Abstract
Considering previous studies on the high-pressure phases and compressibility of Ba–Au alloys with stoichiometries Au2Ba, AuBa, and Au2Ba3, the concentration of the alkaline-earth metal Ba increased, and a particle-swarm optimization algorithm was employed to conduct comprehensive structure searches for the Ba4Au compound at 0, 10, 20, and 50 GPa. First-principles calculations were subsequently carried out to investigate its structural evolution and electronic properties under compression. Enthalpy-difference calculations indicate that the I4/mmm phase of Ba4Au transforms to the Cmmm phase at approximately 0.4 GPa. As pressure increases above 5.7 GPa, the I4/m structure becomes energetically more favorable than Cmmm-Ba4Au, indicating that the Cmmm phase transforms to the I4/m phase at 5.7 GPa. Both phase transitions are first-order and accompanied by discernible volume collapses. Additionally, a comparative analysis of the electronic properties of Ba4Au was performed before and after the phase transitions. In this study, theoretical guidance is provided for the exploration of the high-pressure structural evolution of Ba4Au, and critical insights are offered regarding the changes that occur in its physical and chemical properties under compression.
1. Introduction
Gold-based alloy systems have garnered remarkable attention owing to their enhanced performance, cost-effectiveness, and broad application prospects compared to pure gold. Previous studies have demonstrated that gold-based alloys have extensive applications in catalysis, electronic transport, and optics [1,2,3,4]. For example, Zhao et al. [5] reported a phase-stabilized synthesis method for preparing Au3Cu alloy nanocrystals. The resulting Au–Cu alloy exhibited considerably enhanced catalytic activity in CO2 electroreduction reactions. Ishikawa et al. [6] investigated the properties of Au–In alloys, and Baranov et al. [7] examined the superconductivity of Au–Pb alloys. Both studies found that the superconducting behavior of Au–In and Au–Pb alloys is markedly enhanced compared to that of pure gold [6,7,8]. Lu et al. [9] explored the properties of Au–Al alloys and found that the AuAl2 alloy exhibits a distinctive purple appearance owing to the synergistic effect of plasmons and interband transitions, with implications for its application in jewelry coloration and optical filtering. Refs. [10,11,12] pointed out that some alkali metal aurides, such as KAu, RbAu, and CsAu, exhibit semiconducting behavior despite being composed entirely of metallic elements. Therefore, it is worthwhile to investigate novel gold-based alloy materials that may possess unique properties.
Furthermore, it has been recognized that pressure is an important means of tuning the microstructure of materials. It can induce chemical bond contractions and atomic arrangement reconstruction, thereby altering physical properties or triggering phase transitions in crystal structures [13,14,15,16,17]. Notably, at ambient pressure, the chemical behavior of Ba is governed by its 6s2 valence electrons, resulting in a typical +2 oxidation state and ionic bonding characteristic. However, under high-pressure conditions, its electronic structure undergoes a marked change. When pressure exceeds several tens of GPa, the energy levels of Ba’s inner 5p and 5d orbitals considerably increase owing to compression, enhancing s–p orbital hybridization and potentially driving the oxidation state beyond conventional limits [18,19,20,21,22]. For example, in high-pressure BaFn (n = 3, 4, and 5) compounds, Ba exhibits oxidation states from +3 to +5 by opening its inert 5p shell, with an electronic configuration approaching that of p-block elements [21]. In high-pressure Ba3Ch2 (Ch = S, Se, and Te) compounds, Ba displays mixed +1 and +2 valence states and shows d-block-like magnetic behavior [22]. Li et al. investigated the high-pressure behavior and electronic properties of BaAu compounds, demonstrating that the ambient-pressure Pnma-BaAu structure transformed into a cubic structure with Fd-3m symmetry at approximately 12 GPa [23]. This pressure-induced orbital reconstruction imparts Ba with pseudo-transition metal characteristics, enabling unconventional stoichiometries and bonding motifs in alloy systems. Owing to its unique high-pressure response and strong relativistic effects, the alkaline-earth metal Ba serves as an ideal model for exploring novel alloy compounds under extreme conditions. Therefore, it is meaningful to investigate the high-pressure phases of the Ba–Au alloy system.
In recent years, various algorithms have been developed for predicting material structures, such as minima hopping [24], metadynamics [25], the genetic algorithm [26], particle swarm optimization [27], and the random sampling method [28]. Among these, the CALYPSO structure prediction method, based on the particle swarm optimization algorithm, has been demonstrated as an effective approach for discovering new stable compounds based on known chemical compositions and external conditions. Its success has been validated in predicting the stable structures of a wide range of systems [29,30,31]. Recently, Li et al. [32] performed structure searches on the AuxBay systems (x = 1, 2; y = 1; x = 2, y = 3) at 0, 50, and 100 GPa using the CALYPSO structural prediction method, investigating their thermodynamic stability and compressibility under varying pressures, though they did not address the case of y = 4. Inspired by their work, we increased the Ba content and employed the CALYPSO crystal structure prediction method to identify stable Ba4Au structures at 0, 10, 20, and 50 GPa. Ba4Au is a new Ba-rich compound that does not appear in the Ba−Au system. Compared to combinations with lower Ba content, such as BaAu and Au2Ba, Ba4Au has a much higher Ba content and, consequently, a greater number of Ba–Au bonds. In high-pressure studies of BaAu, significant overlap between Ba 5d and Au 6p orbitals drives charge transfer and orbital hybridization [23]. In Ba4Au, this orbital mixing is expected to be even more extensive and pronounced, potentially leading to more complex electronic-structure evolution and even inducing a metal–semiconductor phase transition. This study focuses on using theoretical calculations and structure prediction to explore the pressure-induced phase transition pathways of Ba4Au in the 0–50 GPa range, revealing the evolution of its crystal structure and electronic properties. This study not only fills the research gap on Ba-rich compounds in the Ba–Au alloy system but also suggests that the Ba4Au structure has the potential to exhibit more prominent properties than structures with lower Ba contents in terms of electronic state control and pressure-induced transition behavior. It is worth noting that this study is purely theoretical and predictive in nature, with no experimental comparisons available at present. However, the results show a clear trend and can provide guidance for future experimental design and verification.
2. Computational Details
To identify the most stable crystal structure of Ba4Au, we employed the CALYPSO crystal structure prediction method, which is based on a particle swarm optimization (PSO) algorithm for global structure searching [33,34,35]. This study conducted variable-cell structure searches for Ba4Au at 0, 10, 20, and 50 GPa. The population size was set to 50 per generation, and the first generation was generated randomly under symmetry constraints. In each generation, 60% of the structures were obtained by the PSO operation of crystal structures with lower enthalpies in the previous generation, while the remaining 40% were produced randomly to ensure structural diversity. Thirty generations were sampled to ensure the convergence of the search. After the CALYPSO search, low-energy candidate phases were selected and further refined using the Vienna Ab initio Simulation Package (VASP5.4.4) [36] for structural optimization and related property calculations. Electron–ion interactions were described using the projector augmented wave method [37], and exchange–correlation effects were treated with the Perdew–Burke–Ernzerhof functional within the generalized gradient approximation [38,39]. A plane-wave cutoff energy of 600 eV and a Monkhorst–Pack k-point grid spacing of 2π × 0.02 Å−1 were employed to ensure total energy convergence within 1 × 10−5 eV/atom. Furthermore, the electron localization function (ELF) [40,41] was calculated using VASP. To assess dynamic stability, phonon dispersion calculations were performed using density functional perturbation theory as implemented in the PHONOPY code [42,43,44,45], with a force convergence threshold of 1 × 10−7 eV/Å.
3. Results and Discussion
To identify potentially stable AuxBay compounds, we extended previous work by increasing the Ba concentration. We performed comprehensive structure searches on the Ba4Au compound (corresponding to x = 1, y = 4) at 0, 10, 20, and 50 GPa. The ground state phase at 0 GPa was I4/mmm-Ba4Au, and that at 10, 20, and 50 GPa was I4/m-Ba4Au. The thermodynamic stability of each Au–Ba compound relative to elemental Au and Ba at a set pressure was quantified by the formation enthalpy [32] (∆H), defined as follows:
Here, H(AuxBay) denotes the total enthalpy of the compound while H(Au) and H(Ba) represent the enthalpies of single Au and Ba atoms in crystals; x and y denote the numbers of Au and Ba atoms in the unit cell, respectively.
Combining the structural data reported in the literature [32], we further refined the convex-hull diagram for AuxBay compounds, as shown in Figure 1. Table S2 presents the formation enthalpies of AuxBay (x = 1, y = 1, 4; x = 2, y = 1, 3) compounds at various pressures. Based on the convex-hull data, structures that exist on the solid line are thermodynamically stable at the corresponding pressure. However, points inside the convex polygon are unstable and can decompose into other compounds or elemental forms. Notably, the Ba4Au structure lies on the solid line at 0 and 50 GPa, indicating its thermodynamic stability under these conditions. This suggests a previously unreported stable Ba4Au phase. The phonon spectra of other predicted stable Ba4Au phases are shown in Figure S1.
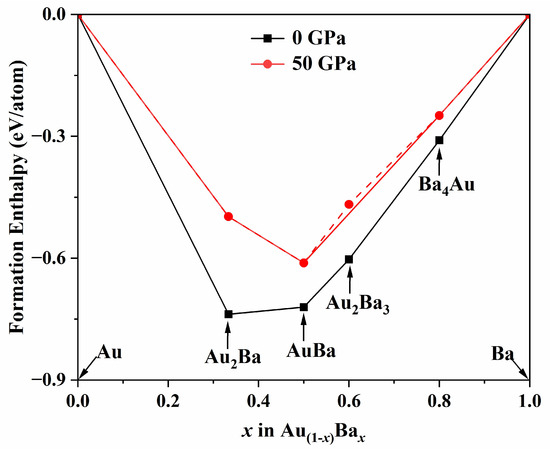
Figure 1.
Phase stabilities of various AuxBay compounds at 0 and 50 GPa. Compounds corresponding to data points located on the convex hull are thermodynamically stable, while those on the dashed lines represent that the alloys are unstable with respect to their decomposition into elements or other stoichiometries.
To investigate the potential crystal structure transitions of Ba4Au in the 0~50 GPa pressure range, we used a swarm-intelligence-based CALYPSO structure search method in conjunction with the VASP code to identify the structures of Ba4Au at pressures of 0, 10, 20, and 50 GPa, respectively. In this study, we selected the 50 lowest enthalpy Ba4Au crystal structures predicted by the CALYPSO (version 6.0) package at each pressure, performed higher-precision structural optimizations, and calculated their elastic constants and phonon spectra, obtaining ten stable structures with the lowest energies (I4/mmm, Pm-3m, Cmmm, P4/mmm, C2, P21/m, Cc, I4/m, R-3c, and Cmcm). Then, we plotted the enthalpy variations in these structures relative to the P21/m-Ba4Au phase as a function of pressure (Figure 2a). This analysis revealed pressure-induced phase transitions among the candidate polymorphs. At ambient pressure, I4/mmm-Ba4Au has the lowest energy. Above 0.4 GPa, the Cmmm phase becomes energetically favorable, indicating that the I4/mmm phase of Ba4Au transforms to the Cmmm phase at approximately 0.4 GPa. As pressure increases, above 5.7 GPa, the I4/m structure becomes energetically more favorable than Cmmm-Ba4Au, indicating that the Cmmm phase transforms to the I4/m phase at 5.7 GPa. Figure 2b presents the volume–pressure curves obtained by fitting a third-order Birch–Murnaghan equation of state. Both the I4/mmm⟶Cmmm and Cmmm⟶I4/m transitions exhibited first-order characteristics, as indicated by significant volume collapses of 8.80% and 1.67%, respectively. The I4/mmm and I4/m phases adopted tetragonal symmetry. In these phases, Au atoms occupy the eight corners and body-center positions of the cell, with Ba atoms arranged around them (Figure 3a,c). In contrast, the Cmmm phase belongs to the orthorhombic system and exhibits distinct layering: each layer of Au atoms is surrounded by several Ba atoms (Figure 3b).
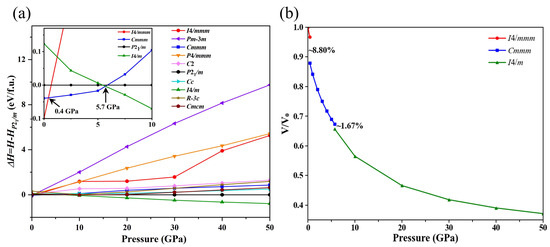
Figure 2.
(a) Enthalpy (relative to P21/m) as a function of external pressure for selected structures of Ba4Au in different symmetries. The inset shows an enlarged view of enthalpy differences for the four lowest-enthalpy phases in the 0–10 GPa region. (b) Equations of state for Ba4Au in the I4/mmm, Cmmm, and I4/m phases.

Figure 3.
Crystal structures of the (a) I4/mmm (0 GPa), (b) Cmmm (5 GPa), and (c) I4/m (10 GPa) phases of Ba4Au (Ba in green atoms and Au in gold atoms).
Table 1 lists detailed structural parameters for the I4/mmm, Cmmm, and I4/m phases of Ba4Au for further study. Table S1 in the Supplementary Materials provides detailed structural information for other Ba4Au compounds. Figure 4 presents the phonon dispersion curves for the I4/mmm, Cmmm, and I4/m phases of Ba4Au. In each case, all phonon eigenfrequencies across the Brillouin zone were positive, confirming the dynamic stability of these structures.

Table 1.
Structural parameters of the I4/mmm, Cmmm, and I4/m phases at varying pressures.
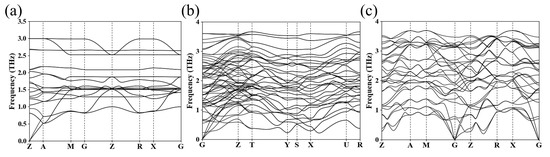
Figure 4.
Phonon spectra of (a) I4/mmm (0 GPa), (b) Cmmm (5 GPa), and (c) I4/m (10 GPa) phases of Ba4Au.
To examine the nature of the chemical bonds in Ba4Au compounds, we calculated the partial density of states for the stable phase at 0, 5, and 10 GPa, as shown in Figure 5. In all three plots, no band gap was observed at the Fermi level. This finding indicates that the electrons at the Fermi level can move freely into the conduction band, showing that the current flow is not limited by a gap. Consequently, all three Ba4Au compounds exhibit electrical conductivity and metallic character, thus confirming their classification as metallic compounds. Further analysis of the electronic states near the Fermi level indicates that electrons in the Ba 5d orbitals play a significant role in the metallic characteristics of Ba4Au. Moreover, we observed a substantial overlap between Ba 5d and Au 6p orbitals, indicating that Ba–Au bonding mainly originates from the hybridization of Ba 5d and Au 6p orbitals. Compared with the ambient-pressure I4/mmm phase (Figure 5a), the Cmmm phase at 5 GPa and the I4/m phase at 10 GPa exhibit a substantial increase in the Au 5d density of states near the Fermi level. This behavior indicates that increasing pressure strengthens the covalent interaction between the Ba 5d and Au 5d orbitals, as evidenced by the enhanced hybridization of these orbitals. Consequently, as external pressure increases, the degree of hybridization among atomic orbitals increases, which enhances the stability of the Ba4Au compound under high-pressure conditions.
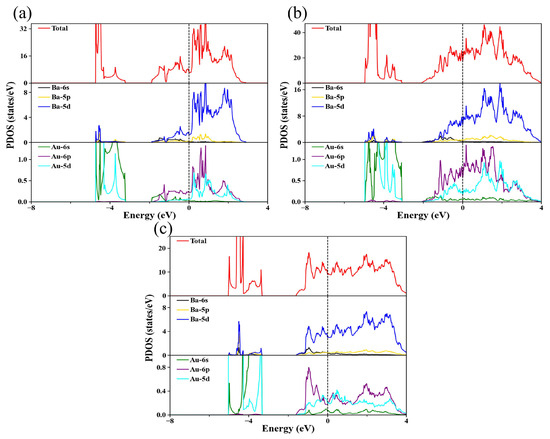
Figure 5.
Total and partial density of states of Ba4Au: (a) I4/mmm phase at 0 GPa, (b) Cmmm phase at 5 GPa, and (c) I4/m phase at 10 GPa. The dashed line at zero indicates Fermi energy.
Moreover, to investigate the chemical bonding in Ba4Au compounds under high pressure, we calculated the ELF of Ba4Au compounds at various pressures. The ELF values range from 0 to 1, where 0 indicates fully delocalized electrons or the typical electron deficiency of ionic bonds, 0.5 corresponds to metallic bonding, and 1 denotes fully localized electrons (indicative of covalent bonds) [41,46,47,48]. In the I4/mmm phase at 0 GPa (Figure 6a), the ELF values between Ba and Au were low, indicating predominantly ionic bonding with charge transfer. The ELF between Ba atoms was approximately 0.5, which is consistent with metallic bonding. At 5 GPa in the Cmmm phase (Figure 6b), the ELF around Ba approached one, reflecting strong electron localization. The ELF value between Ba and Au atoms was approximately 0.4, indicating that the Ba–Au bond remains ionic. Compared with the I4/mmm-Ba4Au structure, the Ba–Au ionic bond in the Cmmm-Ba4Au structure is relatively weaker, with enhanced electron localization between Ba and Au. This behavior correlates with increased hybridization between the Ba 5d and Au 5d orbitals under pressure. Moreover, ELF between Ba atoms reaches approximately 0.7, indicating a weak covalent characteristic. At 10 GPa in the I4/m phase (Figure 6c), the Ba–Au interaction remains ionic, whereas Ba–Ba bonding continues to exhibit a metallic characteristic. In summary, ELF indicates that Ba–Au bonds in all three Ba4Au structures are ionic, while Ba–Ba bonding varies slightly. In the I4/mmm-Ba4Au and I4/m-Ba4Au structures, Ba–Ba bonds are metallic bonds, whereas in the Cmmm-Ba4Au structure, Ba–Ba bonds exhibit weak covalent bonding.

Figure 6.
Two-dimensional electron localization function (ELF) diagram of stable structures of Ba4Au at varying pressures: (a) I4/mmm-Ba4Au at 0 GPa on the (010) plane, (b) Cmmm-Ba4Au at 5 GPa on the (001) plane, and (c) I4/m-Ba4Au at 10 GPa on the (100) plane.
To further investigate the Ba–Au bonding characteristics and charge redistribution, we calculated and analyzed the charge density difference for each phase, as shown in Figure 7. In all three structures, regions of electron depletion and accumulation appeared between Ba and Au atoms, indicating a pronounced charge transfer from Ba to Au. This finding confirms the ionic nature of the Ba–Au bonds in all phases and is consistent with the results from ELF analysis. To quantify the extent of charge transfer between Ba and Au, we conducted Bader charge analysis within the stable pressure ranges of each phase. As shown in Figure 8, in the ambient-pressure I4/mmm phase, the Bader charge transfer from Ba to Au was approximately 1.79 e. At 0.4 GPa, this transfer decreased to approximately 1.75 e. With increasing pressure, the Bader charge on Au decreased significantly. Moreover, as pressure increased, the Ba⟶Au charge transfer in each phase progressively diminished, reflecting weakened ionic characteristics and enhanced covalent interactions in the high-pressure structures. This evolution plays a critical role in the stability of Ba4Au compounds under compression.
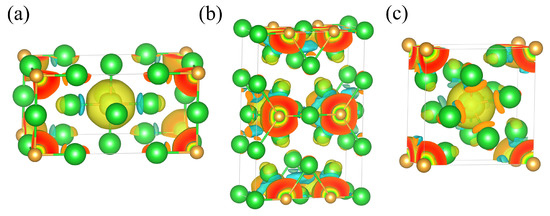
Figure 7.
Charge density differences in the (a) I4/mmm (0 GPa), (b) Cmmm (5 GPa), and (c) I4/m (10 GPa) phases of Ba4Au. Blue and yellow regions indicate electron depletion and accumulation, respectively. (Ba in green atoms and Au in gold atoms).
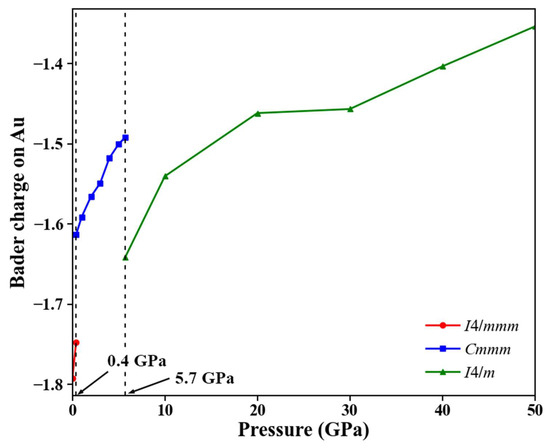
Figure 8.
Pressure dependence of Bader charges on Au in each phase within its stable pressure range.
4. Conclusions
Herein, we employed the PSO algorithm with first-principles calculations to investigate the pressure-induced structural phase transitions and electronic property evolution in Ba4Au. Enthalpy-difference calculations indicated that at ambient pressure, I4/mmm-Ba4Au has the lowest energy. Above 0.4 GPa, the Cmmm phase becomes energetically favorable, indicating that the I4/mmm phase of Ba4Au transforms to the Cmmm phase at approximately 0.4 Gpa. As pressure increases, above 5.7 GPa, the I4/m structure becomes energetically more favorable than Cmmm-Ba4Au, indicating that the Cmmm phase transforms to the I4/m phase at 5.7 GPa. The results of the pressure-induced volume changes revealed that both structural transitions were first-order phase transitions with remarkable volume contractions. The detailed analysis of the Ba4Au electronic properties revealed that its three structures exhibited metallic characteristics across various pressures. Furthermore, with increasing pressure, hybridization between the Ba 5d and Au 5d orbitals progressively strengthened. Bader charge analysis further indicated that as pressure increased, the ionic characteristic between Ba and Au in the high-pressure phases diminished while covalent interactions strengthened. This study not only enriches the high-pressure phase diagram of Ba–Au alloys but also reveals the evolution of the electronic properties of the Ba4Au structure under high pressure, indicating its potential applicability in high-pressure electronic devices and pressure-sensitive components with adjustable conductivity.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/ma18163728/s1. Table S1: Structural parameters of other Ba4Au compounds under various pressure; Figure S1: Phonon spectra of (a) Pm-3m-Ba4Au at 0 GPa, (b) P21/m-Ba4Au at 0 GPa, (c) P4/mmm-Ba4Au at 0 GPa, (d) C2-Ba4Au at 0 GPa, (e) Cc-Ba4Au at 0 GPa, (f) Cmcm-Ba4Au at 10 GPa, and (g) R-3c-Ba4Au at 10 GPa; Table S2: Formation enthalpies of AuxBay (x = 1, y = 1, 4; x = 2, y = 1, 3) compounds under various pressures.
Author Contributions
Conceptualization, Q.W.; Writing—original draft, X.W.; Investigation, X.W., X.J., B.W., and X.Z.; Data curation, J.L. and X.J.; Writing–review and editing, B.W. and M.Z.; Supervision, Q.W.; Project administration, Q.W.; Funding acquisition, B.W. and M.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China (Grant Nos. 11965005 and 11964026) and the Natural Science Basic Research Plan in Shaanxi Province of China (Grant No. 2025JC-YBMS-027).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in this study are included in the article; further inquiries can be directed to the corresponding author.
Acknowledgments
The authors thank the High Performance Computing Center of Xidian University for the use of computing facilities.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Guenther, J.; Mallet-Ladeira, S.; Estevez, L.; Miqueu, K.; Amgoune, A.; Bourissou, D. Activation of Aryl Halides at Gold(I): Practical Synthesis of (P,C) Cyclometalated Gold(III) Complexes. J. Am. Chem. Soc. 2014, 136, 1778–1781. [Google Scholar] [CrossRef]
- Rudolph, M.; Hashmi, A.S.K. Gold catalysis in total synthesis—An update. Chem. Soc. Rev. 2012, 41, 2448–2462. [Google Scholar] [CrossRef]
- Kodiyath, R.; Manikandan, M.; Liu, L.; Ramesh, G.V.; Koyasu, S.; Miyauchi, M.; Sakuma, Y.; Tanabe, T.; Gunji, T.; Dao, T.D. Visible-light photodecomposition of acetaldehyde by TiO2-coated gold nanocages: Plasmon-mediated hot electron transport via defect states. Chem. Commun. 2014, 50, 15553–15556. [Google Scholar] [CrossRef]
- Aragoni, M.C.; Arca, M.; Devillanova, F.A.; Isaia, F.; Lippolis, V.; Pintus, A. Gold(III) Complexes of Asymmetrically ArylSubstituted 1,2-Dithiolene Ligands Featuring Potential-Controlled Spectroscopic Properties: An Insight into the Electronic Properties of bis(Pyren-1-yl-ethylene-1,2-dithiolato) Gold(III). Chem.-Asian J. 2011, 6, 198–208. [Google Scholar] [CrossRef]
- Zhao, W.; Yang, L.; Yin, Y.; Jin, M. Thermodynamic controlled synthesis of intermetallic Au3Cu alloy nanocrystals from Cu microparticles. J. Mater. Chem. 2014, 2, 902–906. [Google Scholar] [CrossRef]
- Ishikawa, T.; Nomura, M.; Kato, K.; Suzuki, N.; Shimizu, K.; Itoh, H. First-principles study on superconductivity of the gold–indium alloy under high pressure. High Press. Res. 2013, 33, 152–157. [Google Scholar] [CrossRef]
- Baranov, D.S.; Vlaic, S.; Baptista, J.; Cofler, E.; Stolyarov, V.S.; Roditchev, D.; Pons, S. Gold Atoms Promote Macroscopic Superconductivity in an Atomic Monolayer of Pb on Si(111). Nano Lett. 2022, 22, 652–657. [Google Scholar] [CrossRef]
- Xing, Y.; Wang, H.; Li, C.K.; Zhang, X.; Liu, J.; Zhang, Y.; Luo, J.; Wang, Z.; Wang, Y.; Ling, L.; et al. Superconductivity in topologically nontrivial material Au2Pb. NPJ Quantum Mater. 2016, 1, 16005. [Google Scholar] [CrossRef]
- Lu, J.; Zhan, M.; Yu, J.; Yu, X.; Duan, Y.; Chen, S.; Xu, M.; Lu, W. Insight on the Electronic, Elastic and Thermal Properties of Au-Al Intermetallic Compounds Based on First-Principles Calculations. J. Electron. Mater. 2024, 53, 3809–3821. [Google Scholar] [CrossRef]
- Ono, S. Two-Dimensional Ionic Crystals: The Cases of IA-VII Alkali Halides and IA-IB CsAu. J. Phys. Soc. Japan 2022, 91, 094606. [Google Scholar] [CrossRef]
- Miao, M.; Brgoch, J.; Krishnapriyan, A.; Goldman, A.; Kurzman, J.A.; Seshadri, R. On the Stereochemical Inertness of the Auride Lone Pair: Ab Initio Studies of AAu (A = K, Rb, Cs). Inorg. Chem. 2013, 52, 8183–8189. [Google Scholar] [CrossRef]
- Aycibin, M.; Dogan, E.K.; Gulebaglan, S.E.; Secuk, M.N.; Erdinc, B.; Akkus, H. Physical properties of RbAu compound. Comput. Condens. Matter 2014, 1, 32–37. [Google Scholar] [CrossRef]
- Lin, J.; Du, X.; Yang, G. Pressure-induced new chemistry. Chin. Phys. B 2019, 28, 106106. [Google Scholar] [CrossRef]
- Lyu, T.; Yang, Q.X.; Li, Z.M.; Zhang, C.; Liu, F.; Li, J.; Hu, L.; Xu, G. High pressure drives microstructure modification and zT enhancement in bismuth telluride-based alloys. ACS Appl. Mater. Interfaces 2023, 15, 19250–19257. [Google Scholar] [CrossRef]
- Miao, M. Caesium in high oxidation states and as a p-block element. Nat. Chem. 2013, 5, 846–852. [Google Scholar] [CrossRef]
- Ma̧czka, M.; Kryś, M.; Sobczak, S.; Vasconcelos, D.L.M.; Freire, P.T.C.; Katrusiak, A. Evidence of Pressure-induced phase transitions and negative linear compressibility in formamidinium manganese-hypophosphite hybrid perovskite. J. Phys. Chem. C 2021, 125, 26958–26966. [Google Scholar] [CrossRef]
- Sun, H.; Huo, M.; Hu, X.; Li, J.; Liu, Z.; Han, Y.; Tang, L.; Mao, Z.; Yang, P.; Wang, B.; et al. Signatures of superconductivity near 80 K in a nickelate under high pressure. Nature 2023, 621, 493–498. [Google Scholar] [CrossRef]
- Nelmes, R.J.; Allan, D.R.; McMahon, M.I.; Belmonte, S.A. Self-Hosting Incommensurate Structure of Barium IV. Phys. Rev. Lett. 1999, 83, 4081. [Google Scholar] [CrossRef]
- Li, P.; Gao, G.; Wang, Y.; Ma, Y. Crystal Structures and Exotic Behavior of Magnesium under Pressure. J. Phys. Chem. C 2010, 114, 21745–21749. [Google Scholar] [CrossRef]
- Rahm, M.; Cammi, R.; Ashcroft, N.W.; Hoffmann, R. Squeezing All Elements in the Periodic Table: Electron Configuration and Electronegativity of the Atoms under Compression. J. Am. Chem. Soc. 2019, 141, 10253–10271. [Google Scholar] [CrossRef]
- Luo, D.; Wang, Y.; Yang, G.; Ma, Y. Barium in High Oxidation States in Pressure-Stabilized Barium Fluorides. J. Phys. Chem. C 2018, 122, 12448–12453. [Google Scholar] [CrossRef]
- Li, F.; Zhang, X.; Fu, Y.; Wang, Y.; Bergara, A.; Yang, G. Ba with Unusual Oxidation States in Ba Chalcogenides under Pressure. J. Phys. Chem. Lett. 2021, 12, 4203–4210. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Wang, J.; Sun, S.; Liu, H. Crystal Structures and Electronic Properties of BaAu Compound under High Pressure. Materials 2022, 15, 7381. [Google Scholar] [CrossRef]
- Amsler, M.; Goedecker, S. Crystal structure prediction using the minima hopping method. J. Chem. Phys. 2010, 133, 224104. [Google Scholar] [CrossRef]
- Laio, A.; Rodriguez-Fortea, A.; Gervasio, F.L.; Ceccarelli, M.; Parrinello, M. Assessing the accuracy of metadynamics. J. Phys. Chem. B 2005, 109, 6714–6721. [Google Scholar] [CrossRef]
- Lonie, D.C.; Zurek, E. XtalOpt: An open-source evolutionary algorithm for crystal structure prediction. Comput. Phys. Commun. 2011, 182, 372–387. [Google Scholar] [CrossRef]
- Wang, Y.; Lv, J.; Zhu, L.; Ma, Y. Crystal structure prediction via particle-swarm optimization. Phys. Rev. B 2010, 82, 094116. [Google Scholar] [CrossRef]
- Pickard, C.J.; Needs, R.J. Ab initio random structure searching. J. Phys. Condens. Matter 2011, 23, 053201. [Google Scholar] [CrossRef]
- Peng, F.; Sun, Y.; Pickard, C.J.; Needs, R.J.; Wu, Q.; Ma, Y. Hydrogen Clathrate Structures in Rare Earth Hydrides at High Pressures: Possible Route to Room-Temperature Superconductivity. Phys. Rev. Lett. 2017, 119, 107001. [Google Scholar] [CrossRef]
- Liu, H.; Naumov, I.I.; Hoffmann, R.; Ashcroft, N.W.; Hemley, R.J. Potential High-Tc Superconducting Lanthanum and Yttrium Hydrides at High Pressure. Proc. Natl. Acad. Sci. USA 2017, 114, 6990–6995. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, Y.; Lv, J.; Li, Q.; Zhang, L.; Ma, Y. CALYPSO Structure Prediction Method and Its Wide Application. Comput. Mater. Sci. 2016, 112, 406–415. [Google Scholar] [CrossRef]
- Li, B.; Liu, H.; Liu, G.; Chen, K. First-principles study on high-pressure phases and compression properties of gold-bearing intermetallic compounds. J. Phys. Condens. Matter 2022, 34, 464001. [Google Scholar] [CrossRef]
- Wang, Y.; Lv, J.; Zhu, L.; Ma, Y. CALYPSO: A method for crystal structure prediction. Comput. Phys. Commun. 2012, 183, 2063–2070. [Google Scholar] [CrossRef]
- Wei, Q.; Yang, J.; Jia, X.; Luo, J.; Zhang, M.; Zhu, X. Crystal structures, mechanical properties, and electronic structure analysis of ternary FeCrAl alloys. Phys. Lett. A 2025, 533, 130228. [Google Scholar] [CrossRef]
- Yan, H.; Zhang, W.; Chen, L.; Zhang, Y.; Wang, H.; Zhang, M.; Wei, Q. Structural, strength and fracture mechanisms of superconducting transition metal nitrides TM3N5 (TM= W and Mo). Phys. Chem. Chem. Phys. 2025, 27, 6134–6145. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef]
- Perdew, J.P.; Zunger, A. Self-interaction correction to density-functional approximations for many-electron systems. Phys. Rev. B 1981, 23, 5048. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef]
- Savin, A.; Jepsen, O.; Flad, J.; Andersen, O.K.; Preuss, H.; Von Schnering, H.G. Electron localization in solid-state structures of the elements: The diamond structure. Angew. Chem. Int. Ed. 1992, 31, 187–188. [Google Scholar] [CrossRef]
- Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Togo, A.; Tanaka, I. First principles phonon calculations in materials science. Scr. Mater. 2015, 108, 1–5. [Google Scholar] [CrossRef]
- Chaput, L.; Togo, A.; Tanaka, I.; Hug, G. Phonon-phonon interactions in transition metals. Phys. Rev. B 2011, 84, 094302. [Google Scholar] [CrossRef]
- Giannozzi, P.; De Gironcoli, S.; Pavone, P.; Baroni, S. Ab initio calculation of phonon dispersions in semiconductors. Phys. Rev. B 1991, 43, 7231. [Google Scholar] [CrossRef]
- Gonze, X.; Lee, C. Dynamical matrices, Born effective charges, dielectric permittivity tensors, and interatomic force constants from density-functional perturbation theory. Phys. Rev. B 1997, 55, 10355. [Google Scholar] [CrossRef]
- Savin, A.; Nesper, R.; Wengert, S.; Fässler, T.F. ELF: The Electron Localization Function. Angew. Chem. Int. Ed. 1997, 36, 1808–1832. [Google Scholar] [CrossRef]
- Wang, Y.X.; Wu, H.; Xie, W.N.; Wang, X.F.; Sun, S.W.; Gu, J.B. Pressure-induced evolution of structures and phase transition of technetium diboride. J. Appl. Phys. 2024, 135, 205901. [Google Scholar] [CrossRef]
- Xie, X.; Wei, Q.; Luo, J.; Jia, X.; Zhang, M. Pressure-induced phase transitions of ZrAl2 from first-principles calculations. Solid State Commun. 2024, 391, 115643. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).