Technetium Immobilization on Carbon Steel Corrosion Products Under Simulated Geological Radioactive Waste Repository Conditions




Abstract
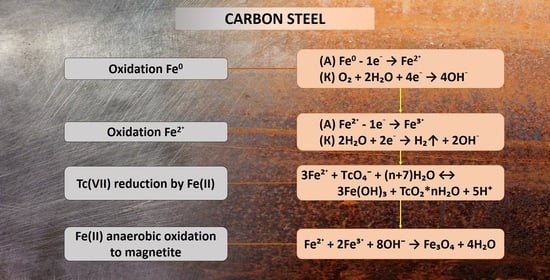
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Experimental Setup
2.2.1. Experiments with Carbon Steel Plates
2.2.2. Sorption Experiments with Synthesized and Commercial Iron Phases
2.2.3. The Effect of Barrier Components on the Immobilization of Technetium
2.3. Analytical Techniques
3. Results
3.1. Interaction of Pertechnetate Ions with Carbon Steel Samples During Their Corrosion
3.2. Characterization of Corrosion Products
3.2.1. Electron Microscopy Analysis
3.2.2. Identification of Corrosion Products by XPS
3.3. Evaluation of the Oxidation State of Technetium on a Steel Surface
3.4. Assessing the Role of Corrosion Products in Technetium Immobilization
3.5. Analysis of Technetium Forms on Corrosion Products by the Stepwise Desorption Method
3.6. The Influence of Engineering Safety Barrier Components on the Immobilization of Tc on Corrosion Products
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ZVI | Zero-valent iron |
| DGR | Deep geological repository |
| XPS | X-ray photoelectron spectroscopy |
| SEM | Scanning electron microscopy |
| Eh | Oxidation Reduction Potential |
| S:L | Solid-to-liquid |
References
- Desmet, G.; Myttenaere, C. (Eds.) Technetium in the Environment; Topics in Health Physics and Radiation Protection; Springer Science & Business Media: London, UK, 1986; Volume 10102. [Google Scholar]
- Yoshihara, K. Technetium in the Environment. In Technetium and Rhenium: Their Chemistry and Its Applications; Yoshihara, K., Omori, T., Eds.; Springer: Berlin/Heidelberg, Germany, 2005; pp. 17–35. [Google Scholar]
- Icenhower, J.P.; Qafoku, N.P.; Zachara, J.M.; Martin, W.J. The Biogeochemistry of Technetium: A Review of the Behavior of an Artificial Element in the Natural Environment. Am. J. Sci. 2010, 310, 721–752. [Google Scholar] [CrossRef]
- Meena, A.H.; Arai, Y. Environmental Geochemistry of Technetium. Environ. Chem. Lett. 2017, 15, 241–263. [Google Scholar] [CrossRef]
- Pegg, I.L. Behavior of Technetium in Nuclear Waste Vitrification Processes. J. Radioanal. Nucl. Chem. 2015, 305, 287–292. [Google Scholar] [CrossRef]
- Isaacs, M.; Lange, S.; Deissmann, G.; Bosbach, D.; Milodowski, A.E.; Read, D. Retention of Technetium-99 by Grout and Backfill Cements: Implications for the Safe Disposal of Radioactive Waste. Appl. Geochem. 2020, 116, 104580. [Google Scholar] [CrossRef]
- Totskiy, Y.; Knappett, C.; Lloyd, A.W.; Graham, J.; Smith, K.; Harrison, M.T.; Law, G.T.W. A Contribution from Fundamental and Applied Technetium Chemistry to the Nuclear Waste Disposal Safety Case. In Key Topics in Deep Geological Disposal; Conference Report; EURADWASTE: Luxembourg, 2015; Volume 7696. [Google Scholar]
- Singh, B.K.; Um, W.; Catalano, J.G.; Gartman, B.N.; Pearce, C.I.; Zavarin, M.; Szecsody, J.E.; N’Diaye, A.T.; Crum, J.V.; Kerisit, S.N. Design and Application of Materials for Sequestration and Immobilization of 99Tc. Environ. Sci. Technol. 2023, 57, 6776–6798. [Google Scholar] [CrossRef]
- Um, W.; Chang, H.-S.; Icenhower, J.P.; Lukens, W.W.; Serne, R.J.; Qafoku, N.P.; Westsik, J.H.; Buck, E.C.; Smith, S.C. Immobilization of 99-Technetium (VII) by Fe (II)-Goethite and Limited Reoxidation. Environ. Sci. Technol. 2011, 45, 4904–4913. [Google Scholar] [CrossRef]
- Asmussen, R.M.; Matyas, J.; Qafoku, N.P.; Neeway, J.J.; Lawter, A.R.; Sevigny, G.J. Getters for Improved Technetium Containment in Cementitious Waste Forms. J. Hazard. Mater. 2018, 341, 238–247. [Google Scholar] [CrossRef]
- Westsik, J.H.; Serne, R.J.; Lindberg, M.J. Technetium Immobilization Forms Literature Survey; PNNL-23268; Pacific Northwest National Laboratory: Richland, WA, USA, 2014.
- Lukens, W.W.; Bucher, J.J.; Shuh, D.K.; Edelstein, N.M. Evolution of Technetium Speciation in Reducing Grout. Environ. Sci. Technol. 2005, 39, 8064–8070. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez Hernandez, D.M. Technetium Environmental Chemistry. Mechanisms for the Surface-Mediated Reduction of Tc (VII). Ph.D. Thesis, Technische Universität Dresden, Dresden, Germany, 2021. [Google Scholar]
- Schulte, E.H.; Scoppa, P. Sources and Behavior of Technetium in the Environment. Sci. Total Environ. 1987, 64, 163–179. [Google Scholar] [CrossRef] [PubMed]
- Um, W.; Chang, H.-S.; Icenhower, J.P.; Lukens, W.W.; Serne, R.J.; Qafoku, N.P.; Westsik, J.H. Immobilization and Limited Reoxidation of Technetium-99 by Fe (II)-Goethite; PNNL-19833; Pacific Northwest National Laboratory: Richland, WA, USA, 2010.
- Fan, D.; Anitori, R.P.; Tebo, B.M.; Tratnyek, P.G.; Lezama Pacheco, J.S.; Kukkadapu, R.K.; Engelhard, M.H.; Bowden, M.E.; Kovarik, L.; Washton, N.M. Oxidative Remobilization of Technetium Sequestered by Sulfide-Transformed Nano Zerovalent Iron. Environ. Sci. Technol. 2014, 48, 7409–7417. [Google Scholar] [CrossRef] [PubMed]
- King, F. Container Materials for the Storage and Disposal of Nuclear Waste. Corrosion 2013, 69, 986–1011. [Google Scholar] [CrossRef] [PubMed]
- Ahn, T.M. Multiple Lines of Evidence for Performance of the Canister and Waste Form in Long-Term Nuclear Waste Disposal: Reviews. Prog. Nucl. Energy 2016, 93, 343–350. [Google Scholar] [CrossRef]
- Naish, C.C.; Balkwill, P.H.; O’Brien, T.M.; Taylor, K.J.; Marsh, G.P. The Anaerobic Corrosion of Carbon Steel in Concrete; Report EUR-13663; European Commission: Brussels, Belgium, 1991.
- Platts, N.; Blackwood, D.J.; Naish, C.C. Anaerobic Oxidation of Carbon Steel in Granitic Groundwaters: A Review of the Relevant Literature; Technical Report TR-94-01; Svensk Kärnbränslehantering AB (SKB): Stockholm, Sweden, 1994. [Google Scholar]
- Smart, N.R.; Blackwood, D.J.; Werme, L. Anaerobic Corrosion of Carbon Steel and Cast Iron in Artificial Groundwaters: Part 1—Electrochemical Aspects. Corrosion 2002, 58, 547–559. [Google Scholar] [CrossRef]
- Linnenbom, V.J. The Reaction Between Iron and Water in the Absence of Oxygen. J. Electrochem. Soc. 1958, 105, 322. [Google Scholar] [CrossRef]
- Rodwell, W.R.; Harris, A.W.; Horseman, S.T.; Lalieux, P.; Müller, W.; Ortiz Amaya, L.; Pruess, K. Gas Migration and Two-Phase Flow Through Engineered and Geological Barriers for a Deep Repository for Radioactive Waste; Report EUR-19122 EN; European Commission: Brussels, Belgium, 1999.
- Druteikienė, R.; Lukšienė, B.; Pečiulytė, D.; Mažeika, K.; Gudelis, A.; Baltrūnas, D. Behaviour of 99Tc in aqueous solutions in the presence of iron oxides and microorganisms. Appl. Radiat. Isot. 2014, 89, 85–94. [Google Scholar] [CrossRef]
- Boglaienko, D.; Levitskaia, T.G. The Abiotic Reductive Removal and Subsequent Incorporation of Tc(IV) into Iron Oxides: A Frontier Review. Environ. Sci. Nano 2019, 6, 3492–3500. [Google Scholar] [CrossRef]
- Laverov, N.P.; Yudintsev, S.V.; Kochkin, B.T.; Malkovsky, V.I. The Russian Strategy of Using Crystalline Rock as a Repository for Nuclear Waste. Elements 2016, 12, 253–256. [Google Scholar] [CrossRef]
- Petrov, V.G.; Zakharova, E.V.; Kalmykov, S.N.; Petrov, V.G.; Orelovich, O.L.; Maslakov, K.I.; Tukumova, A.S. Preferential Sorption of Radionuclides on Different Mineral Phases Typical for Host Rocks at the Site of the Future Russian High Level Waste Repository. Appl. Geochem. 2019, 100, 90–95. [Google Scholar] [CrossRef]
- Tyupina, E.A.; Kozlov, P.P.; Krupskaya, V.V. Application of Cement-Based Materials as a Component of an Engineered Barrier System at Geological Disposal Facilities for Radioactive Waste—A Review. Energies 2023, 16, 605. [Google Scholar] [CrossRef]
- Morozov, I.; Zakusin, S.; Kozlov, P.; Zakusina, O.; Roshchin, M.; Chernov, M.; Boldyrev, K.; Zaitseva, T.; Tyupina, E.; Krupskaya, V. Bentonite–Concrete Interactions in Engineered Barrier Systems during the Isolation of Radioactive Waste Based on the Results of Short-Term Laboratory Experiments. Appl. Sci. 2022, 12, 3074. [Google Scholar] [CrossRef]
- GOST R 380-2005; Common Quality Carbon Steel Grades. Standardinform: Moscow, Russia, 2008; 11p. (In Russian)
- Rozov, K.B.; Rumynin, V.G.; Nikulenkov, A.M.; Leskova, P.G. Sorption of 137Cs, 90Sr, Se, 99Tc, 152(154)Eu, 239(240)Pu on Fractured Rocks of the Yeniseysky Site (Nizhne-Kansky Massif, Krasnoyarsk Region, Russia). J. Environ. Radioact. 2018, 192, 513–523. [Google Scholar] [CrossRef]
- Tyurina, M.V.; Chekulaev, M.A.; Avdeev, Y.G.; Kuznetsov, Y.I. Protection of Low-Carbon Steel in Solutions of Mineral Acids by Inhibitor of the IFKhAN-29 Series. Prot. Met. Phys. Chem. Surf. 2022, 58, 1284–1289. [Google Scholar] [CrossRef]
- Yoshinaga, N.; Kanasaki, N. Synthesis of ferrihydrite and feroxyhyte. Clay Sci. 1993, 9, 43–51. [Google Scholar] [CrossRef]
- Abramova, E.; Popova, N.; Artemiev, G.; Zharkova, V.; Zakharova, E.; Safonov, A. Characteristics and Rates of Microbial Processes in Clays of Different Mineral and Elemental Composition in Relation to Safety Prediction for ESB Clay Materials. Appl. Sci. 2022, 12, 1843. [Google Scholar] [CrossRef]
- Martynov, K.V.; Zakharova, E.V. Leaching of the Matrix with Radioactive Waste under Disposal Conditions on the Example of Model Phosphate Glass. Radiochemistry 2021, 63, 107–118. [Google Scholar] [CrossRef]
- Tessier, A.; Campbell, P.G.C.; Bisson, M. Sequential Extraction Procedure for the Speciation of Particulate Trace Metals. Anal. Chem. 1979, 51, 844–851. [Google Scholar] [CrossRef]
- Keith-Roach, M.J.; Morris, K.; Dahlgaard, H. An Investigation into Technetium Binding in Sediments. Mar. Chem. 2003, 81, 149–162. [Google Scholar] [CrossRef]
- Makarov, A.; Safonov, A.; Konevnik, Y.; Teterin, Y.; Maslakov, K.; Teterin, A.Y.; Karaseva, Y.; German, K.; Zakharova, E. Activated Carbon Additives for Technetium Immobilization in Bentonite-Based Engineered Barriers for Radioactive Waste Repositories. J. Hazard. Mater. 2021, 401, 123436. [Google Scholar] [CrossRef]
- Shirley, D.A. High-Resolution X-Ray Photoemission Spectrum of the Valence Bands of Gold. Phys. Rev. B 1972, 5, 4709. [Google Scholar] [CrossRef]
- Rong, Z.; Tang, X.; Wu, L.; Chen, X.; Dang, W.; Wang, Y. A Novel Method to Synthesize Scorodite Using Ferrihydrite and Its Role in Removal and Immobilization of Arsenic. J. Mater. Res. Technol. 2020, 9, 5848–5857. [Google Scholar] [CrossRef]
- Li, S.; Qin, G.W.; Zhang, Y.; Pei, W.; Zuo, L.; Esling, C. Anisotropic Growth of Iron Oxyhydroxide Nanorods and Their Photocatalytic Activity. Adv. Eng. Mater. 2010, 12, 1082–1085. [Google Scholar] [CrossRef]
- Li, X.; Wang, Y.; Zhou, Q.; Qi, T.; Liu, G.H.; Peng, Z.H.; Wang, H.Y. Transformation of Hematite in Diasporic Bauxite During Reductive Bayer Digestion and Recovery of Iron. Trans. Nonferrous Met. Soc. China 2017, 27, 2715–2726. [Google Scholar] [CrossRef]
- Chirita, M.; Banica, R.; Sfirloaga, P.; Ieta, A.; Grozescu, I. A Short Route of Micrometric Magnetite Synthesis via Fe-EDTA Thermal Decomposition. In Proceedings of the 2010 International Semiconductor Conference (CAS), Sinaia, Romania, 11–13 October 2010; Volume 2, pp. 507–510. [Google Scholar] [CrossRef]
- Yonemochi, Y.; Iijima, M.; Tsukada, M.; Jiang, H.; Kauppinen, E.I.; Kimata, M.; Hasegawa, M.; Kamiya, H. Microstructure of Iron Particles Reduced from Silica-Coated Hematite in Hydrogen. Adv. Powder Technol. 2005, 16, 621–637. [Google Scholar] [CrossRef]
- Supattarasakda, K.; Petcharoen, K.; Permpool, T.; Sirivat, A.; Lerdwijitjarud, W. Control of Hematite Nanoparticle Size and Shape by the Chemical Precipitation Method. Powder Technol. 2013, 249, 353–359. [Google Scholar] [CrossRef]
- Grosvenor, A.P.; Kobe, B.A.; Biesinger, M.C.; McIntyre, N.S. Investigation of multiplet splitting of Fe 2p XPS spectra and bonding in iron compounds. Surf. Interface Anal. 2004, 36, 1564–1574. [Google Scholar] [CrossRef]
- Panov, A.P. SPRO Spectrum Processing Software Package and the SL Programming Language; Preprint IAE-6019/15; Institute of Atomic Energy: Moscow, Russia, 1997; 31p. (In Russian)
- Maslakov, K.I.; Teterin, A.Y.; Safonov, A.V.; Makarov, A.V.; Artemiev, G.D.; Teterin, Y.A.; Dvoriak, S.V. Xps determination of the oxidation state of 99Tc isotope absorbed on the surface of pyrrhotite FenSn+1 and stibnite Sb2S3. Radiochemistry 2024, 66, 145–155. [Google Scholar] [CrossRef]
- Usman, M.; Byrne, J.M.; Chaudhary, A.; Orsetti, S.; Hanna, K.; Ruby, C.; Kappler, A.; Haderlein, S.B. Magnetite and Green Rust: Synthesis, Properties, and Environmental Applications of Mixed-Valent Iron Minerals. Chem. Rev. 2018, 118, 3251–3304. [Google Scholar] [CrossRef]
- Usman, M.; Hanna, K.; Abdelmoula, M.; Zegeye, A.; Faure, P.; Ruby, C. Formation of green rust via mineralogical transformation of ferric oxides (ferrihydrite, goethite and hematite). Appl. Clay Sci. 2012, 64, 38–43. [Google Scholar] [CrossRef]
- Smart, N.R.; Reddy, B.; Rance, A.P.; Nixon, D.J.; Diomidis, N. The Anaerobic Corrosion of Carbon Steel in Saturated Compacted Bentonite in the Swiss Repository Concept. Corros. Eng. Sci. Technol. 2017, 52, 113–126. [Google Scholar] [CrossRef]
- Abramova, E.; Gavrilov, S.; Boldyrev, K.; Dushik, V.; Klyukina, A.; Podosokorskaya, O.; Elizarov, I.; Grafov, O.; Shapagina, N.; Safonov, A. Bioinduced Corrosion of Carbon and Alloyed Steel by Thermophilic Microorganisms in the Presence of Uranyl Ions under Anaerobic Conditions. J. Nucl. Mater. 2025, 603, 155380. [Google Scholar] [CrossRef]
- Boldyrev, K.A.; Safonov, A.V.; Abramova, E.S.; Gladkikh, N.A.; Kryuchkov, D.V. Research on the St3 Carbon Steel Corrosion in the Presence of Microorganisms Isolated from the Groundwater at the Yeniseyskiy Site. Radioact. Waste 2021, 3, 84–92. [Google Scholar] [CrossRef]
- Ishikawa, T.; Kondo, Y.; Yasukawa, A.; Kandori, K. Formation of magnetite in the presence of ferric oxyhydroxides. Corros. Sci. 1998, 40, 1239–1251. [Google Scholar] [CrossRef]
- McBeth, J.M.; Lear, G.; Lloyd, J.R.; Livens, F.R.; Morris, K.; Burke, I.T. Technetium Reduction and Reoxidation in Aquifer Sediments. Geomicrobiol. J. 2007, 24, 189–197. [Google Scholar] [CrossRef]
- McBeth, J.M.; Lloyd, J.R.; Law, G.T.W.; Livens, F.R.; Burke, I.T.; Morris, K. Redox Interactions of Technetium with Iron-Bearing Minerals. Mineral. Mag. 2018, 75, 2419–2430. [Google Scholar] [CrossRef]
- Lu, C.; Samper, J.; Fritz, B.; Clement, A.; Montenegro, L. Interactions of corrosion products and bentonite: An extended multicomponent reactive transport model. Phys. Chem. Earth 2011, 36, 1661–1668. [Google Scholar] [CrossRef]
- Samper, J.; Naves, A.; Montenegro, L.; Mon, A. Reactive transport modelling of the long-term interactions of corrosion products and compacted bentonite in a HLW repository in granite: Uncertainties and relevance for performance assessment. Appl. Geochem. 2016, 67, 42–51. [Google Scholar] [CrossRef]
- Su Sun, Y.; Zhou, Y.; Wei, X.; Dong, J.; Chen, N.; Ren, Q.; Wang, J.; Ke, W. Effects of Iron Corrosion Products on the Degradation of Bentonite Structure and Properties. NPJ Mater. Degrad. 2023, 7, 66. [Google Scholar] [CrossRef]
- Kaufhold, S.; Hassel, A.W.; Sanders, D.; Dohrmann, R. About the Corrosion Mechanism of Metal Iron in Contact with Bentonite. ACS Earth Space Chem. 2020, 4, 711–721. [Google Scholar] [CrossRef]
- Yan, M.; Sun, C.; Xu, J.; Dong, J.; Ke, W. Role of Fe Oxides in Corrosion of Pipeline Steel in a Red Clay Soil. Corros. Sci. 2014, 80, 309–317. [Google Scholar] [CrossRef]
- Yang, J.; Kukkadapu, R.K.; Dong, H.; Shelobolina, E.S.; Zhang, J.; Kim, J. Effects of Redox Cycling of Iron in Nontronite on Reduction of Technetium. Chem. Geol. 2012, 291, 206–216. [Google Scholar] [CrossRef]
- Bishop, M.E.; Dong, H.; Kukkadapu, R.K.; Liu, C.; Edelmann, R.E. Bioreduction of Fe-Bearing Clay Minerals and Their Reactivity toward Pertechnetate (Tc-99). Geochim. Cosmochim. Acta 2011, 75, 5229–5246. [Google Scholar] [CrossRef]
- Kaufhold, S.; Hassel, A.W.; Sanders, D.; Dohrmann, R. Corrosion of High-Level Radioactive Waste Iron-Canisters in Contact with Bentonite. J. Hazard. Mater. 2015, 285, 464–473. [Google Scholar] [CrossRef]
- Bennett, D.G.; Gens, R. Overview of European Concepts for High-Level Waste and Spent Fuel Disposal with Special Reference Waste Container Corrosion. J. Nucl. Mater. 2008, 379, 1–8. [Google Scholar] [CrossRef]
- Pownceby, M.I.; Hapugoda, S.; Manuel, J.; Webster, N.A.S.; MacRae, C.M. Characterisation of phosphorus and other impurities in goethite-rich iron ores—Possible P incorporation mechanisms. Miner. Eng. 2019, 142, 106022. [Google Scholar] [CrossRef]
- Tyupina, E.; Kozlov, P.; Boldyrev, K.; Krupskaya, V. Prediction of Filtration Properties of Portland Cement Concrete Based on the Results of Calibration of the Basic Hydration Simulation. Open Chem. Eng. J. 2025, 19, e18742106284569. [Google Scholar] [CrossRef]
- Alexey, M.; Alexey, S.; Anastasiia, S.; Konstantin, M.; Elena, Z.; Sergey, K. Clay and carbon materials-based engineered barriers for technetium immobilization. Prog. Nucl. Energy 2022, 152, 104398. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Point | O | S | Ca | Mn | Fe | Tc |
|---|---|---|---|---|---|---|
| 1 | 23.01 | 0.69 | 68.57 | 7.73 | ||
| 2 | 23.42 | 1.00 | 0.16 | 58.05 | 17.37 | |
| 3 | 26.73 | 28.08 | 45.19 | |||
| 4 | 22.41 | 64.50 | 13.09 | |||
| 5 | 22.34 | 0.39 | 70.66 | 6.61 |
| Conditions | Aerobic Conditions | Anaerobic Conditions | ||
|---|---|---|---|---|
| Sample | Degree of Immobilization, % | Kd, cm3/g | Degree of Immobilization, % | Kd, cm3/g |
| Corrosion product powder | 96.6 | 568 | 34.2 | 134.8 |
| FeO | 98.9 | 1798 | 97.8 | 889 |
| Ferrihydrite (Fe3+10O14(OH)2), freshly precipitated | 98.8 | 1646 | 98.6 | 1409 |
| Magnetite (Fe3O4) | 3.1 | 0.6 | 0 | 0 |
| Goethite (FeO(OH)) | 4.0 | 0.8 | 0 | 0 |
| Hematite (Fe2O3) | 5.1 | 1.1 | 0 | 0 |
| Sample | A/A0, % | Kd, cm3/g | ||||
|---|---|---|---|---|---|---|
| 0 | 1 h | 4 h | 24 h | 168 h | ||
| MW | 100 | 58.1 | 27.8 | 3.4 | 3.4 | 568 |
| MWG | 100 | 57.3 | 28.5 | 6.9 | 6.8 | 474 |
| MWC | 100 | 56.6 | 27.3 | 4.8 | 4.7 | 406 |
| MWB | 100 | 65.9 | 33.8 | 13.3 | 13.0 | 134 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abramova, E.; Artemiev, G.; German, K.; Safonov, A. Technetium Immobilization on Carbon Steel Corrosion Products Under Simulated Geological Radioactive Waste Repository Conditions. Materials 2025, 18, 5220. https://doi.org/10.3390/ma18225220
Abramova E, Artemiev G, German K, Safonov A. Technetium Immobilization on Carbon Steel Corrosion Products Under Simulated Geological Radioactive Waste Repository Conditions. Materials. 2025; 18(22):5220. https://doi.org/10.3390/ma18225220
Chicago/Turabian StyleAbramova, Elena, Grigoriy Artemiev, Konstantin German, and Alexey Safonov. 2025. "Technetium Immobilization on Carbon Steel Corrosion Products Under Simulated Geological Radioactive Waste Repository Conditions" Materials 18, no. 22: 5220. https://doi.org/10.3390/ma18225220
APA StyleAbramova, E., Artemiev, G., German, K., & Safonov, A. (2025). Technetium Immobilization on Carbon Steel Corrosion Products Under Simulated Geological Radioactive Waste Repository Conditions. Materials, 18(22), 5220. https://doi.org/10.3390/ma18225220