
Adsorption and Dissociation of 2-Chlorophenols on the 2D ZnO Monolayer Decorated with Al Atoms: A DFT Study


Abstract
1. Introduction
2. Calculation Methods and Models
3. Results and Discussion
3.1. Adsorption of 2-Chlorophenol on Pristine ZnO Monolayers
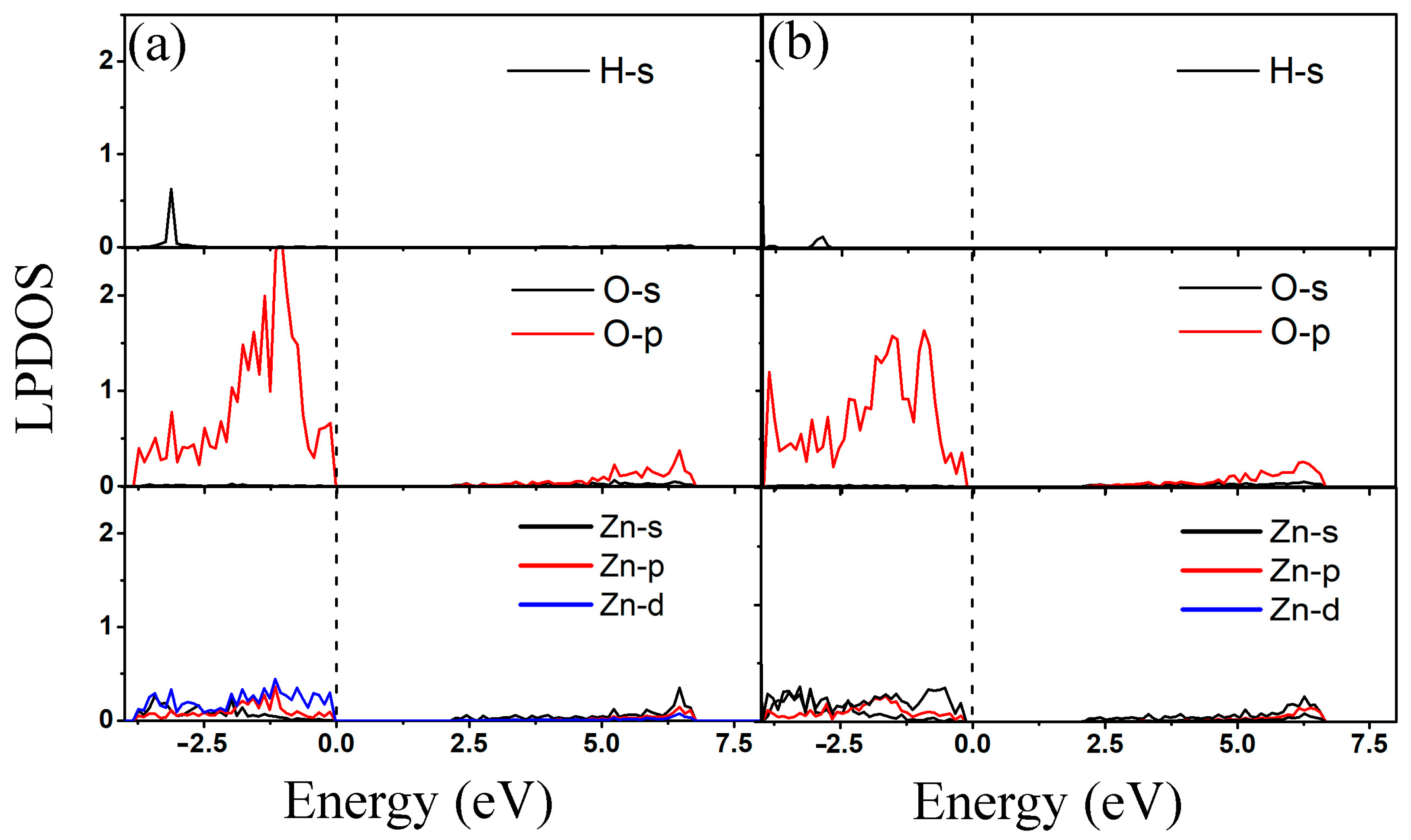
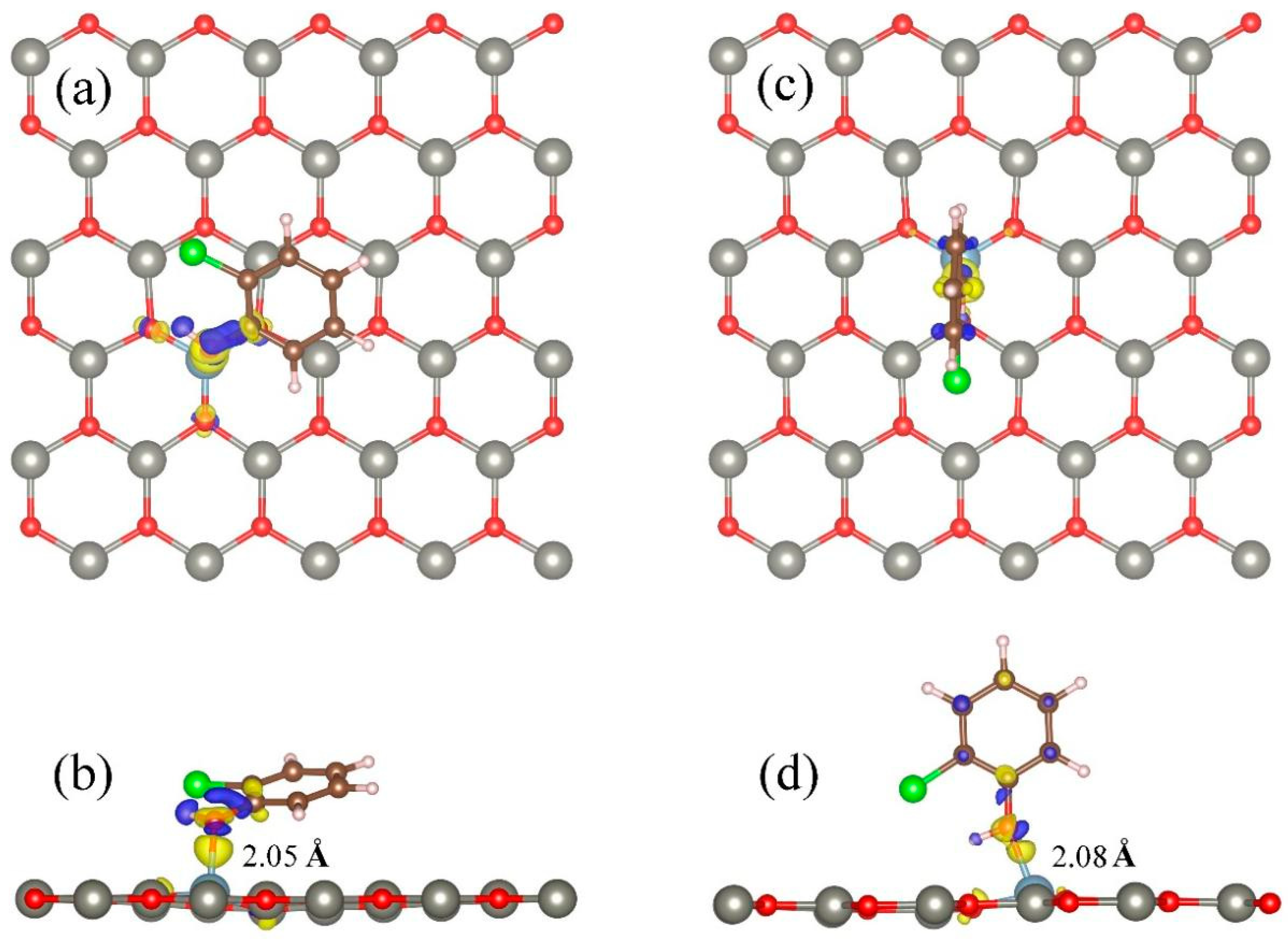
3.2. Adsorption of 2-Chlorophenol on Al-Decorated ZnO Monolayers
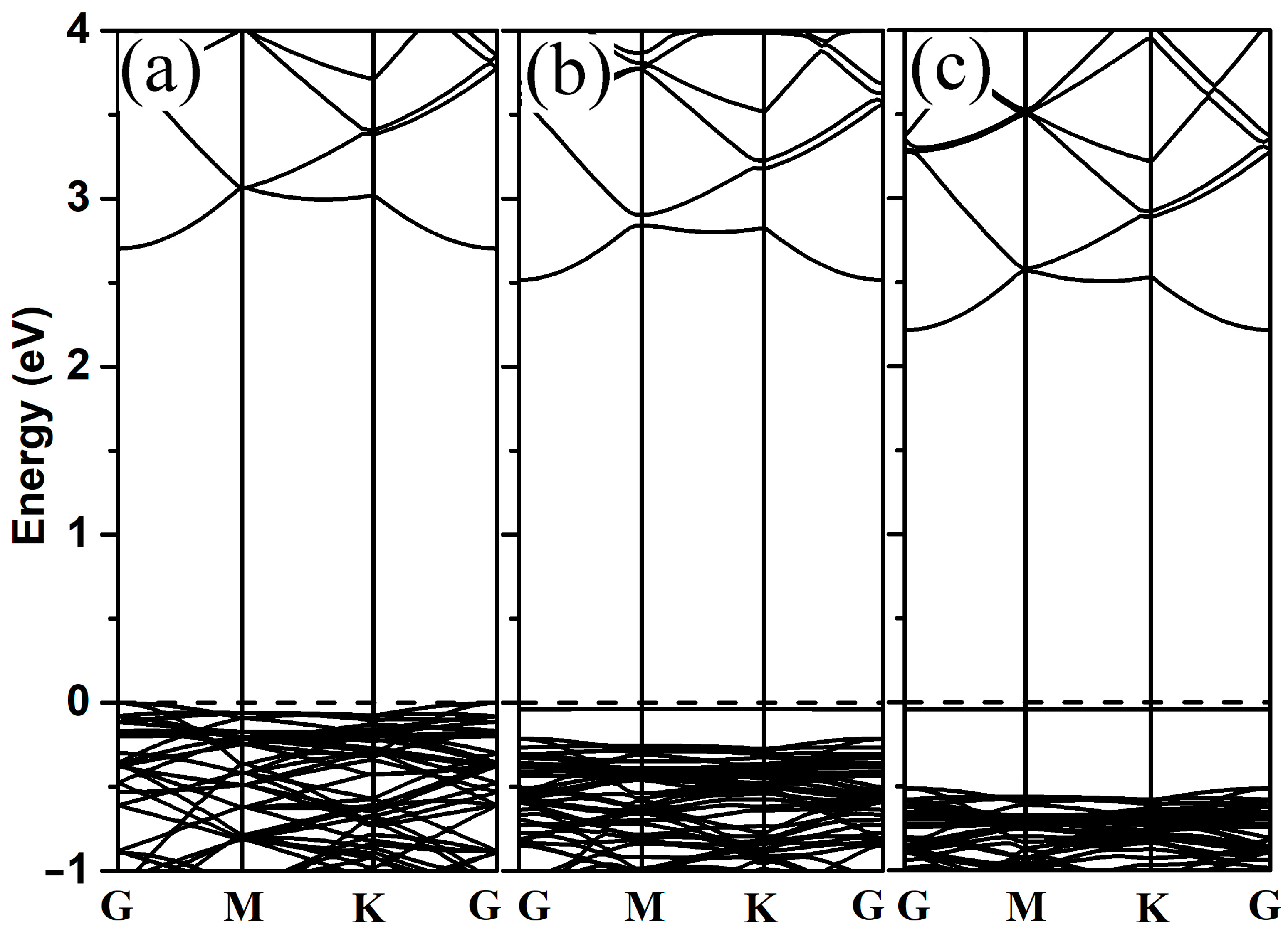
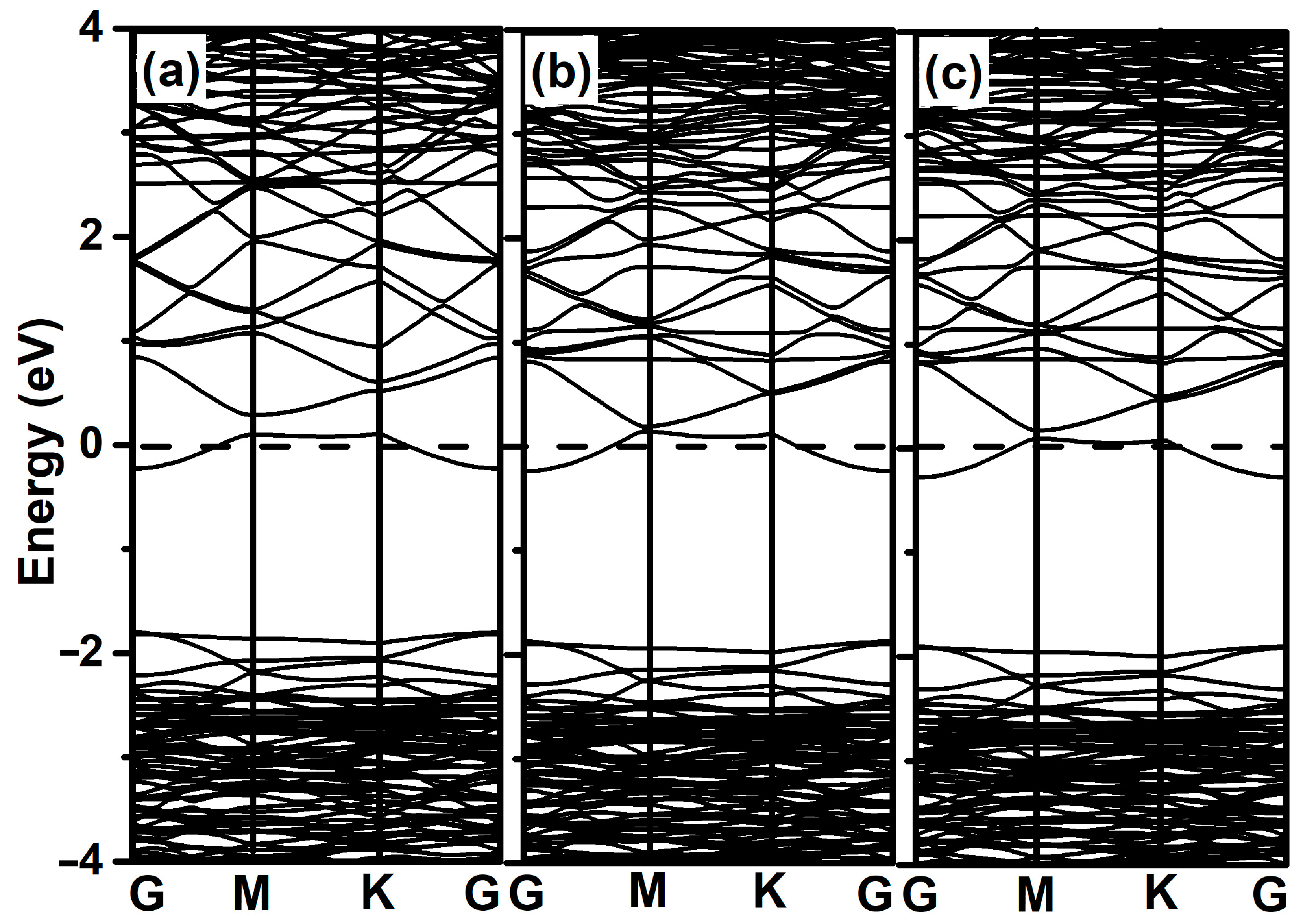
3.3. Dissociation of 2-Chlorophenol on Pristine and Al-Decorated ZnO Monolayers
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Khachatryan, L.; Asatryan, R.; Dellinger, B. An Elementary Reaction Kinetic Model of the Gas-Phase Formation of Polychlorinated Dibenzofurans from Chlorinated Phenols. J. Phys. Chem. A 2004, 108, 9567–9572. [Google Scholar] [CrossRef]
- Zhang, Q.; Li, S.; Qu, X.; Shi, X.; Wang, W. A Quantum Mechanical Study on the Formation of PCDD/Fs from 2-Chlorophenol as Precursor. Environ. Sci. Technol. 2008, 42, 7301–7308. [Google Scholar] [CrossRef] [PubMed]
- Altarawneh, M.; Dlugogorski, B.Z.; Kennedy, E.M.; Mackie, J.C. Mechanisms for formation, chlorination, dechlorination and destruction of polychlorinated dibenzo-p-dioxins and dibenzofurans (PCDD/Fs). Prog. Energy Combust. Sci. 2009, 35, 245–274. [Google Scholar] [CrossRef]
- Pan, W.; Zhang, D.; Han, Z.; Zhan, J.; Liu, C. New Insight into the Formation Mechanism of PCDD/Fs from 2-Chlorophenol Precursor. Environ. Sci. Technol. 2013, 47, 8489–8498. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, D.; Gao, J.; Zhan, J.; Liu, C. New Understanding of the Formation of PCDD/Fs from Chlorophenol Precursors: A Mechanistic and Kinetic Study. J. Phys. Chem. A 2014, 118, 449–456. [Google Scholar] [CrossRef]
- Pan, W.; Fu, J.; Zhang, A. Theoretical study on the formation mechanism of pre-intermediates for PXDD/Fs from 2-Bromophenol and 2-Chlorophenol precursors via radical/molecule reactions. Environ. Pollut. 2017, 225, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.; Chang, J.; Liu, X.; Xue, Q.; Fu, J.; Zhang, A. Interfacial formation of environmentally persistent free radicals—A theoretical investigation on pentachlorophenol activation on montmorillonite in PM2.5. Ecotoxicol. Environ. Saf. 2019, 169, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lv, J.; Ying, Y.; Ma, Y.; Wu, A.; Lin, X.; Cao, A.; Li, X.; Yan, J. A new insight into the CaO-induced inhibition pathways on PCDD/F formation: Metal passivation, dechlorination and hydroxide substitution. Sci. Total Environ. 2023, 885, 163782. [Google Scholar] [CrossRef] [PubMed]
- Altarawneh, M.; Radny, M.W.; Smith, P.V.; Mackie, J.C.; Kennedy, E.M.; Dlugogorski, B.Z.; Soon, A.; Stampfl, C. A first-principles density functional study of chlorophenol adsorption on Cu2O (110): CuO. J. Chem. Phys. 2009, 130, 184505. [Google Scholar] [CrossRef] [PubMed]
- Altarawneh, M.; Radny, M.W.; Smith, P.V.; Mackie, J.C.; Kennedy, E.M.; Dlugogorski, B.Z.; Soon, A.; Stampfl, C. Adsorption of 2-chlorophenol on Cu2O (111)–Cu CUS: A first-principles density functional study. Appl. Surf. Sci. 2010, 256, 4764–4770. [Google Scholar] [CrossRef]
- Gao, J.; Teplyakov, A.V. Thermal transformations of 2-chlorophenol on a surface of ZnO powder catalyst. Catal. Today 2014, 238, 111–117. [Google Scholar] [CrossRef]
- Mosallanejad, S.; Dlugogorski, B.Z.; Kennedy, E.M.; Stockenhuber, M. Adsorption of 2-chlorophenol on the Surface of Silica- and Alumina- supported Iron Oxide: An FTIR and XPS Study. ChemCatChem 2017, 9, 481–491. [Google Scholar] [CrossRef]
- Assaf, N.W.; Altarawneh, M.; Radny, M.W.; Al-Nu’airat, J.; Dlugogorski, B.Z. Formation of environmentally-persistent free radicals (EPFR) on α-Al2 O3 clusters. RSC Adv. 2017, 7, 52672–52683. [Google Scholar] [CrossRef]
- Ahmed, O.H.; Altarawneh, M.; Harahsheh, M.A.; Jiang, Z.; Dlugogorski, B.Z. Formation of phenoxy-type Environmental Persistent Free Radicals (EPFRs) from dissociative adsorption of phenol on Cu/Fe and their partial oxides. Chemosphere 2020, 240, 124921. [Google Scholar] [CrossRef] [PubMed]
- Altarawneh, M.; Assaf, N.W.; Hussain, H.M.; Dlugogorski, B.Z. Structural properties of alumina surfaces and their roles in the synthesis of environmentally persistent free radicals (EPFRs). Nanotechnol. Rev. 2023, 12, 20220536. [Google Scholar] [CrossRef]
- Vejerano, E.P.; Rao, G.; Khachatryan, L.; Cormier, S.A.; Lomnicki, S. Environmentally Persistent Free Radicals: Insights on a New Class of Pollutants. Environ. Sci. Technol. 2018, 52, 2468–2481. [Google Scholar] [CrossRef]
- Pan, B.; Li, H.; Lang, D.; Xing, B. Environmentally persistent free radicals: Occurrence, formation mechanisms and implications. Environ. Pollut. 2019, 248, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Freeman, C.L.; Claeyssens, F.; Allan, N.L. Graphitic Nanofilms as Precursors to Wurtzite Films: Theory. Phys. Rev. Lett. 2006, 96, 066102. [Google Scholar] [CrossRef] [PubMed]
- Tusche, C.; Meyerheim, H.L.; Kirschner, J. Observation of Depolarized ZnO(0001) Monolayers:Formation of Unreconstructed Planar Sheets. Phys. Rev. Lett. 2007, 99, 026102. [Google Scholar] [CrossRef]
- Weirum, G.; Barcaro, G.; Fortunelli, A.; Weber, F.; Schennach, R.; Surnev, S.; Netzer, F.P. Growth and Surface Structure of Zinc Oxide Layers on a Pd(111) Surface. J. Phys. Chem. C 2010, 114, 15432–15439. [Google Scholar] [CrossRef]
- Peng, Q.; Han, L.; Wen, X.; Liu, S.; Chen, Z.; Lian, J.; De, S. Mechanical properties and stabilities of g-ZnS monolayers. RSC Adv. 2015, 5, 11240. [Google Scholar] [CrossRef]
- Hong, H.K.; Jo, J.; Hwang, D.; Lee, J.; Kim, N.Y.; Son, S.; Kim, J.H.; Jin, M.J.; Jun, Y.C.; Erni, R.; et al. Atomic scale study on growth and heteroepitaxy of ZnO monolayer on grapheme. Nano. Lett. 2017, 17, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Son, S.; Cho, Y.; Hong, H.K.; Lee, J.; Kim, J.H.; Kim, K.; Lee, Y.; Yoon, A.; Shin, H.J.; Lee, Z. Spontaneous Formation of a ZnO Monolayer by the Redox Reaction of Zn on Graphene Oxide. ACS Appl. Mater. Interfaces 2020, 12, 54222–54229. [Google Scholar] [CrossRef] [PubMed]
- Chaurasiya, R.; Dixit, A.; Pandey, R. Strain-driven thermodynamic stability and electronic transitions in ZnX (X=O, S, Se, and Te) monolayers. J. Appl. Phys. 2019, 125, 082540. [Google Scholar] [CrossRef]
- Topsakal, M.; Cahangirov, S.; Bekaroglu, E.; Ciraci, S. First-principles study of zinc oxide honeycomb structures. Phys. Rev. B 2009, 80, 235119. [Google Scholar] [CrossRef]
- Wang, H.; Yang, J.; Xu, C.; Huang, H.; Min, Q.; Xiong, Y.; Luo, S. Investigations on Structural, Electronic and Optical Properties of ZnO in Two-dimensional Configurations By First-principles Calculations. J. Phys. Condens. Matter 2023, 35, 014002. [Google Scholar] [CrossRef] [PubMed]
- Ta, H.Q.; Zhao, L.; Pohl, D.; Pang, J.; Trzebicka, B.; Rellinghaus, B.; Pribat, D.; Gemming, T.; Liu, Z.; Bachmatiuk, A.; et al. Graphene-Like ZnO: A Mini Review. Crystals 2016, 6, 100. [Google Scholar] [CrossRef]
- Tu, Z.C. First-Principles Study on Physical Properties of a Single ZnO Monolayer with Graphene-Like Structure. J. Comput. Theor. Nanosci. 2010, 7, 1182–1186. [Google Scholar] [CrossRef]
- Chen, H.; Tan, C.; Zhang, K.; Zhao, W.; Tian, X.; Huang, Y. Enhanced photocatalytic performance of ZnO monolayer for water splitting via biaxial strain and external electric field. Appl. Surf. Sci. 2019, 481, 1064–1071. [Google Scholar] [CrossRef]
- Lang, R.; Du, X.; Huang, Y.; Jiang, X.; Zhang, Q.; Guo, Y.; Liu, K.; Qiao, B.; Wang, A.; Zhang, T. Single-Atom Catalysts Based on the Metal−Oxide Interaction. Chem. Rev. 2020, 120, 11986–12043. [Google Scholar] [CrossRef]
- Kaiser, S.K.; Chen, Z.; Akl, D.F.; Mitchell, S.; Ramírez, J. Single-Atom Catalysts across the Periodic Table. Chem. Rev. 2020, 120, 11703–11809. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Barbar, A.; Najam, T.; Javed, M.S.; Shen, J.; Tsiakaras, P.; Cai, X. Single noble metal atoms doped 2D materials for catalysis. Appl. Catal. B Environ. 2021, 297, 120389. [Google Scholar] [CrossRef]
- Liang, X.; Fu, N.; Yao, S.; Li, Z.; Li, Y. The Progress and Outlook of Metal Single-Atom-Site Catalysis. J. Am. Chem. Soc. 2022, 144, 18155–18174. [Google Scholar] [CrossRef]
- Ren, J.; Zhang, H.; Cheng, X. Electronic and Magnetic Properties of all 3d Transition-metal Doped ZnO Monolayers. Int. J. Quantum Chem. 2013, 113, 2243–2250. [Google Scholar] [CrossRef]
- Lei, J.; Xu, M.; Hu, S. Transition metal decorated graphene-like zinc oxide monolayer: A first-principles investigation. J. Appl. Phys. 2015, 118, 104302. [Google Scholar] [CrossRef]
- Zhang, M.; Shi, X.; Wang, X.; Li, T.; Huttula, M.; Luo, Y.; Cao, W. Transition Metal Adsorbed-Doped ZnO Monolayer: 2D Dilute Magnetic Semiconductor, Magnetic Mechanism, and Beyond 2D. ACS Omega 2017, 2, 1192–1197. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhang, X.; Xiong, Z.; Liu, Y.; Cui, Y.; Liu, B. Noble metal dopants modified two-dimensional zinc oxide: Electronic structures and magnetic properties. J. Alloys Compd. 2019, 798, 149–157. [Google Scholar] [CrossRef]
- Li, Y.; Yang, X. Effects of Fe doping on the magnetic and absorption spectrum of graphene-like ZnO monolayer from first-principles calculations. Chem. Phys. 2023, 565, 111742. [Google Scholar] [CrossRef]
- Qu, Y.; Ding, J.; Fu, H.; Peng, J.; Chen, H. Adsorption of CO, NO, and NH3 on ZnO monolayer decorated with noble metal (Ag, Au). Appl. Surf. Sci. 2020, 508, 145202. [Google Scholar] [CrossRef]
- Lalroliana, B.; Tiwari, R.C.; Madaka, R. Transition metal decorated ZnO monolayer for CO and NO sensing: A DFT +U study with vdW correction. App. Surf. Sci. 2022, 604, 154570. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, Y.; Wei, Z.; Wang, Q.; Liang, Z.; Yuan, T. Ni-Decorated ZnO Monolayer for Sensing CO and HCHO in Dry-Type Transformers: A First-Principles Theory. Chemosensors 2022, 10, 307. [Google Scholar] [CrossRef]
- Chen, L.; Xiong, Z.; Cui, Y.; Luo, H.; Gao, Y. Adsorption of C6H6 and C7H8 onto pristine and metal (Pd, Pt)-mediated ZnO monolayers: Electronic and gas sensing properties. Appl. Surf. Sci. 2021, 542, 148767. [Google Scholar] [CrossRef]
- Ma, D.; Wang, Q.; Li, T.; Tang, Z.; Yang, G.; He, C.; Lu, Z. CO catalytic oxidation on Al-doped graphene-like ZnO monolayer sheets: A first-principles study. J. Mater. Chem. C 2015, 3, 9964–9972. [Google Scholar] [CrossRef]
- Nguyen, D.C.; Phung, T.K.; Vo, D.N.; Le, T.H.; Khieu, D.Q.; Pham, T.L.M. Unraveling the effect of Al doping on CO adsorption at ZnO(10-10). RSC Adv. 2020, 10, 40663–40672. [Google Scholar] [CrossRef]
- Altarawneh, M.; Radny, M.W.; Smith, P.V.; Mackie, J.C.; Kennedy, E.M.; Dlugogorski, B.Z. 2-Chlorophenol adsorption on Cu(100): First-principles density functional study. Surf. Sci. 2008, 602, 1554–1562. [Google Scholar] [CrossRef]
- Altarawneh, M.; Radny, M.W.; Smith, P.V.; Mackie, J.C.; Kennedy, E.M.; Dlugogorski, B.Z. Adsorption of chlorophenol on the Cu(1 1 1) surface: A first-principles density functional theory study. Appl. Surf. Sci. 2008, 254, 4218–4224. [Google Scholar] [CrossRef]
- Katoorani, P.; Ebrahimi, S. Adsorption of acetone onto the pristine and Al-doped ZnO nanotubes: A dispersion corrected DFT study. Mater. Sci. Semicond. Process. 2021, 136, 106141. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Dudarev, S.L.; Botton, G.A.; Savrasov, S.Y.; Humphreys, C.J.; Sutton, A.P. Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+U study. Phys. Rev. B 1998, 57, 1505–1509. [Google Scholar] [CrossRef]
- Henkelman, G.; Uberuaga, B.P.; Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Orientation | Position | Ead (eV) | h (Å) | Q (e) |
|---|---|---|---|---|
| parallel | TZn | −0.68 | 2.56 | 0.063 |
| H | −0.61 | 2.91 | 0.055 | |
| TO | −0.57 | 3.40 | 0.044 | |
| vertical | TZn | −0.47 | 2.62 | 0.036 |
| H | −0.46 | 2.95 | 0.031 | |
| TO | −0.45 | 3.44 | 0.025 |
| Orientation | Position | Ead (eV) | h (Å) | Q (e) |
|---|---|---|---|---|
| parallel | TAl | −1.12 | 2.05 | 0.153 |
| TZn | −0.70 | 2.55 | 0.073 | |
| H | −0.62 | 2.90 | 0.064 | |
| TO | −0.58 | 3.39 | 0.050 | |
| vertical | TAl | −0.97 | 2.08 | 0.103 |
| TZn | −0.48 | 2.60 | 0.043 | |
| H | −0.47 | 2.92 | 0.041 | |
| TO | −0.46 | 3.42 | 0.030 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zong, Z.; Wang, C.; Zhao, M.; Chen, W.; Jia, Y. Adsorption and Dissociation of 2-Chlorophenols on the 2D ZnO Monolayer Decorated with Al Atoms: A DFT Study. Materials 2025, 18, 813. https://doi.org/10.3390/ma18040813
Zong Z, Wang C, Zhao M, Chen W, Jia Y. Adsorption and Dissociation of 2-Chlorophenols on the 2D ZnO Monolayer Decorated with Al Atoms: A DFT Study. Materials. 2025; 18(4):813. https://doi.org/10.3390/ma18040813
Chicago/Turabian StyleZong, Zhengjun, Changqing Wang, Miaomiao Zhao, Weiguang Chen, and Yu Jia. 2025. "Adsorption and Dissociation of 2-Chlorophenols on the 2D ZnO Monolayer Decorated with Al Atoms: A DFT Study" Materials 18, no. 4: 813. https://doi.org/10.3390/ma18040813
APA StyleZong, Z., Wang, C., Zhao, M., Chen, W., & Jia, Y. (2025). Adsorption and Dissociation of 2-Chlorophenols on the 2D ZnO Monolayer Decorated with Al Atoms: A DFT Study. Materials, 18(4), 813. https://doi.org/10.3390/ma18040813