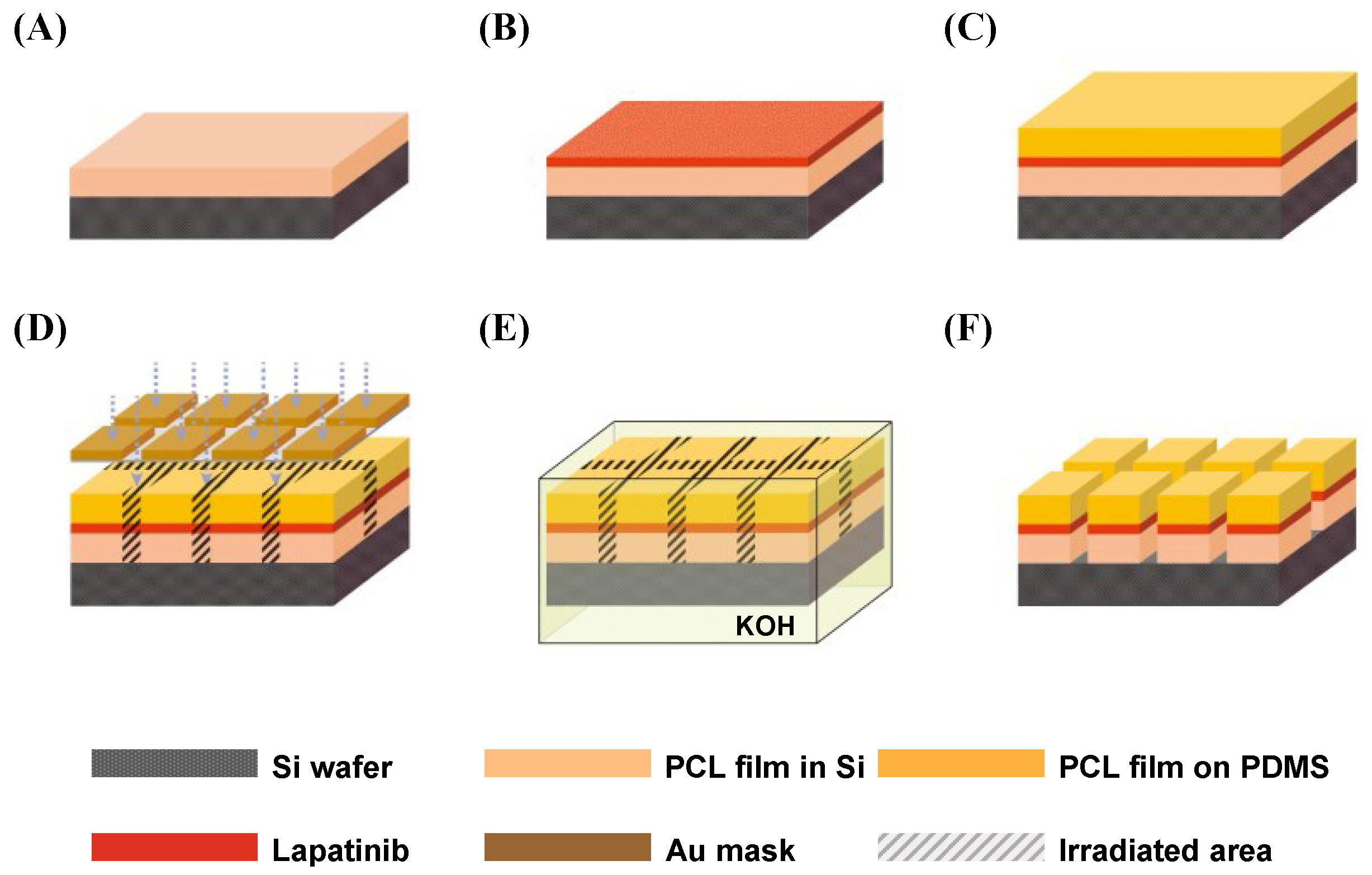

2.2. Evaluation of Lapatinib Powder-Entrapped Microstructure Activity
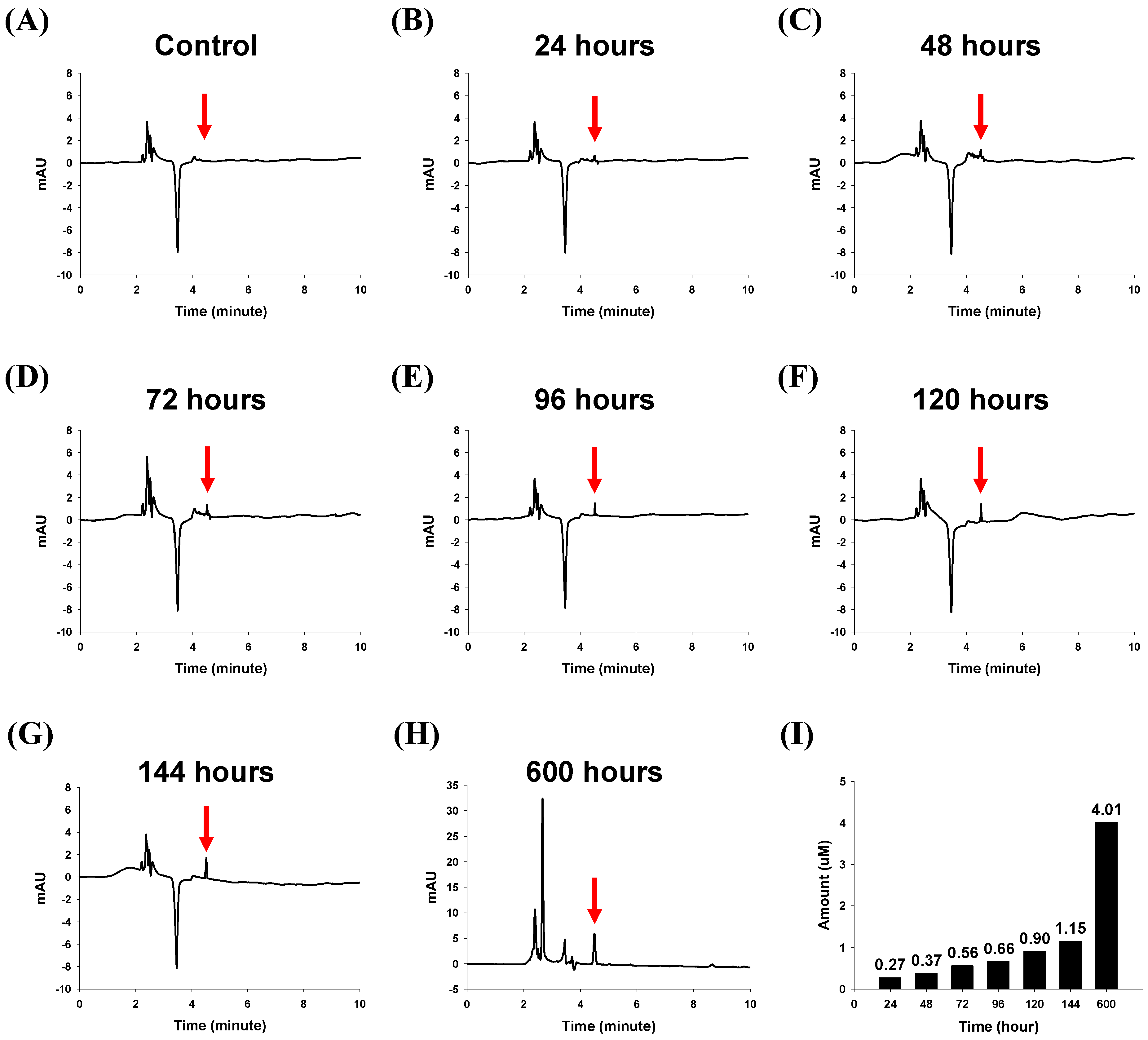
2.2.1. In Vitro Drug Release Characteristics of the Lapatinib Powder-Entrapped Microstructures

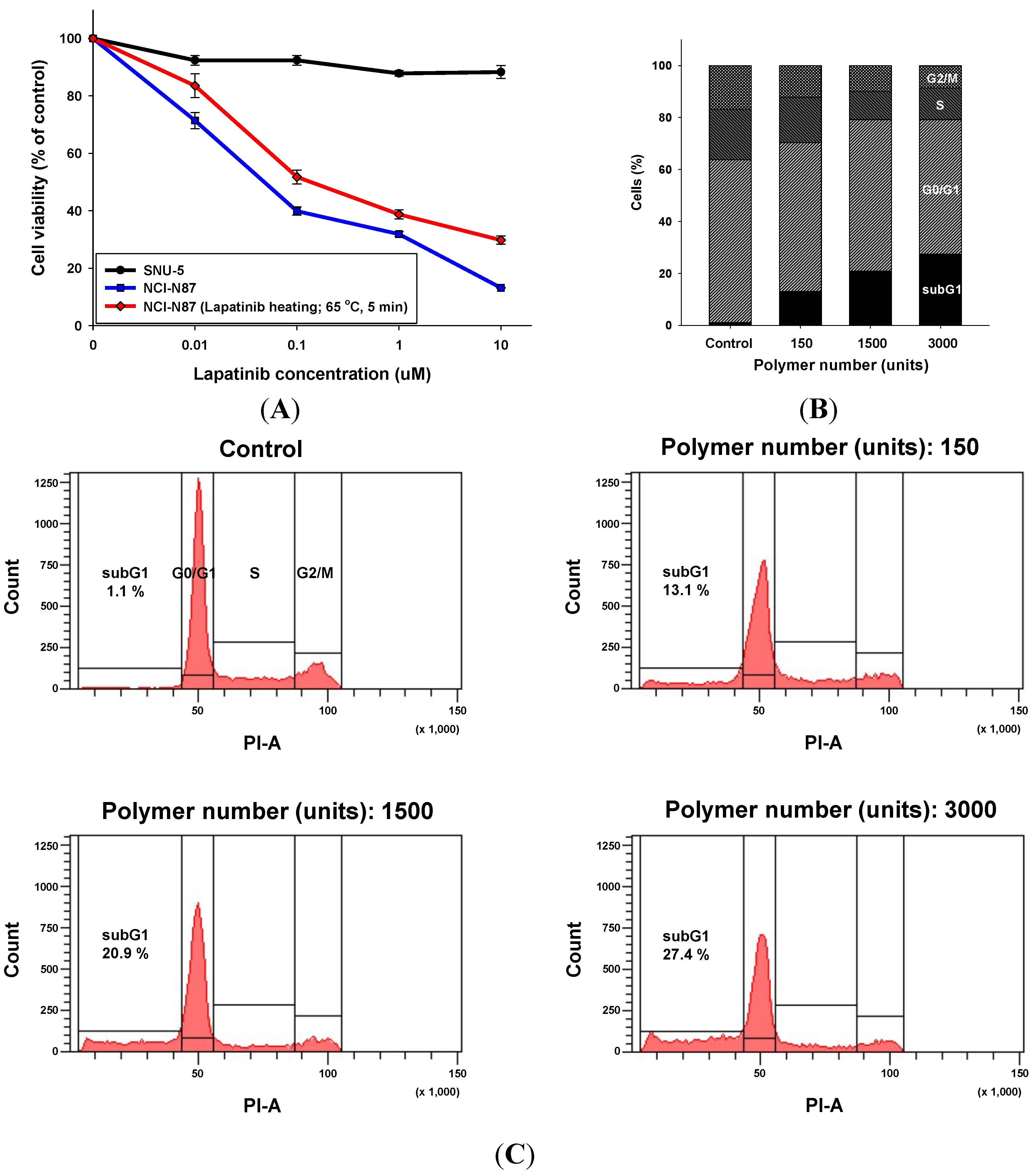
2.2.2. Growth Inhibitory Activity in HER2-Amplified Gastric Cancer Cell Lines by Lapatinib after the Lamination Process
2.2.3. Apoptotic Cell Death by the Lapatinib Powder-Entrapped Microstructures

2.3. Drug Release Efficiency of the Lapatinib Powder-Entrapped Microstructures
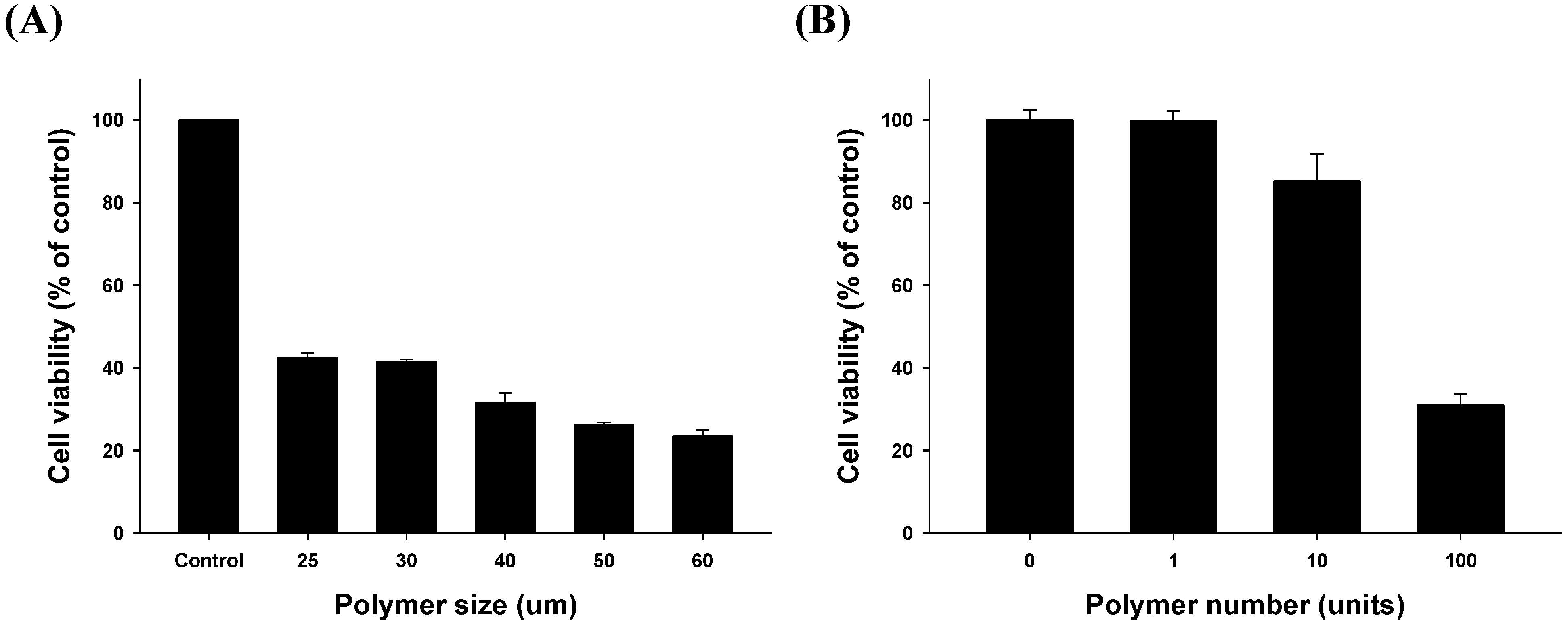
2.3.1. Size- and Dose-Related Drug Release Efficiency Test
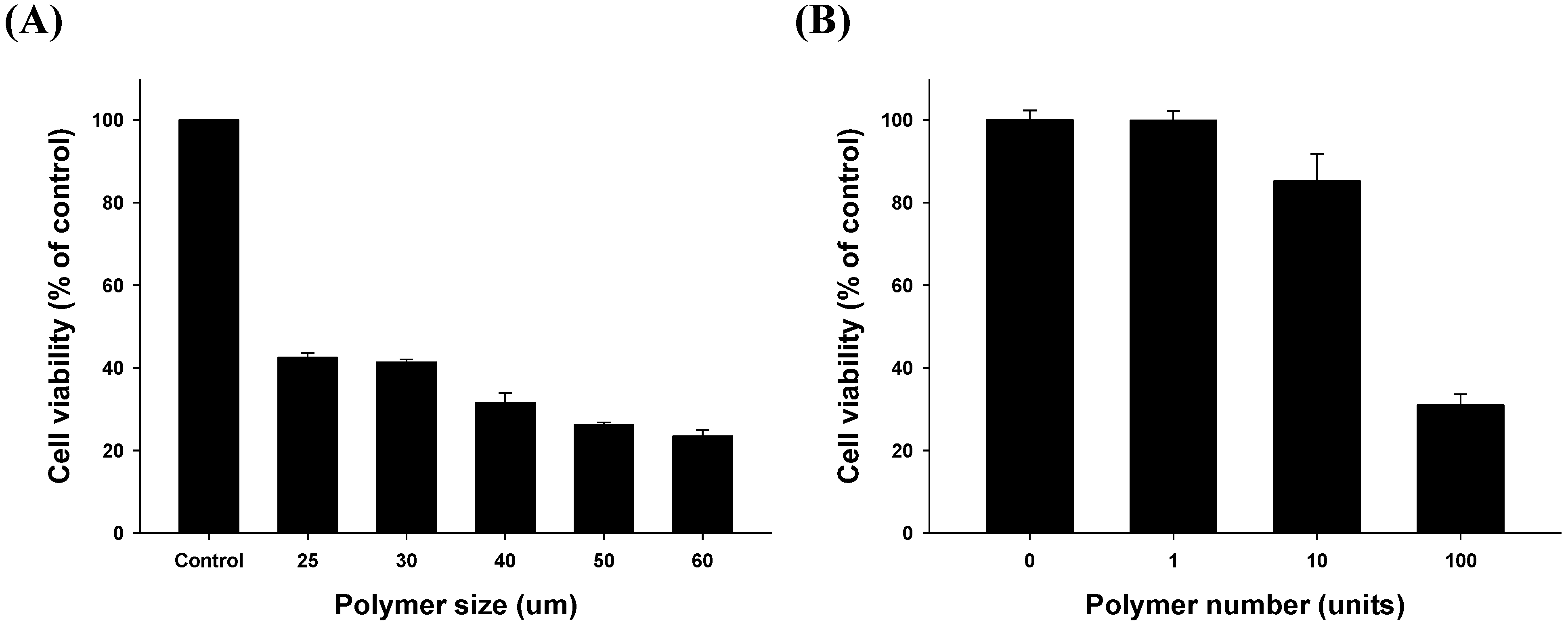
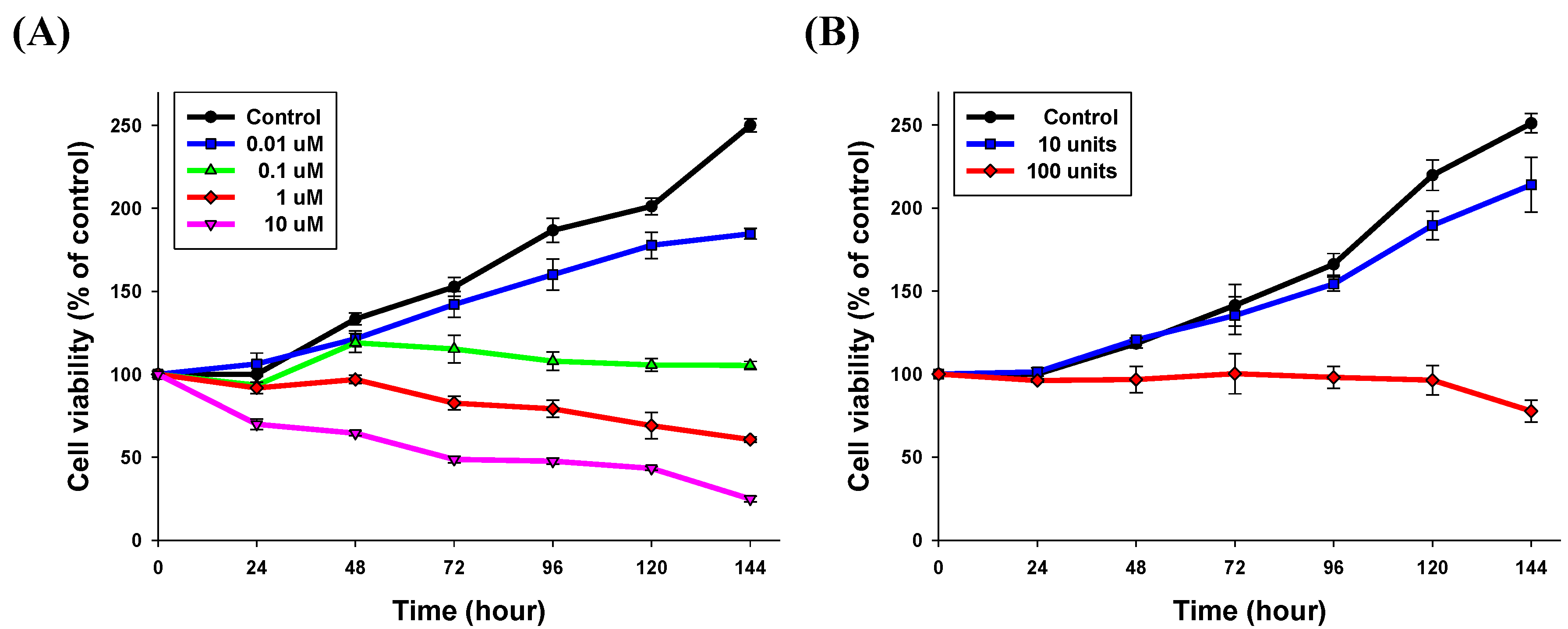
2.3.2. Time-Related Drug Release Efficiency Test

2.4. Applications of Lapatinib Powder-Entrapped Microstructures
3. Experimental Section
3.1. Biodegradable Polymers and Molecular Targeted Anti-Cancer Drug for the DDS
3.2. X-ray Lithography-Based Microfabrication Method
3.3. High Performance Liquid Chromatography for the in Vitro Drug Release Test
3.4. Cell Culture
3.5. Cell Cycle Analysis
3.6. Growth Inhibition Assays
3.7. Statistics
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gerber, D.E. Targeted therapies: A new generation of cancer treatments. Am. Fam. Physician 2008, 77, 311–319. [Google Scholar] [PubMed]
- Zhang, J.; Yang, P.L.; Gray, N.S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Cobleigh, M.A.; Vogel, C.L.; Tripathy, D.; Robert, N.J.; Scholl, S.; Fehrenbacher, L.; Wolter, J.M.; Paton, V.; Shak, S.; Lieberman, G.; et al. Multinational study of the efficacy and safety of humanized anti-HER2 monoclonal antibody in women who have HER2-overexpressing metastatic breast cancer that has progressed after chemotherapy for metastatic disease. J. Clin. Oncol. 1999, 17, 2639–2648. [Google Scholar] [PubMed]
- Vogel, C.L.; Cobleigh, M.A.; Tripathy, D.; Gutheil, J.C.; Harris, L.N.; Fehrenbacher, L.; Slamon, D.J.; Murphy, M.; Novotny, W.F.; Burchmore, M.; et al. Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J. Clin. Oncol. 2002, 20, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.V.; Haber, D.A.; Settleman, J. Cell line-based platforms to evaluate the therapeutic efficacy of candidate anticancer agents. Nat. Rev. Cancer 2010, 10, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Fukuoka, M.; Yano, S.; Giaccone, G.; Tamura, T.; Nakagawa, K.; Douillard, J.Y.; Nishiwaki, Y.; Vansteenkiste, J.; Kudoh, S.; Rischin, D.; et al. Multi-institutional randomized phase II trial of gefitinib for previously treated patients with advanced non-small-cell lung cancer (the ideal 1 trial). J. Clin. Oncol. 2003, 21, 2237–2246. [Google Scholar] [CrossRef] [PubMed]
- Kris, M.G.; Natale, R.B.; Herbst, R.S.; Lynch, T.J., Jr.; Prager, D.; Belani, C.P.; Schiller, J.H.; Kelly, K.; Spiridonidis, H.; Sandler, A.; et al. Efficacy of gefitinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in symptomatic patients with non-small cell lung cancer: A randomized trial. JAMA 2003, 290, 2149–2158. [Google Scholar] [CrossRef] [PubMed]
- Dagher, R.; Cohen, M.; Williams, G.; Rothmann, M.; Gobburu, J.; Robbie, G.; Rahman, A.; Chen, G.; Staten, A.; Griebel, D.; et al. Approval summary: Imatinib mesylate in the treatment of metastatic and/or unresectable malignant gastrointestinal stromal tumors. Clin. Cancer Res. 2002, 8, 3034–3038. [Google Scholar] [PubMed]
- Demetri, G.D.; von Mehren, M.; Blanke, C.D.; van den Abbeele, A.D.; Eisenberg, B.; Roberts, P.J.; Heinrich, M.C.; Tuveson, D.A.; Singer, S.; Janicek, M.; et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N. Engl. J. Med. 2002, 347, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Rusnak, D.W.; Affleck, K.; Cockerill, S.G.; Stubberfield, C.; Harris, R.; Page, M.; Smith, K.J.; Guntrip, S.B.; Carter, M.C.; Shaw, R.J.; et al. The characterization of novel, dual ErbB-2/EGFR, tyrosine kinase inhibitors: Potential therapy for cancer. Cancer Res. 2001, 61, 7196–7203. [Google Scholar] [PubMed]
- Rusnak, D.W.; Lackey, K.; Affleck, K.; Wood, E.R.; Alligood, K.J.; Rhodes, N.; Keith, B.R.; Murray, D.M.; Knight, W.B.; Mullin, R.J.; et al. The effects of the novel, reversible epidermal growth factor receptor/ErbB-2 tyrosine kinase inhibitor, GW2016, on the growth of human normal and tumor-derived cell lines in vitro and in vivo. Mol. Cancer Ther. 2001, 1, 85–94. [Google Scholar] [PubMed]
- Konecny, G.E.; Pegram, M.D.; Venkatesan, N.; Finn, R.; Yang, G.; Rahmeh, M.; Untch, M.; Rusnak, D.W.; Spehar, G.; Mullin, R.J.; et al. Activity of the dual kinase inhibitor lapatinib (GW572016) against HER-2-overexpressing and trastuzumab-treated breast cancer cells. Cancer Res. 2006, 66, 1630–1639. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Li, S.; Hu, Y.P.; Wang, J.; Hauser, J.; Conway, A.N.; Vinci, M.A.; Humphrey, L.; Zborowska, E.; Willson, J.K.; et al. Blockade of EGFR and ErbB2 by the novel dual EGFR and ErbB2 tyrosine kinase inhibitor GW572016 sensitizes human colon carcinoma GEO cells to apoptosis. Cancer Res. 2006, 66, 404–411. [Google Scholar] [CrossRef] [PubMed]
- Moy, B.; Kirkpatrick, P.; Kar, S.; Goss, P. Lapatinib. Nat. Rev. Drug Dis. 2007, 6, 431–432. [Google Scholar] [CrossRef]
- Slamon, D.J.; Clark, G.M.; Wong, S.G.; Levin, W.J.; Ullrich, A.; McGuire, W.L. Human breast cancer: Correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987, 235, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Godolphin, W.; Jones, L.A.; Holt, J.A.; Wong, S.G.; Keith, D.E.; Levin, W.J.; Stuart, S.G.; Udove, J.; Ullrich, A.; et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 1989, 244, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Press, M.F.; Pike, M.C.; Chazin, V.R.; Hung, G.; Udove, J.A.; Markowicz, M.; Danyluk, J.; Godolphin, W.; Sliwkowski, M.; Akita, R.; et al. HER-2/neu expression in node-negative breast cancer: Direct tissue quantitation by computerized image analysis and association of overexpression with increased risk of recurrent disease. Cancer Res. 1993, 53, 4960–4970. [Google Scholar] [PubMed]
- Yonemura, Y.; Ninomiya, I.; Yamaguchi, A.; Fushida, S.; Kimura, H.; Ohoyama, S.; Miyazaki, I.; Endou, Y.; Tanaka, M.; Sasaki, T. Evaluation of immunoreactivity for ErbB-2 protein as a marker of poor short term prognosis in gastric cancer. Cancer Res. 1991, 51, 1034–1038. [Google Scholar] [PubMed]
- Orita, H.; Maehara, Y.; Emi, Y.; Kakeji, Y.; Baba, H.; Korenaga, D.; Sugimachi, K. C-erbB-2 expression is predictive for lymphatic spread of clinical gastric carcinoma. Hepato-gastroenterology 1997, 44, 294–298. [Google Scholar] [PubMed]
- Takehana, T.; Kunitomo, K.; Kono, K.; Kitahara, F.; Iizuka, H.; Matsumoto, Y.; Fujino, M.A.; Ooi, A. Status of c-erbB-2 in gastric adenocarcinoma: A comparative study of immunohistochemistry, fluorescence in situ hybridization and enzyme-linked immuno-sorbent assay. Int. J. Cancer 2002, 98, 833–837. [Google Scholar] [CrossRef] [PubMed]
- Im, S.A.; Lee, K.E.; Nam, E.; Kim, D.Y.; Lee, J.H.; Han, H.S.; Seoh, J.Y.; Park, H.Y.; Cho, M.S.; Han, W.S.; et al. Potential prognostic significance of p185(HER2) overexpression with loss of pten expression in gastric carcinomas. Tumori 2005, 91, 513–521. [Google Scholar] [PubMed]
- Tanner, M.; Hollmen, M.; Junttila, T.T.; Kapanen, A.I.; Tommola, S.; Soini, Y.; Helin, H.; Salo, J.; Joensuu, H.; Sihvo, E.; et al. Amplification of HER-2 in gastric carcinoma: Association with topoisomerase iialpha gene amplification, intestinal type, poor prognosis and sensitivity to trastuzumab. Ann. Oncol. 2005, 16, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Kim, H.P.; Im, S.A.; Kang, S.; Hur, H.S.; Yoon, Y.K.; Oh, D.Y.; Kim, J.H.; Lee, D.S.; Kim, T.Y.; et al. The growth inhibitory effect of lapatinib, a dual inhibitor of EGFR and HER2 tyrosine kinase, in gastric cancer cell lines. Cancer Lett. 2008, 272, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.P.; Yoon, Y.K.; Kim, J.W.; Han, S.W.; Hur, H.S.; Park, J.; Lee, J.H.; Oh, D.Y.; Im, S.A.; Bang, Y.J.; et al. Lapatinib, a dual EGFR and HER2 tyrosine kinase inhibitor, downregulates thymidylate synthase by inhibiting the nuclear translocation of EGFR and HER2. PLoS One 2009, 4. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.P.; Han, S.W.; Song, S.H.; Jeong, E.G.; Lee, M.Y.; Hwang, D.; Im, S.A.; Bang, Y.J.; Kim, T.Y. Testican-1-mediated epithelial-mesenchymal transition signaling confers acquired resistance to lapatinib in HER2-positive gastric cancer. Oncogene 2014, 33, 3334–3341. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Shah, N.H.; Malick, A.W.; Rhodes, C.T. Controlled drug delivery by biodegradable poly(ester) devices: Different preparative approaches. Drug Dev. Ind. Pharm. 1998, 24, 703–727. [Google Scholar] [CrossRef] [PubMed]
- Razzacki, S.Z.; Thwar, P.K.; Yang, M.; Ugaz, V.M.; Burns, M.A. Integrated microsystems for controlled drug delivery. Adv. Drug Deli. Rev. 2004, 56, 185–198. [Google Scholar] [CrossRef]
- Huang, H.M.H.; Yang, H. Preparation of Polymorphic Form of Lapatinib Ditosylate. U.S. Patent 12/806,466, 14 August 2010. [Google Scholar]
- Langer, R. Drug delivery and targeting. Nature 1998, 392, 5–10. [Google Scholar] [PubMed]
- Allen, T.M.; Cullis, P.R. Drug delivery systems: Entering the mainstream. Science 2004, 303, 1818–1822. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, D.K.; Fong, L.S.; Zhang, Y. Nanoparticles in photodynamic therapy: An emerging paradigm. Adv. Drug Deli. Rev. 2008, 60, 1627–1637. [Google Scholar] [CrossRef]
- Serrano, M.C.; Pagani, R.; Vallet-Regı, M.; Pena, J.; Ramila, A.; Izquierdo, I.; Portoles, M.T. In vitro biocompatibility assessment of poly (ε-caprolactone) films using L929 mouse fibroblasts. Biomaterials 2004, 25, 5603–5611. [Google Scholar] [CrossRef] [PubMed]
- Cho, D.I.; Yoo, H.J. Microfabrication methods for biodegradable polymeric carriers for drug delivery system applications: A review. IEEE J. Microelectromech. Syst. 2015, 24, 10–18. [Google Scholar]
- Shea, L.D.; Wang, D.; Franceschi, R.T.; Mooney, D.J. Engineered bone development from a pre-osteoblast cell line on three-dimensional scaffolds. Tissue Eng. 2000, 6, 605–617. [Google Scholar] [CrossRef] [PubMed]
- Jackson, J.K.; Gleave, M.E.; Yago, V.; Beraldi, E.; Hunter, W.L.; Burt, H.M. The suppression of human prostate tumor growth in mice by the intratumoral injection of a slow-release polymeric paste formulation of paclitaxel. Cancer Res. 2000, 60, 4146–4151. [Google Scholar] [PubMed]
- Cam, D.; Hyon, S.-H.; Ikada, Y. Degradation of high molecular weight poly (l-lactide) in alkaline medium. Biomaterials 1995, 16, 833–843. [Google Scholar] [CrossRef] [PubMed]
- Varshney, L.; Dodke, P. Radiation effect studies on anticancer drugs, cyclophosphamide and doxorubicin for radiation sterilization. Radiat. Phys. Chem. 2004, 71, 1103–1111. [Google Scholar] [CrossRef]
- Pohang Accelerator Laboratory. Available online: http://pal.postech.ac.kr (accessed on 30 January 2015).
- Ku, J.L.; Park, J.G. Biology of snu cell lines. Cancer Res. Treat. 2005, 37, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Park, J.G.; Frucht, H.; LaRocca, R.V.; Bliss, D.P., Jr.; Kurita, Y.; Chen, T.R.; Henslee, J.G.; Trepel, J.B.; Jensen, R.T.; Johnson, B.E.; et al. Characteristics of cell lines established from human gastric carcinoma. Cancer Res. 1990, 50, 2773–2780. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeong, E.-G.; Yoo, H.J.; Song, B.; Kim, H.-P.; Han, S.-W.; Kim, T.-Y.; Cho, D.-I.D. Evaluation of Lapatinib Powder-Entrapped Biodegradable Polymeric Microstructures Fabricated by X-Ray Lithography for a Targeted and Sustained Drug Delivery System. Materials 2015, 8, 519-534. https://doi.org/10.3390/ma8020519
Jeong E-G, Yoo HJ, Song B, Kim H-P, Han S-W, Kim T-Y, Cho D-ID. Evaluation of Lapatinib Powder-Entrapped Biodegradable Polymeric Microstructures Fabricated by X-Ray Lithography for a Targeted and Sustained Drug Delivery System. Materials. 2015; 8(2):519-534. https://doi.org/10.3390/ma8020519
Chicago/Turabian StyleJeong, Eun-Goo, Hyung Jung Yoo, Byeonghwa Song, Hwang-Phill Kim, Sae-Won Han, Tae-You Kim, and Dong-Il Dan Cho. 2015. "Evaluation of Lapatinib Powder-Entrapped Biodegradable Polymeric Microstructures Fabricated by X-Ray Lithography for a Targeted and Sustained Drug Delivery System" Materials 8, no. 2: 519-534. https://doi.org/10.3390/ma8020519
APA StyleJeong, E.-G., Yoo, H. J., Song, B., Kim, H.-P., Han, S.-W., Kim, T.-Y., & Cho, D.-I. D. (2015). Evaluation of Lapatinib Powder-Entrapped Biodegradable Polymeric Microstructures Fabricated by X-Ray Lithography for a Targeted and Sustained Drug Delivery System. Materials, 8(2), 519-534. https://doi.org/10.3390/ma8020519