Cellulose Isolation Methodology for NMR Analysis of Cellulose Ultrastructure

Abstract
:1. Introduction
2. Results and Discussion
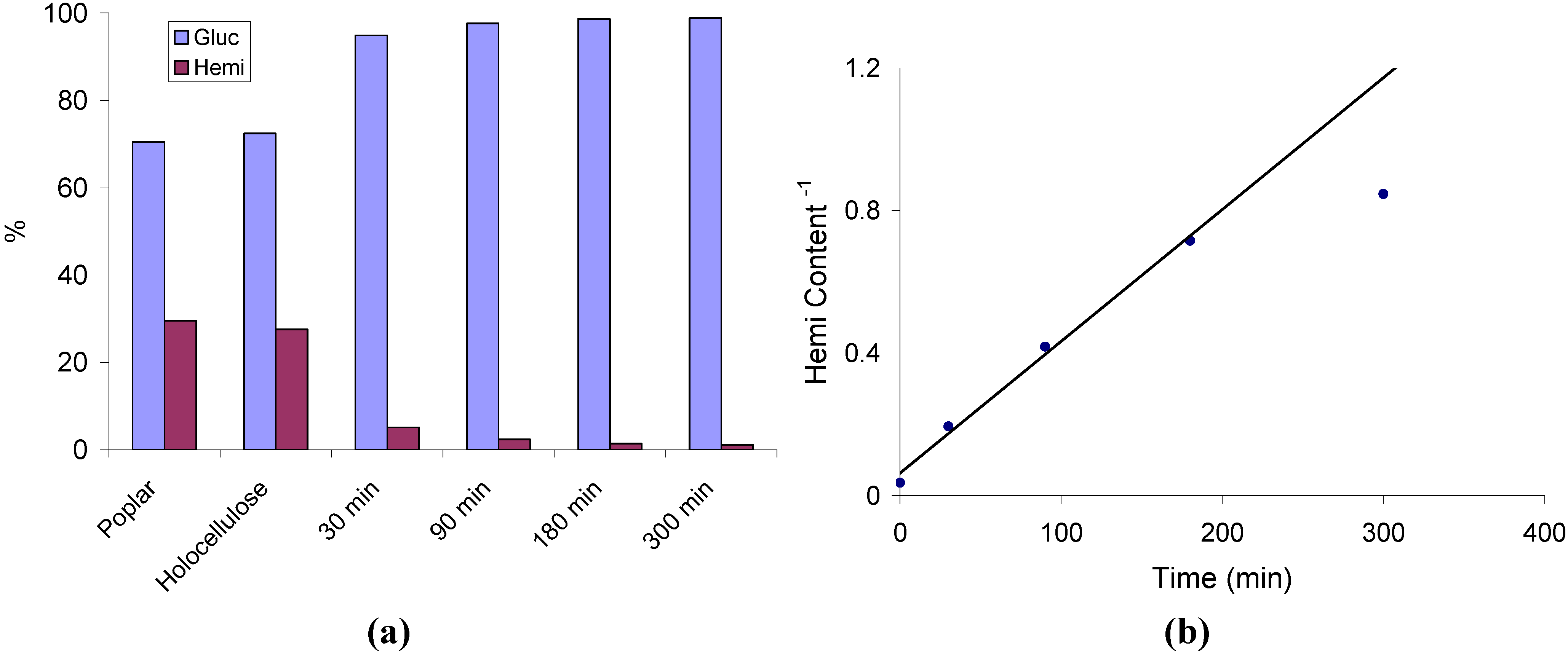
2.1. Hemicellulose Removal
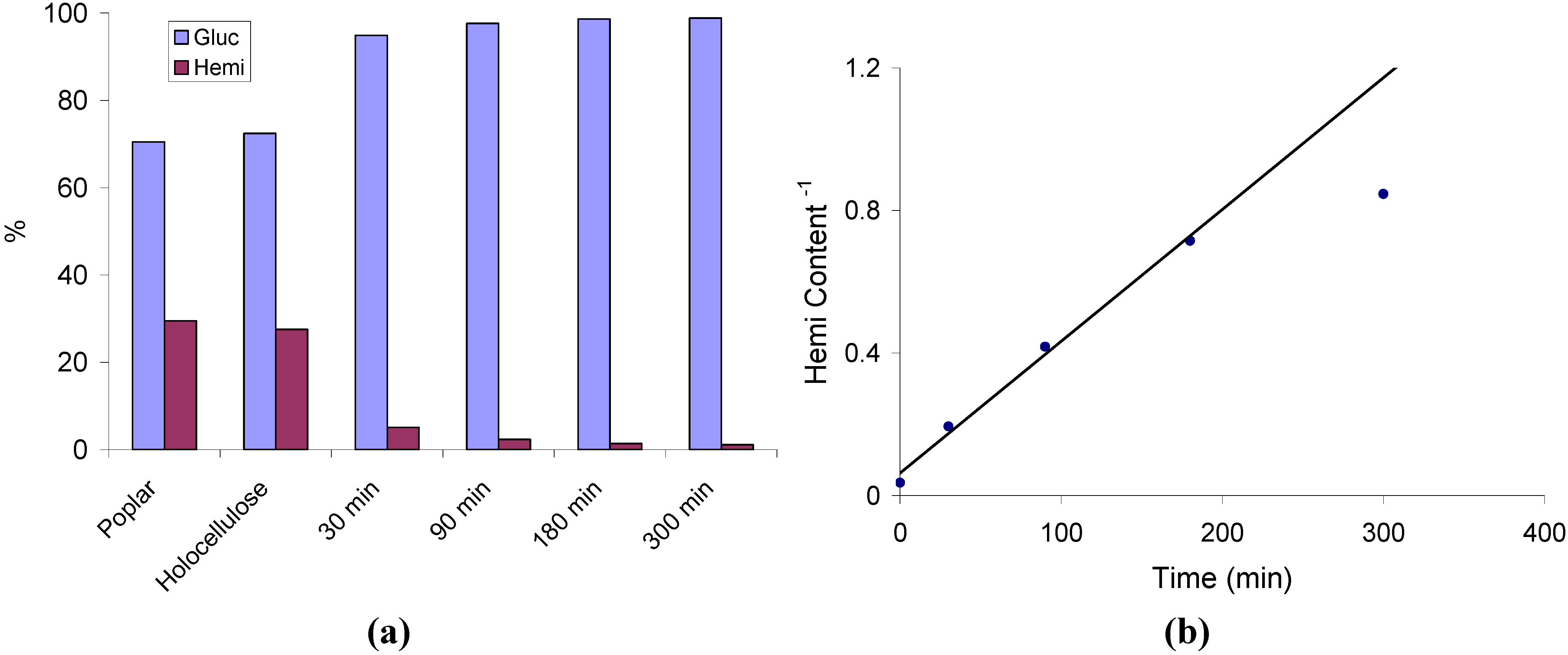
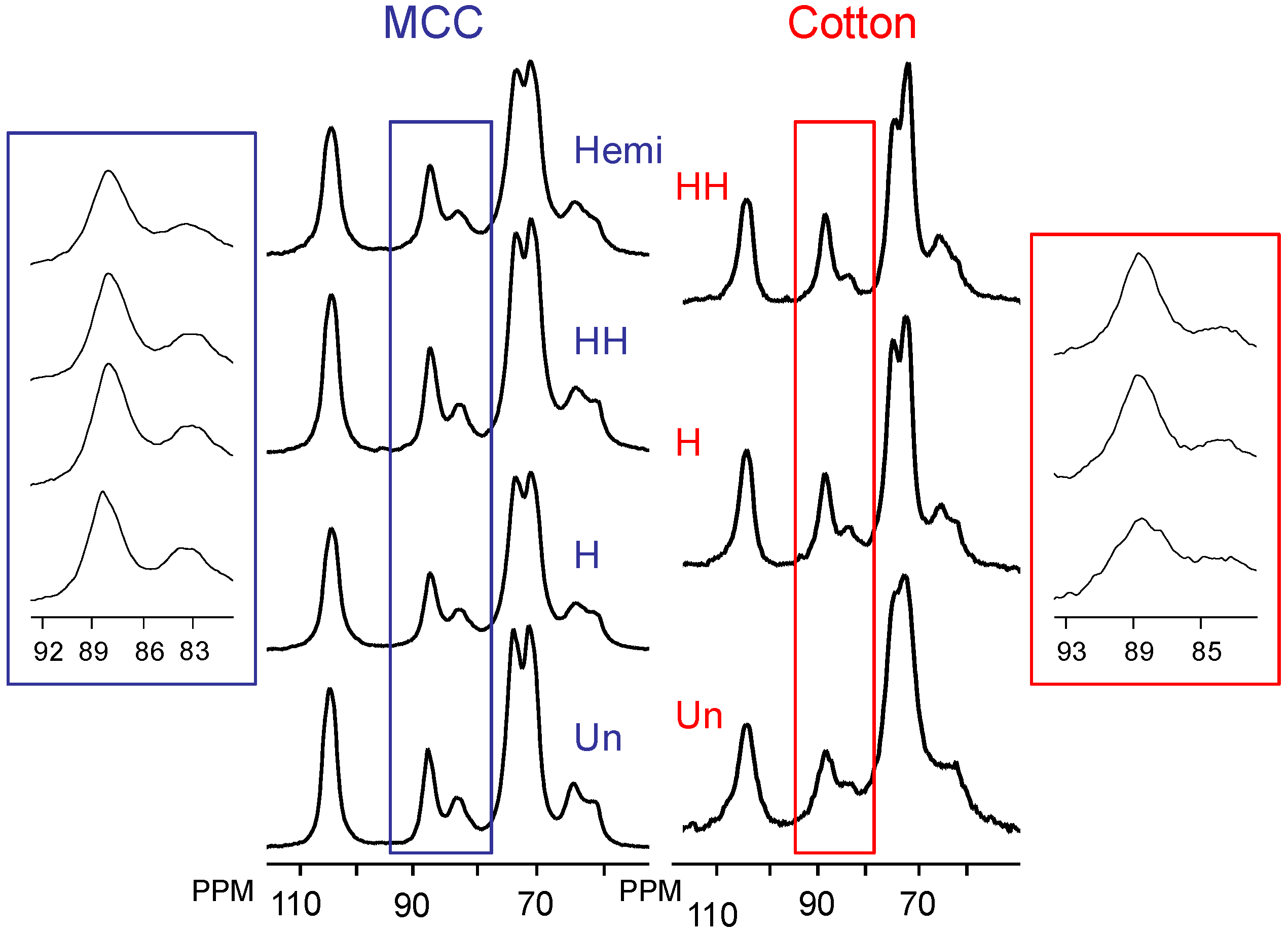
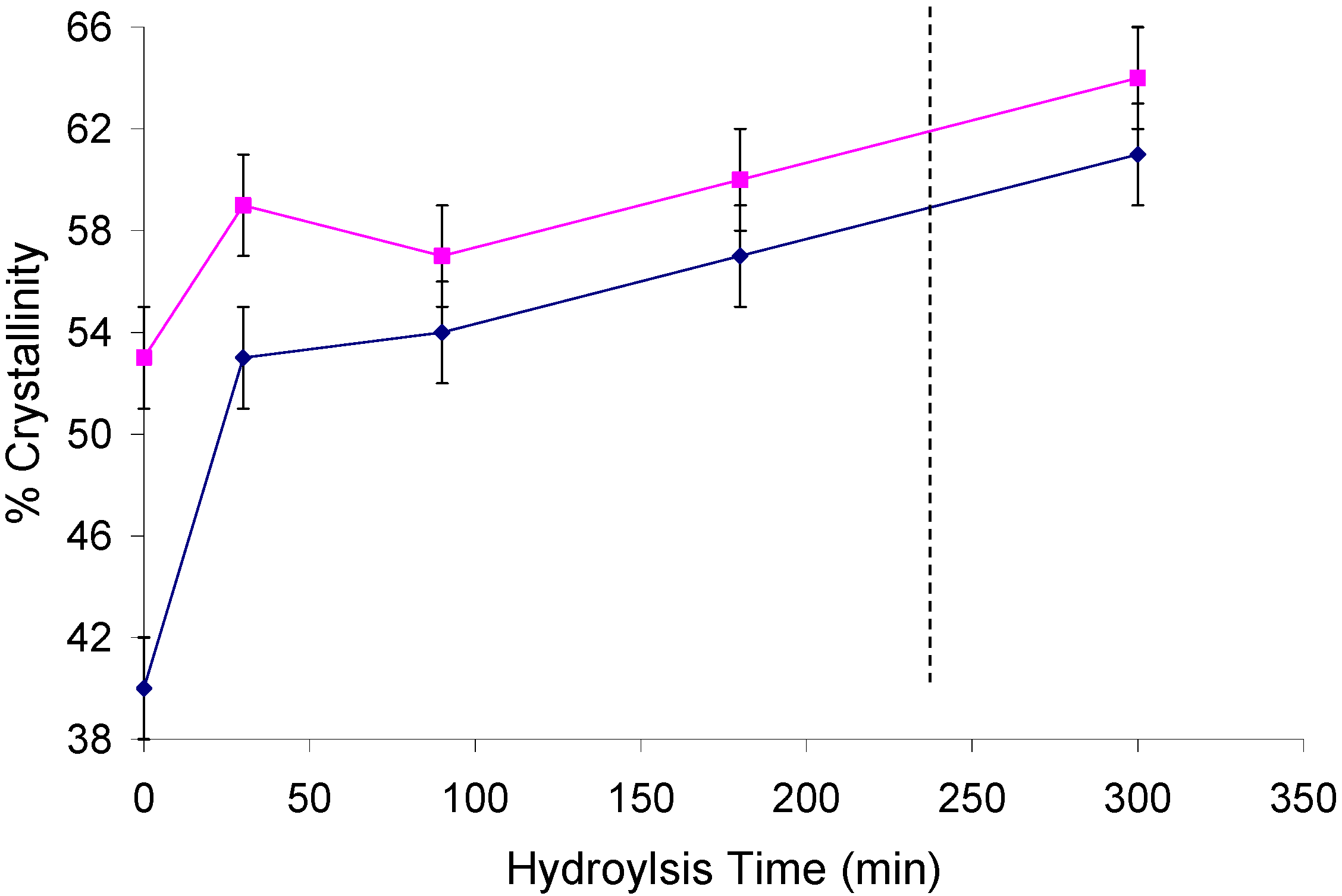
2.2. Changes in Ultrastructure and Crystallinity
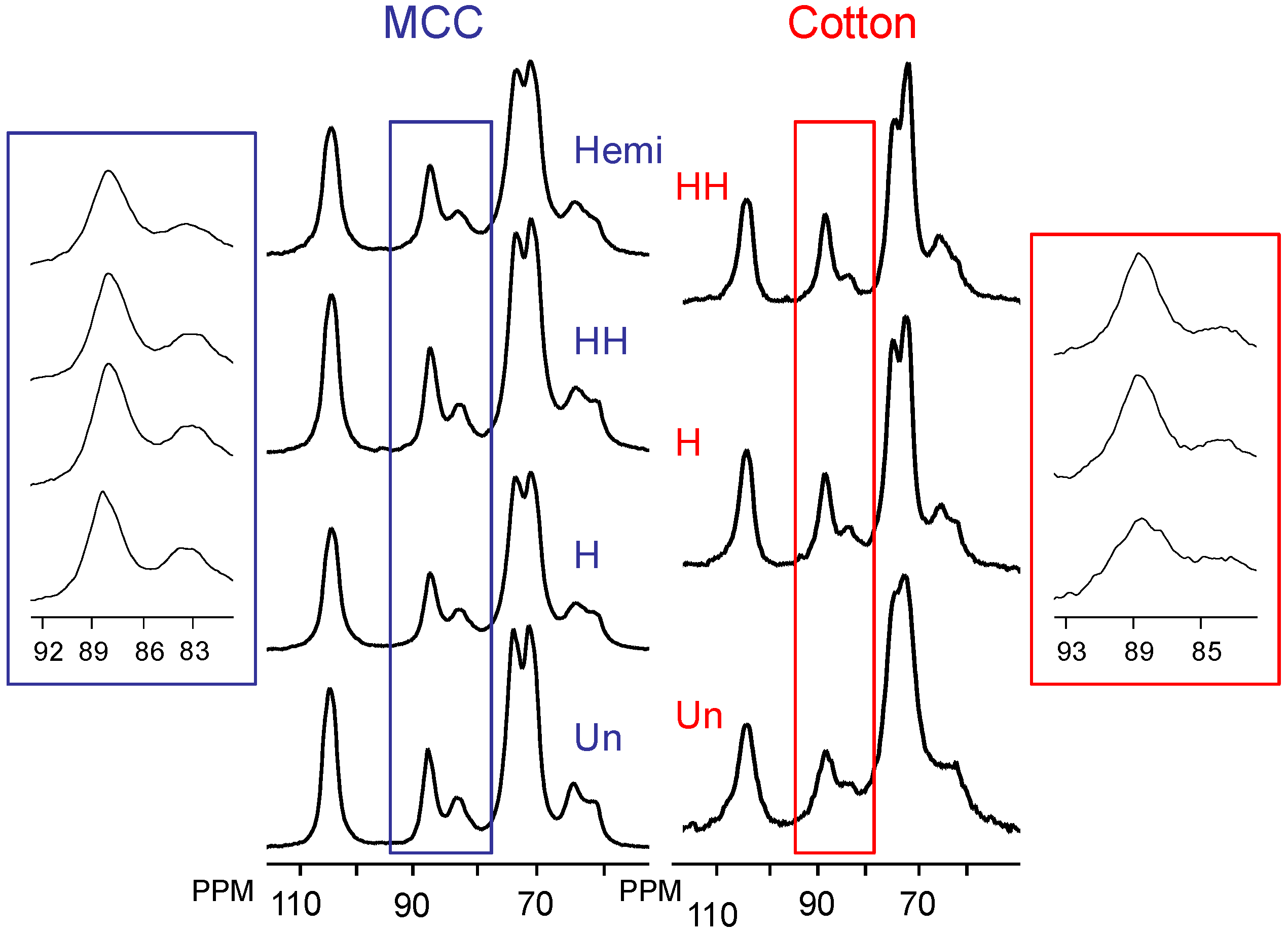
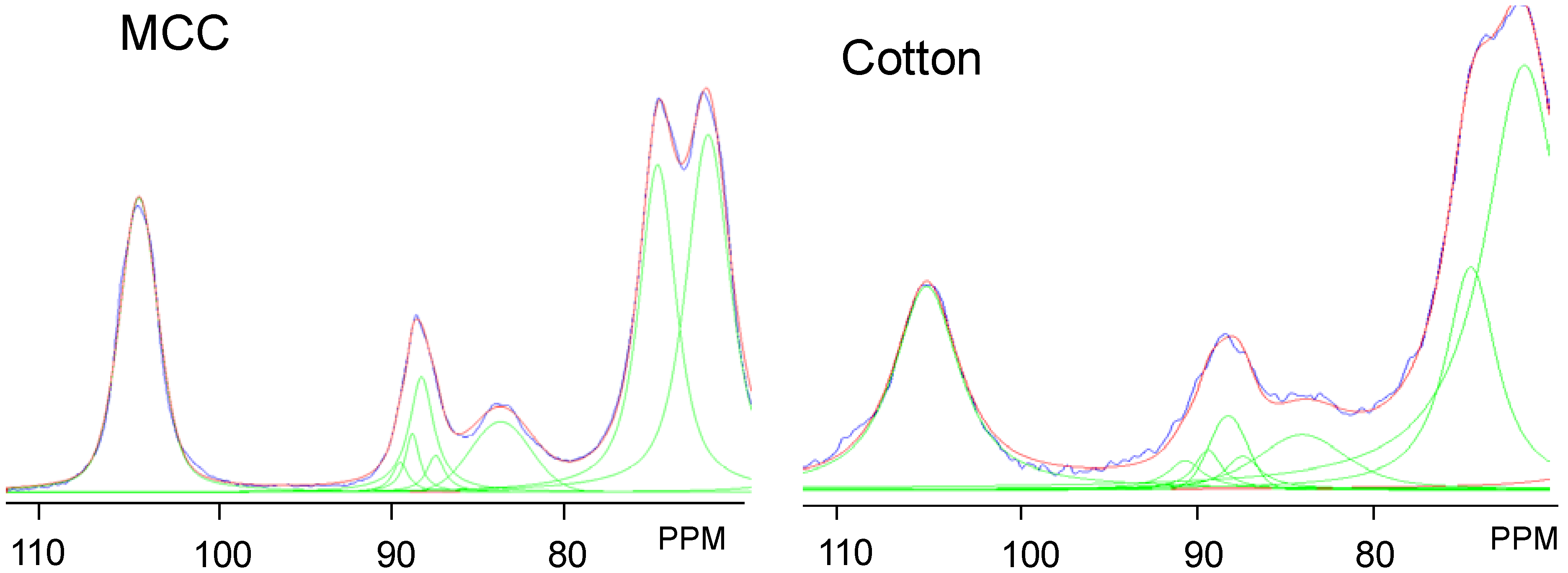

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Iα (%) ±3 | Iα+β (%) ±4 | Para (%) ±6 | Iβ (%) ±6 | CrI (%) ±2 | LFD (nm) ±0.5 |
|---|---|---|---|---|---|---|
| Un MCC | 8 | 11 | 35 | 8 | 62 | 5.2 |
| H MCC | 7 | 8 | 35 | 10 | 60 | 4.9 |
| HH MCC | 6 | 10 | 42 | 11 | 68 | 6.3 |
| Hemi MCC | 7 | 11 | 40 | 19 | 66 | 5.9 |
| Un cotton | 9 | 9 | 35 | 9 | 62 | 5.2 |
| H cotton | 6 | 13 | 40 | 6 | 66 | 5.9 |
| HH cotton | 6 | 14 | 49 | 6 | 79 | 9.9 |
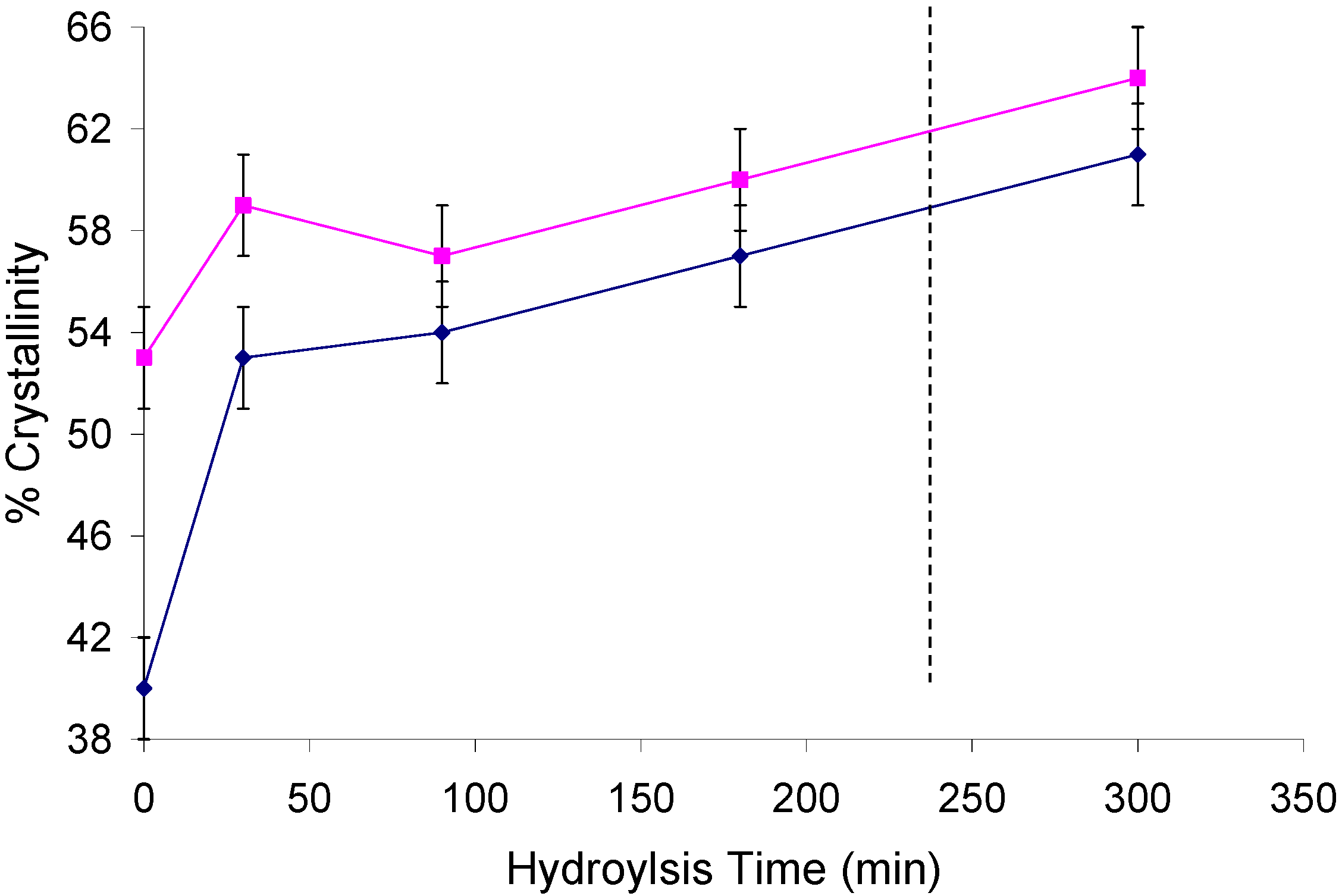
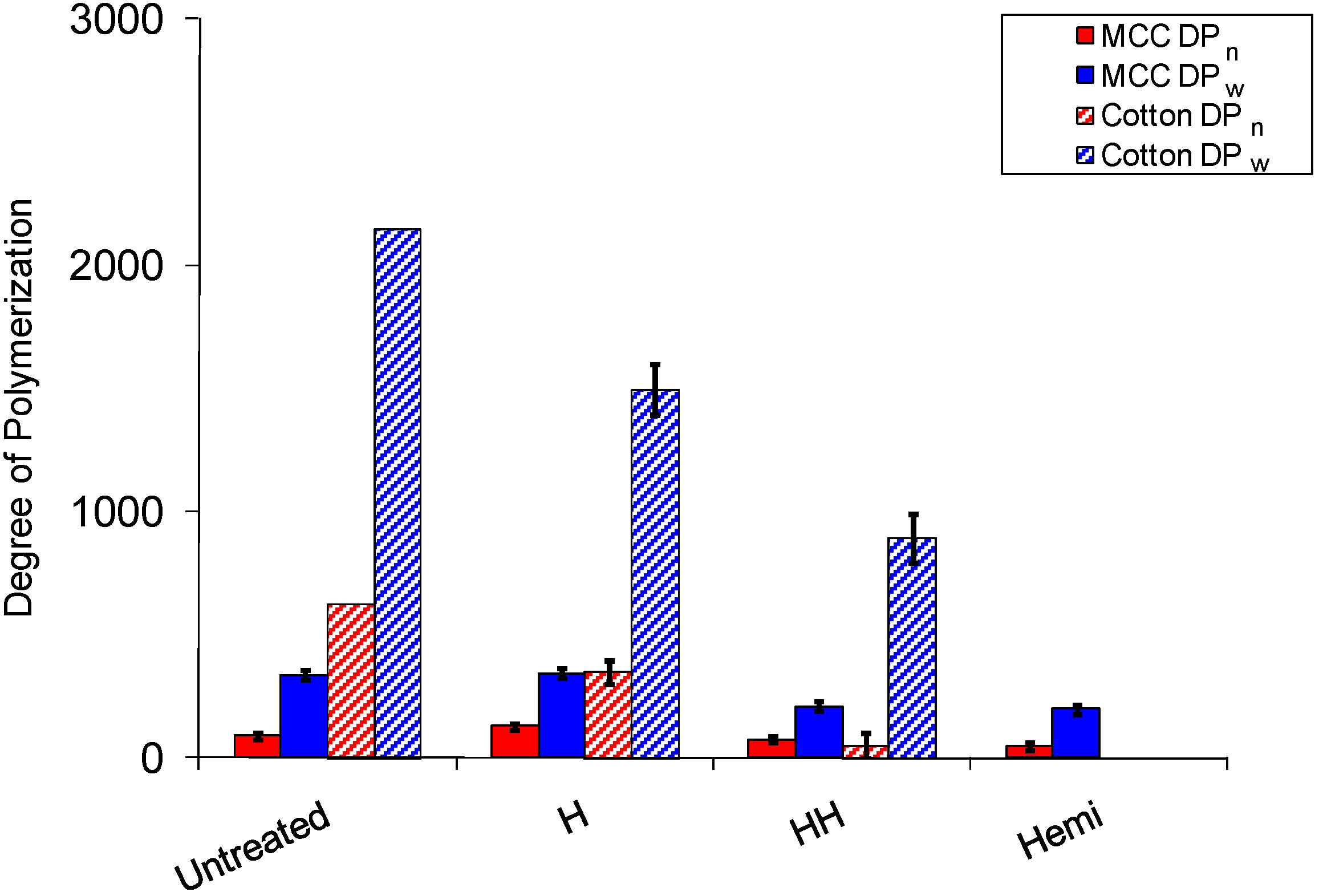
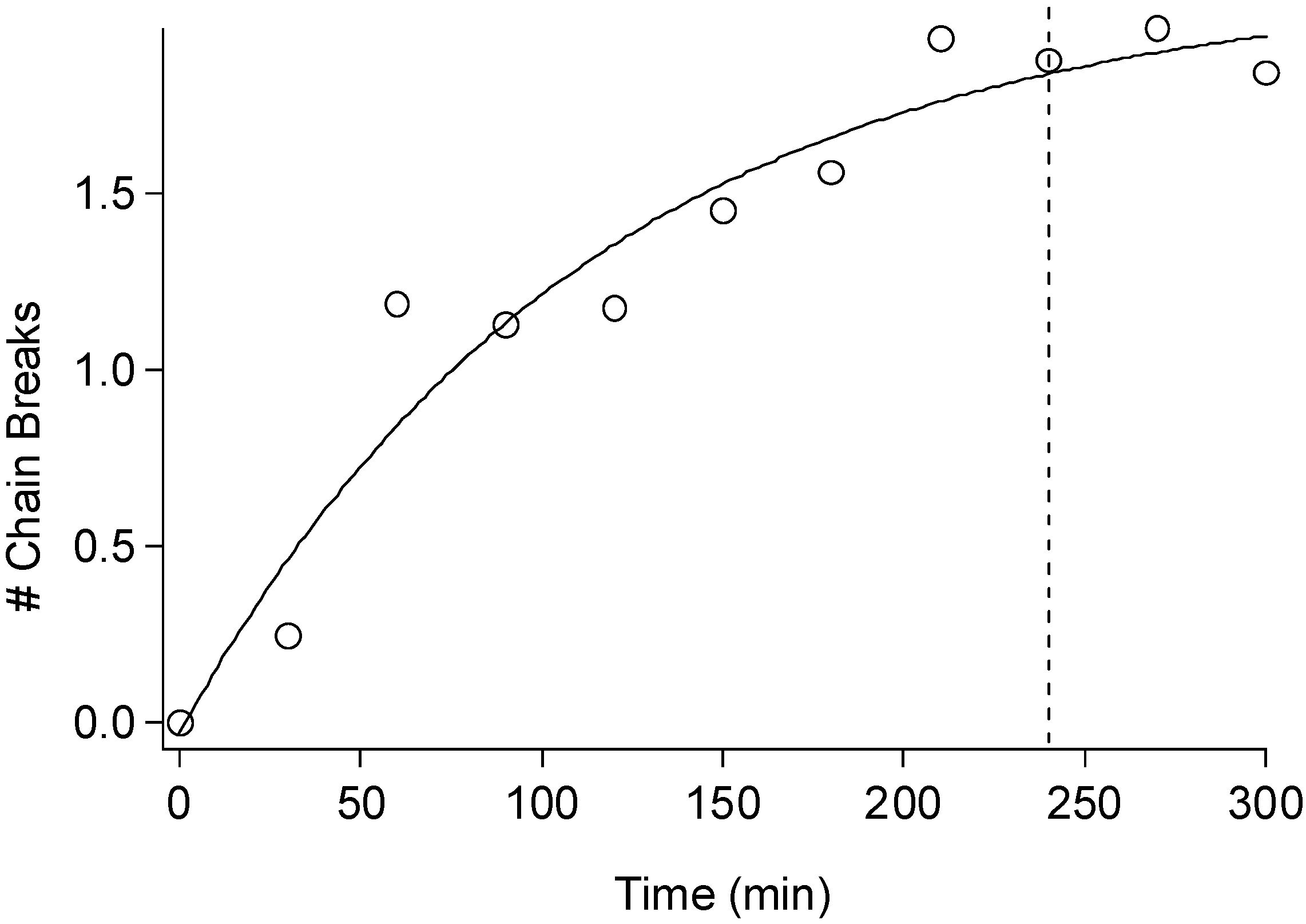
2.3. Cellulose Degradation
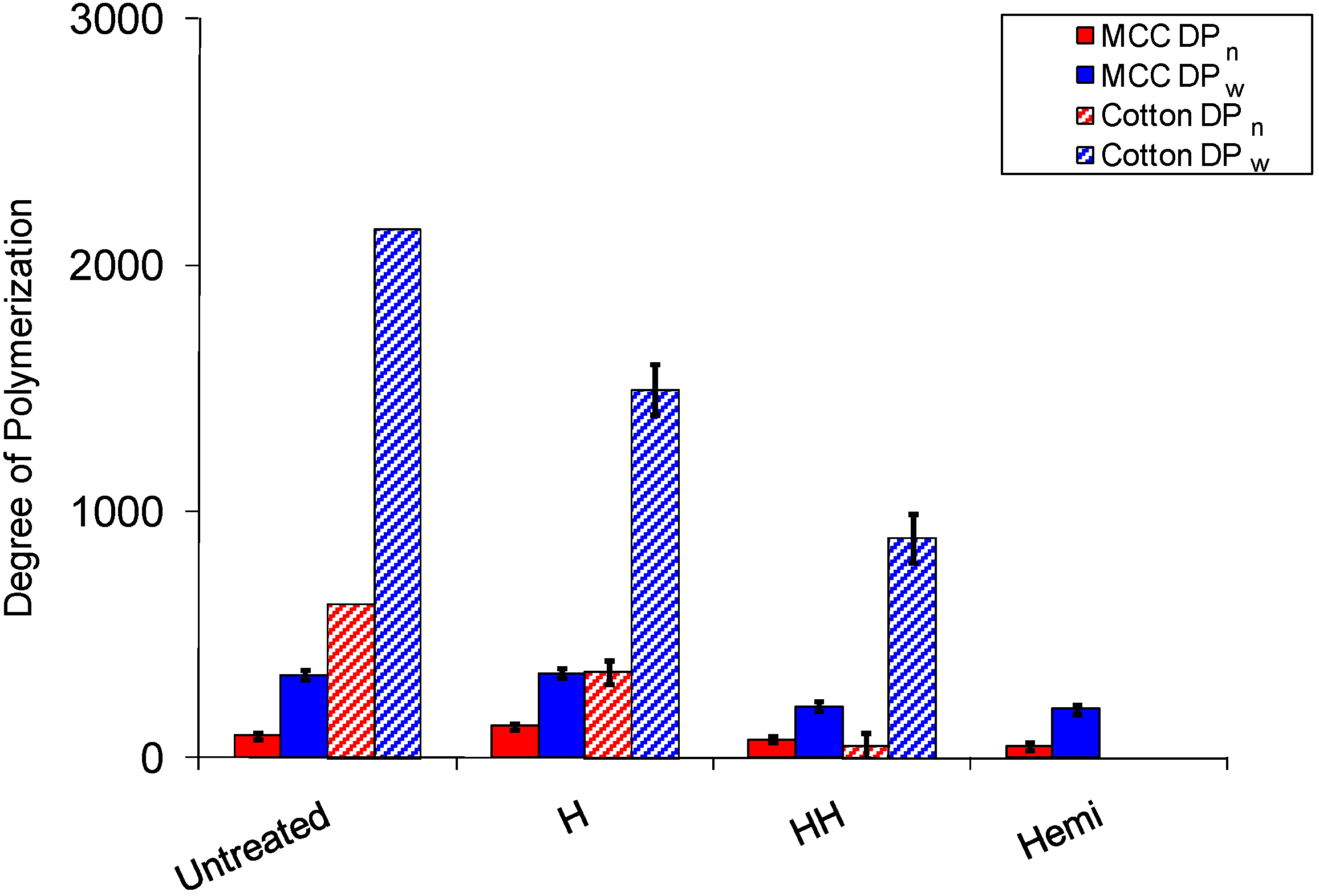
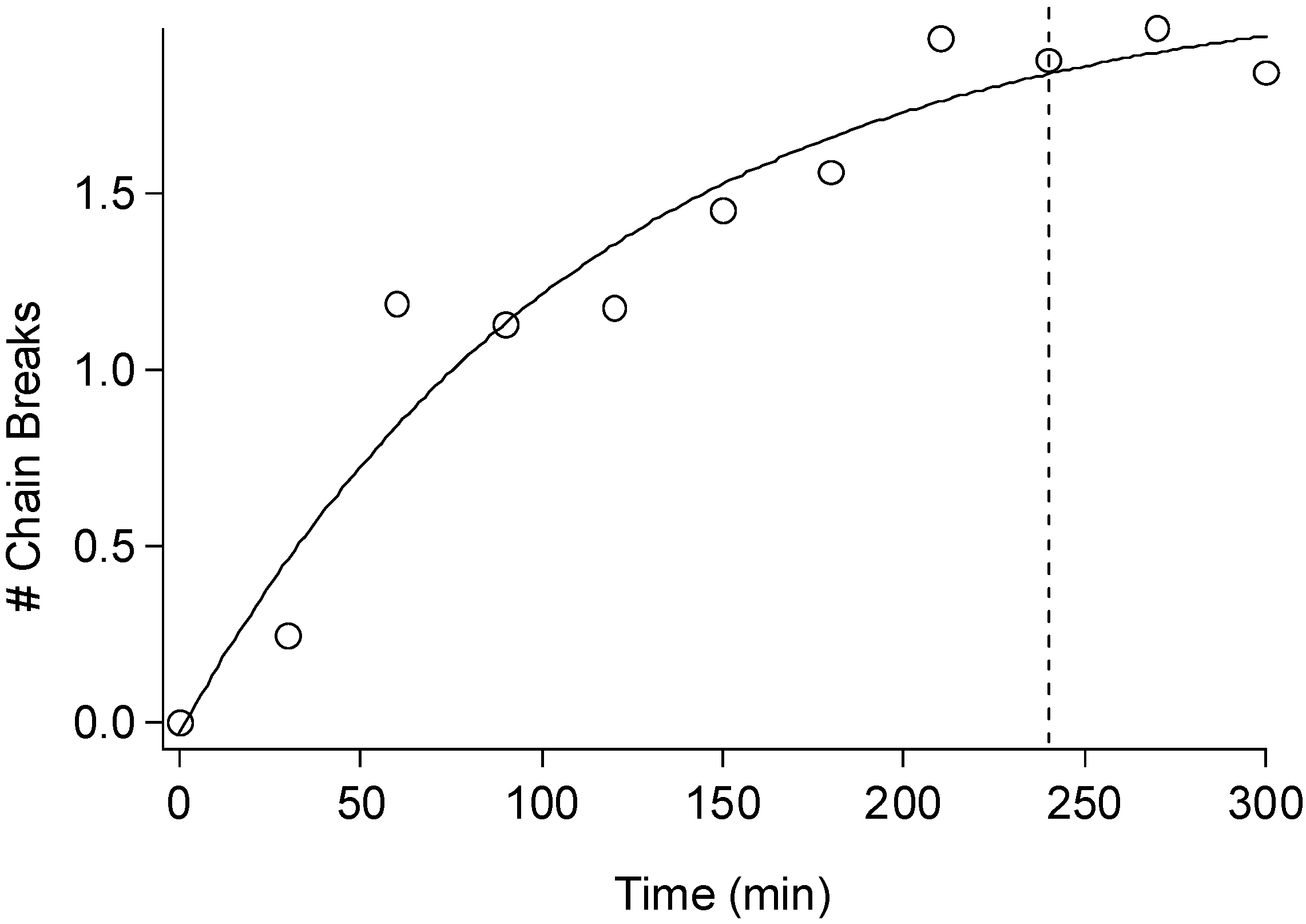
3. Experimental Section
3.1. Substrates
3.2. Carbohydrates and Klason Lignin Analysis
3.3. Holocellulose Pulping
3.4. Cellulose Isolation for GPC Analysis
3.5. Gel Permeation Chomatography (GPC) Analysis of Cellulose
3.6. Cellulose Isolation for NMR Analysis
3.7. NMR Analysis
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgements
References and Notes
- Pu, Y.; Zhang, D.; Singh, P.M.; Ragauskas, A.J. The new forestry biofuels sector. Biofuels Bioprod. Bioref. 2008, 2, 58–73. [Google Scholar] [CrossRef]
- Ragauskas, A.J.; Williams, C.K.; Davison, B.H.; Britovsek, G.; Cairney, J.; Eckert, C.A.; Frederick, W.J., Jr.; Hallett, J.P.; Leak, D.J.; Liotta, C.L.; et al. The path forward for biofuels and biomaterials. Science 2006, 311, 474–479. [Google Scholar] [CrossRef]
- Himmel, M.E.; Ding, S.-Y.; Johnson, D.K.; Adney, W.S.; Nimlos, M.R.; Brady, J.W.; Foust, T.D. Biomass recalcitrance: Engineering plants and enzymes for biofuels production. Science 2007, 315, 804–807. [Google Scholar] [CrossRef] [PubMed]
- Hsu, T.A.; Ladisch, M.R.; Tsao, T. Alcohol from cellulose. Chem. Technol. 1980, 10, 315–319. [Google Scholar]
- Lee, S.B.; Kim, I.H.; Ryu, D.D.; Taguchi, H. Structural properties of cellulose and cellulase reaction mechanism. Biotechnol. Bioeng. 1983, 25, 33–51. [Google Scholar] [CrossRef] [PubMed]
- Foston, M.; Ragauskas, A.J. Changes in lignocellulosic supramolecular and ultrastructure during dilute acid pretreatment of Populus and switchgrass. Biomass Bioenergy 2010, 34, 1885–1898. [Google Scholar] [CrossRef]
- Foston, M.; Ragauskas, A.J. Changes in the structure of the cellulose fiber wall during dilute acid pretreatment in Populus studied by 1H and 2H NMR. Energy Fuels 2010, 24, 5677–5685. [Google Scholar] [CrossRef]
- Joeh, T.; Ishizawa, C.I.; Davis, M.F.; Himmel, M.E.; Adney, W.S.; Johnson, D.K. Cellulase digestibility of pretreated biomass is limited by cellulose accessibility. Biotechnol. Bioeng. 2007, 98, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Thompson, D.N.; Chen, H.C.; Grethlein, H.E. Comparison of pretreatment methods on the basis of available surface area. Bioresour. Technol. 1992, 39, 155–163. [Google Scholar] [CrossRef]
- Kumar, P.; Barrett, D.M.; Delwiche, M.J.; Stroeve, P. Methods for pretreatment of lignocellulosic biomass for efficient hydrolysis and biofuel production. Ind. Eng. Chem. Res. 2009, 48, 3713–3729. [Google Scholar] [CrossRef]
- Mosier, N.; Wyman, C.; Dale, B.; Elander, R.; Lee, Y.Y.; Holtzapple, M.; Ladisch, M. Features of promising technologies for pretreatment of lignocellulosic biomass. Bioresour. Technol. 2005, 96, 673–686. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Cheng, J. Hydrolysis of lignocellulosic materials for ethanol production: A review. Bioresour. Technol. 2002, 83, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Cohen, R.; Jensen, K.A., Jr.; Houtman, C.J.; Hammel, K.E. Significant levels of extracellular reactive oxygen species produced by brown rot basidomycetes on cellulose. FEBS Lett. 2002, 531, 473–478. [Google Scholar] [CrossRef]
- Sinitsyn, A.P.; Gusakov, A.V.; Vlasenko, E. Effect of structural and physico-chemical features of cellulosic substrates on the efficiency of enzymatic hydrolysis. Appl. Biochem. Biotechnol. 1991, 30, 44–59. [Google Scholar] [CrossRef]
- Pu, Y.; Ziemer, C.; Ragauskas, A.J. CP/MAS 13C NMR analysis of cellulase treated bleached softwood kraft pulp. Carbohydr. Res. 2006, 337, 591–597. [Google Scholar] [CrossRef]
- Zhang, Y.H.; Lynd, L.R. Toward an aggregated understanding of enzymatic hydrolysis of cellulose: Noncomplexed cellulase systems. Biotechnol. Bioeng. 2004, 88, 797–824. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, A.C. Cellulose: The structure slowly unravels. Cellulose 1997, 4, 173–207. [Google Scholar] [CrossRef]
- Atalla, R.H. Cellulose. In Comprehensive Natural Products Chemistry; Barton, D.N.K., Meth-Cohn, O., Eds.; Elsevier: Amsterdam, The Netherlands, 1999; Volume 3. [Google Scholar]
- VanderHart, D.L.; Atalla, R.H. 13C NMR spectra of cellulose polymorphs. J. Am. Chem. Soc. 1980, 109, 3249–3250. [Google Scholar]
- VanderHart, D.L.; Atalla, R.H. Observations by high-resolution carbon-13 nuclear magnetic resonance of cellulose I related to morphology and crystal structure. Macromolecules 1981, 14, 570–574. [Google Scholar] [CrossRef]
- Larsson, P.T.; Westermark, U.; Iversen, T. Determination of the cellulose Iα allomorph content in a tunicate cellulose by CP/MAS 13C NMR spectroscopy. Carbohydr. Res. 1995, 278, 339–344. [Google Scholar] [CrossRef]
- Larsson, P.T.; Wickholm, K.; Iversen, T. A CP/MAS 13C NMR investigation of molecular ordering in celluloses. Carbohydr. Res. 1997, 302, 19–25. [Google Scholar] [CrossRef]
- Larsson, P.T.; Hult, E.L.; Wickholm, K.; Pettersson, E.; Iversen, T. CP/MAS 13C-NMR spectroscopy applied to structure and interaction studies on cellulose I. Solid State Nucl. Magn. Reson. 1999, 15, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Wickholm, K.; Larsson, P.T.; Iversen, T. Assignment of non-crystalline forms in cellulose I by CP/MAS 13C NMR spectroscopy. Carbohydr. Res. 1998, 312, 123–129. [Google Scholar] [CrossRef]
- Wickholm, K.; Hult, E.L.; Larsson, P.T.; Iversen, T.; Lennholm, H. Quantification of cellulose forms in complex cellulose materials: A chemometric model. Cellulose 2001, 8, 139–147. [Google Scholar] [CrossRef]
- VanderHart, D.L.; Atalla, R.H. Studies of microstructure in native celluloses using solid-state carbon-13 NMR. Macromolecules 1984, 17, 1455–1462. [Google Scholar] [CrossRef]
- VanderHart, D.L.; Atalla, R.H. A composite of two distinct crystalline forms. Science 1984, 223, 283–285. [Google Scholar] [CrossRef] [PubMed]
- Newman, R.H. Estimation of the lateral dimensions of cellulose crystallites using 13C NMR signal strengths. Solid State Nucl. Magn. Reson. 1999, 15, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Newman, R.H.; Hemmingson, J.A. Carbon-13 NMR distinction between categories of molecular order and disorder in cellulose. Cellulose 1994, 2, 95–110. [Google Scholar] [CrossRef]
- Maunu, S.L.; Littia, T.; Kauliomaki, S.; Hortling, B.; Sundquist, J. 13C CPMAS NMR investigations of cellulose polymorphs in different pulps. Cellulose 2000, 7, 146–159. [Google Scholar] [CrossRef]
- Lennholm, H.; Larsson, P.T.; Iversen, T. Determination of cellulose Iα and Iβ in lignocellulosic materials. Carbohydr. Res. 1994, 261, 119–131. [Google Scholar] [CrossRef]
- Liitiä, T. Application of Modern NMR Spectroscopic Techniques to Structural Studies of Wood and Pulp Components; University of Helsinki: Helsinki, Finland, 2002. [Google Scholar]
- Huex, L.; Dinand, E.; Vignon, M.R. Structural aspects in ultrathin cellulose microfibrils followed by 13C CP-MAS NMR. Carbohydr. Polym. 1999, 40, 115–124. [Google Scholar] [CrossRef]
- Hult, E.L.; Larsson, P.T.; Iversen, T. A comparative CP/MAS 13C-NMR study of cellulose structure in spruce wood and kraft pulp. Cellulose 2000, 7, 35–55. [Google Scholar] [CrossRef]
- Hult, E.L.; Larsson, P.T.; Iversen, T. Cellulose fibril aggregation—An inherent property of kraft pulps. Polymer 2001, 42, 3309–3314. [Google Scholar] [CrossRef]
- Hult, E.L.; Larsson, P.T.; Iversen, T. A comparative CP/MAS 13C-NMR study of the supermolecular structure of polysaccharides in sulphite and kraft pulps. Holzforschung 2002, 56, 179–184. [Google Scholar] [CrossRef]
- Hult, E.L.; Liitia, T.; Maunu, S.L.; Hortling, B.; Iversen, T. A CP/MAS 13C-NMR study of cellulose structure on the surface of refined kraft pulp fibers. Carbohydr. Polym. 2002, 49, 231–234. [Google Scholar] [CrossRef]
- Hult, E.L.; Iversen, T.; Sugiyama, J. Characterization of the supermolecular structure of cellulose in wood pulp fibres. Cellulose 2003, 10, 103–110. [Google Scholar] [CrossRef]
- Newman, R.H.; Hemmingson, J.A. Determination of the degree of cellulose crystallinity in wood by Carbon-13 nuclear magnetic resonance spectroscopy. Holzforschung 1990, 44, 351–355. [Google Scholar] [CrossRef]
- Leary, G.; Morgan, K.; Newman, R.H. A 13C CP/MAS NMR comparison of wood fractions from spruce. Holzforschung 1986, 40, 221–224. [Google Scholar] [CrossRef]
- Park, S.; Johnson, D.K.; Ishizawa, C.; Parilla, P.; Davis, M. Measuring the crystallinity index of cellulose by solid state 13C nuclear magnetic resonance. Cellulose 2009, 16, 641–646. [Google Scholar] [CrossRef]
- Ding, S.; Himmel, M. The maize primary cell wall microfibril: A new model derived from direct visualization. J. Agric. Food Chem. 2006, 54, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Wang, Y. High resolution solid-state NMR spectroscopy of starch polysaccharides. Mod. Magn. Reson. 2006, 3, 1761–1769. [Google Scholar]
- Rowland, S.P.; Roberts, E.J.; Bose, J.L.; Wade, C.P. Assessment of the state of order of hydroxyl groups and of molecular segments in hydrocellulose. J. Polym. Sci. Part A 1971, 9, 1623–1633. [Google Scholar] [CrossRef]
- Kolpak, F.; Weih, M.; Blackwell, J. Assessment of the state of order of hydroxyl groups and of molecular segments in hydrocellulose. Polymer 1978, 19, 123–131. [Google Scholar] [CrossRef]
- Langan, P.; Nishiyama, Y.; Chanzy, H. X-ray structure of mercerized cellulose II at 1Å resolution. Biomacromolecules 2001, 2, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Baker, J.O.; Himmel, M.; Parilla, P.A.; Johnson, D.K. Cellulose crystallinity index: Measurement techniques and their impact on interpreting cellulase performance. Biotechnol. Biofuels 2010, 3, 10:1–10:10. [Google Scholar] [CrossRef]
- Heux, L.; Dinand, E.; Vignon, M.R. Structural aspects in ultra-thin cellulose microfibrils followed by 13C CP-MAS NMR. Carbohydr. Polym. 1999, 40, 115–124. [Google Scholar] [CrossRef]
- Newman, R.H. Evidence of assignment of 13C NMR signals to cellulose crystallite surfaces in wood, pulp and isolated cellulose. Holzforschung 1998, 52, 157–159. [Google Scholar] [CrossRef]
- Teleman, A.; Larsson, P.T.; Iversen, T. On the accessibility and structure of xylan in birch kraft pulp. Cellulose 2001, 8, 209–215. [Google Scholar] [CrossRef]
- Hemmingson, J.A.; Newnan, R. A CP/MAS 13C NMR study of the effect of steam explosion processes on wood composition and structure. J. Wood Chem. Technol. 1985, 5, 159–188. [Google Scholar] [CrossRef]
- Zhang, Y.; Lynd, L. Determination of the number-average degree of polymerization of cellodextrins and cellulose with application to enzymatic hydrolysis. Biomacromolecules 2005, 6, 1510–1515. [Google Scholar] [CrossRef] [PubMed]
- Hubbell, C.; Ragauskas, A.J. Effect of acid-chlorite delignification on cellulose degree of polymerization. Bioresour. Technol. 2010, 101, 7370–7375. [Google Scholar] [CrossRef]
- Wood, B.F.; Conner, A.H.; Hill, C.G., Jr. The effect of precipitation on the molecular weight distribution of cellulose tricarbanilate. J. Appl. Polym. Sci. 1986, 32, 3703–3712. [Google Scholar] [CrossRef]
- Stephens, C.; Whitmore, P.; Morris, H.; Bier, M. Hydrolysis of the amorphous cellulose in cotton-based paper. Biomacromolecules 2008, 9, 1093–1099. [Google Scholar] [CrossRef] [PubMed]
- Holtman, K. An Investigation of the Milled Wood Lignin Isolation Procedure by Solution- and Solid-State NMR Spectroscopy. Ph.D. Dissertation, North Carolina State University, Raleigh, NC, USA, 2003. [Google Scholar]
- Davis, M. A rapid modified method for compositional carbohydrate analysis of lignocellulosics by high pH anion-exchange chomatography with pulsed amperometric detection (HPAEC/PAD). J. Wood Chem. Technol. 1998, 18, 235–252. [Google Scholar] [CrossRef]
- NutsPro—NMR Utility Transform Software—Professional; Acorn NMR: Livermore, CA, USA, 1993. Available online: http://www.acornnmr.com (accessed on 30 September 2011).
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Foston, M.B.; Hubbell, C.A.; Ragauskas, A.J. Cellulose Isolation Methodology for NMR Analysis of Cellulose Ultrastructure. Materials 2011, 4, 1985-2002. https://doi.org/10.3390/ma4111985
Foston MB, Hubbell CA, Ragauskas AJ. Cellulose Isolation Methodology for NMR Analysis of Cellulose Ultrastructure. Materials. 2011; 4(11):1985-2002. https://doi.org/10.3390/ma4111985
Chicago/Turabian StyleFoston, Marcus B., Chistopher A. Hubbell, and Art J. Ragauskas. 2011. "Cellulose Isolation Methodology for NMR Analysis of Cellulose Ultrastructure" Materials 4, no. 11: 1985-2002. https://doi.org/10.3390/ma4111985
APA StyleFoston, M. B., Hubbell, C. A., & Ragauskas, A. J. (2011). Cellulose Isolation Methodology for NMR Analysis of Cellulose Ultrastructure. Materials, 4(11), 1985-2002. https://doi.org/10.3390/ma4111985