
A Bipyridine-Ester Dual-Modified 2,2,6,6-Tetramethylpiperidin-1-oxyl Derivative for Aqueous Organic Redox Flow Batteries



Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
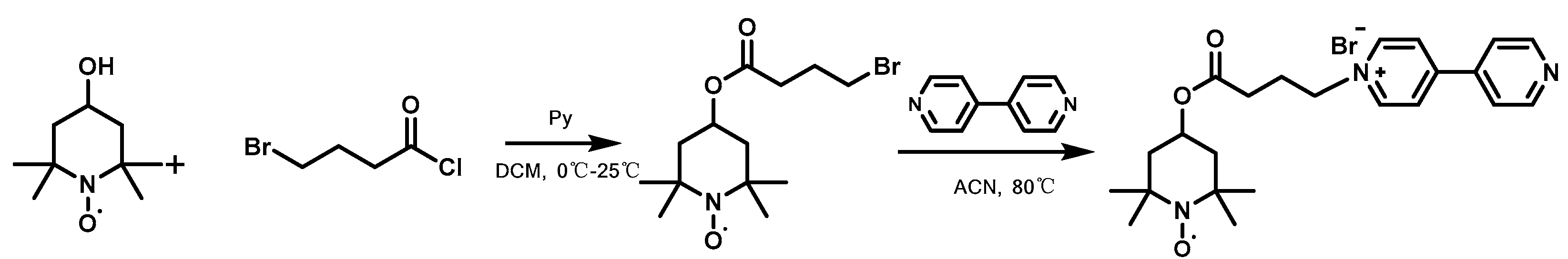
2.2. Synthesis of TEMP-BPy
2.3. Material Characterization
2.4. Electrochemical Characterization
2.5. Flow Battery Tests
3. Results and Discussion
3.1. Chemical Characterization of TEMP-BPy
3.1.1. Structure and Composition of TEMP-BPy
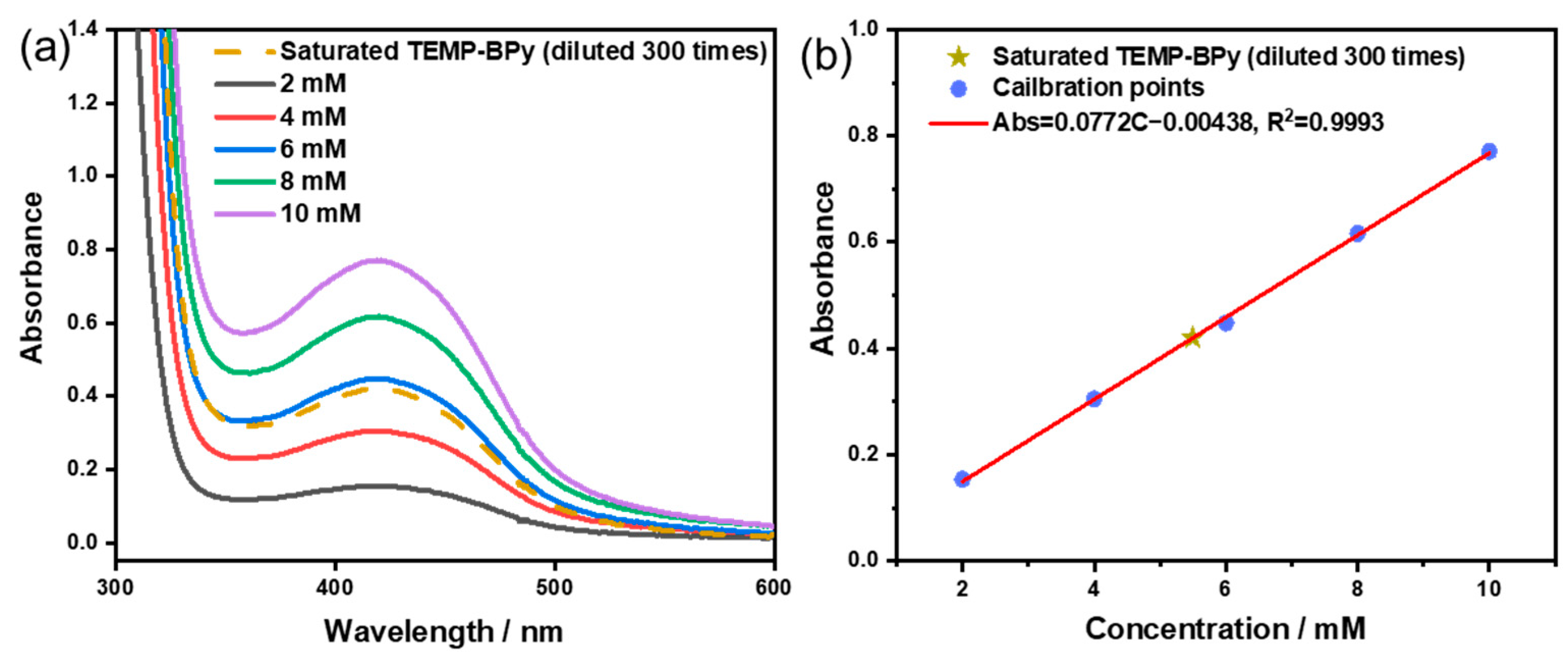
3.1.2. Solubility and Viscosity of TEMP-BPy
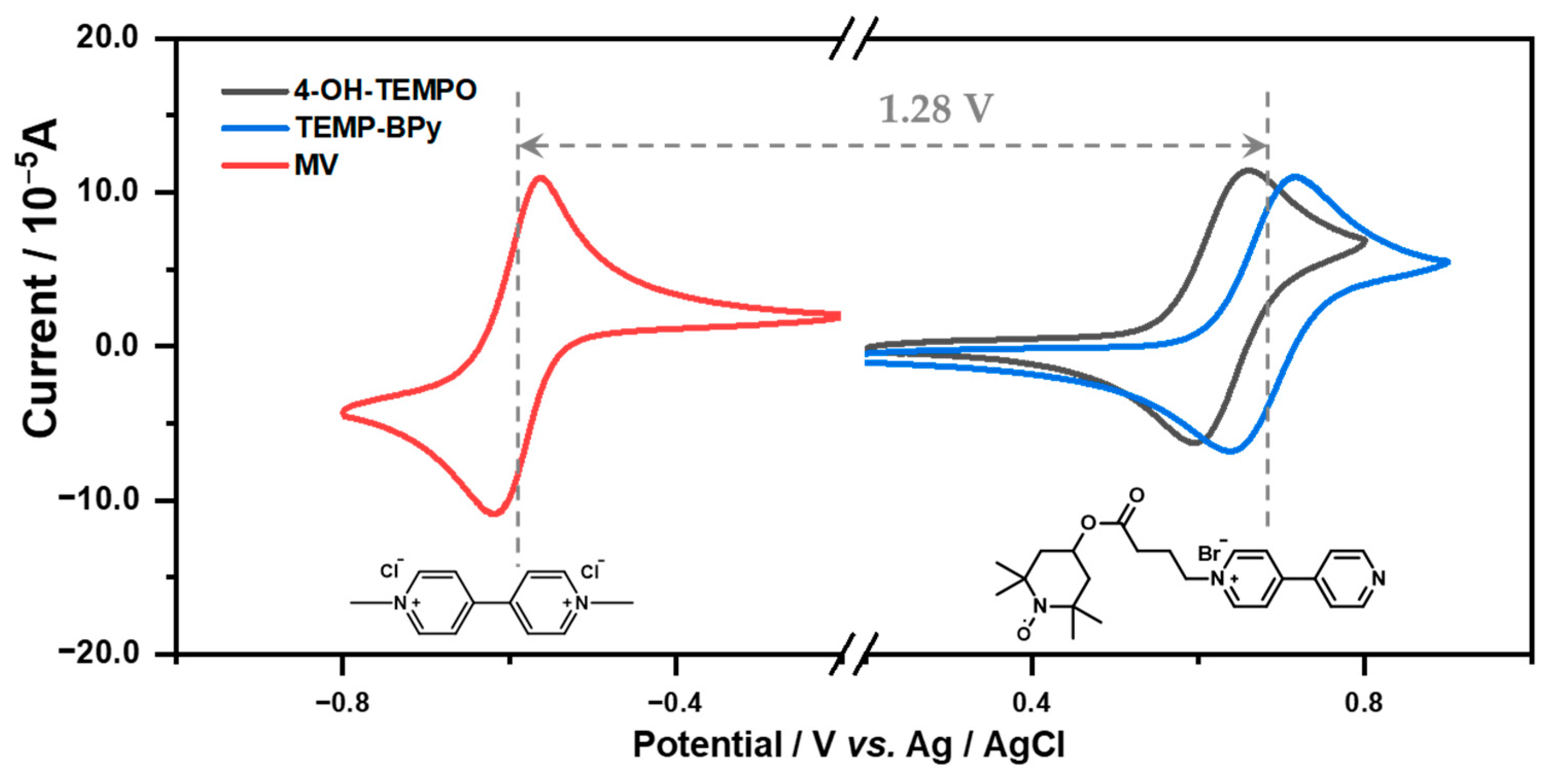
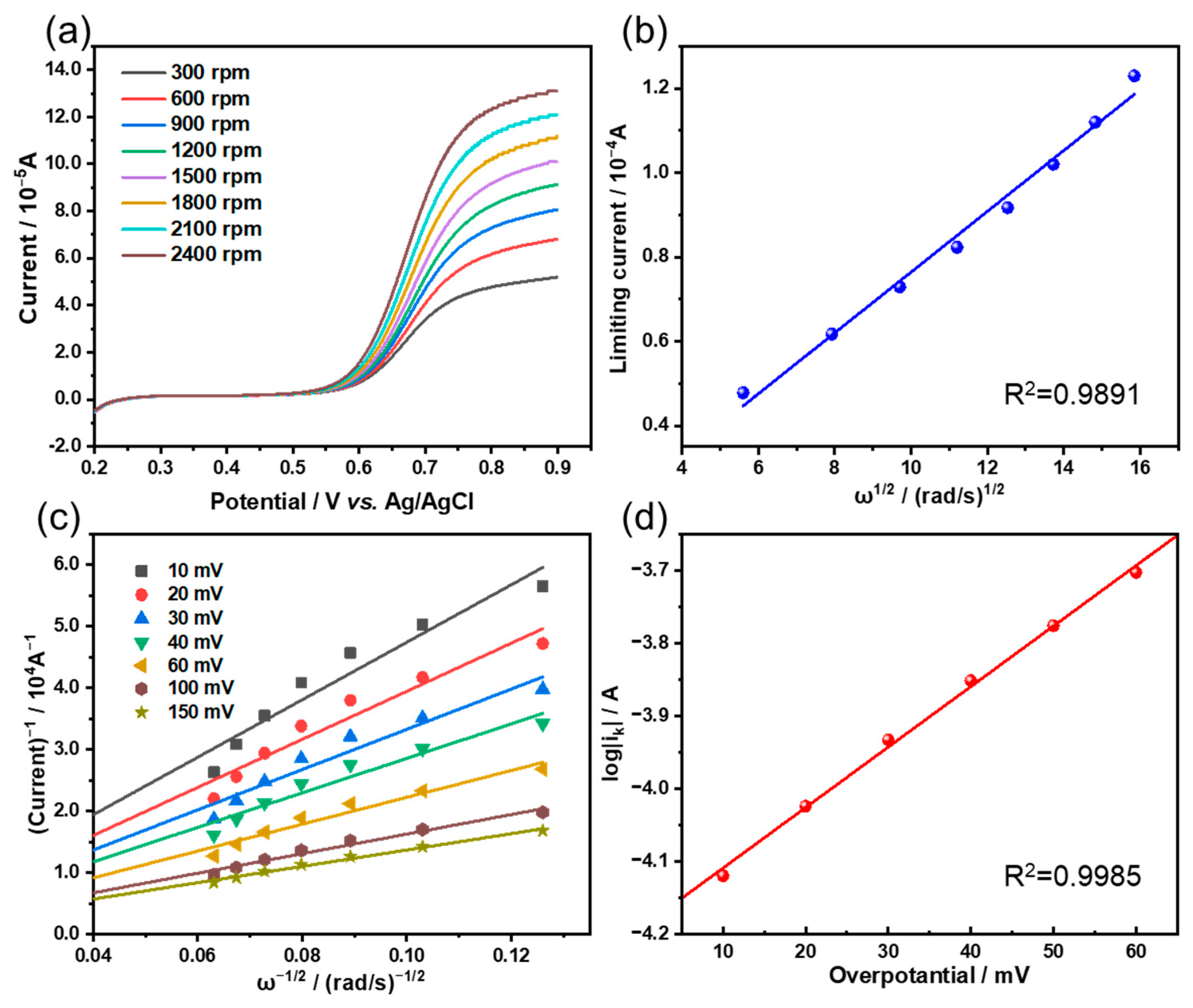
3.2. Electrochemical Performance
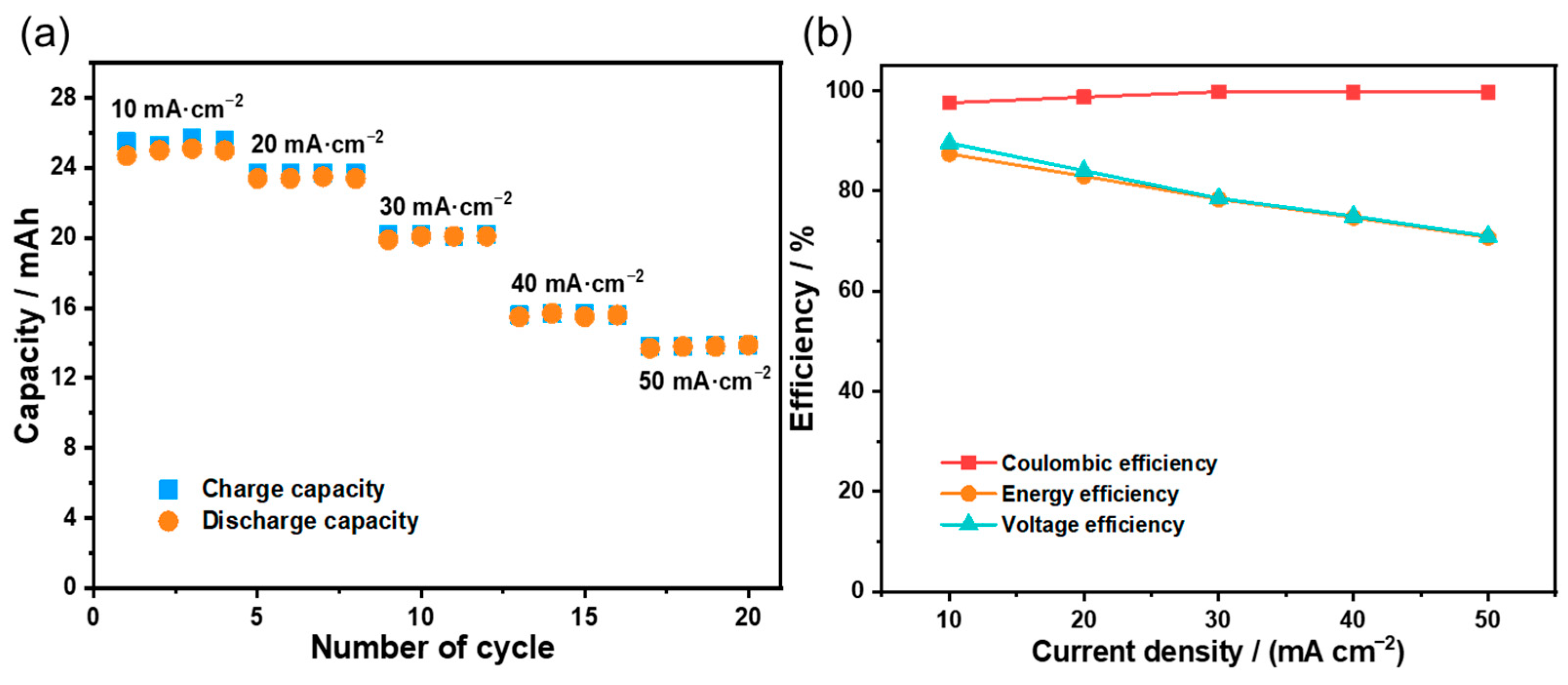
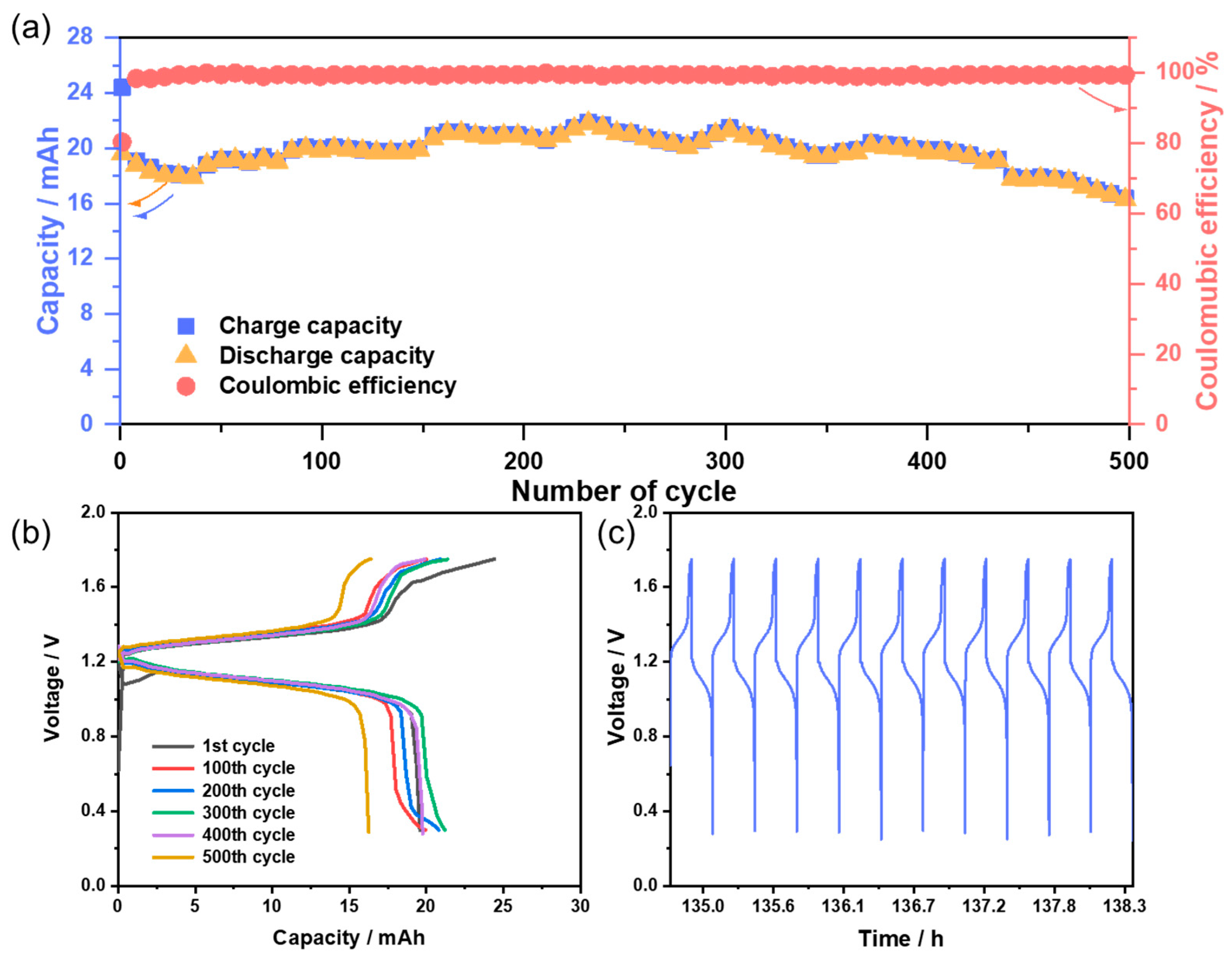
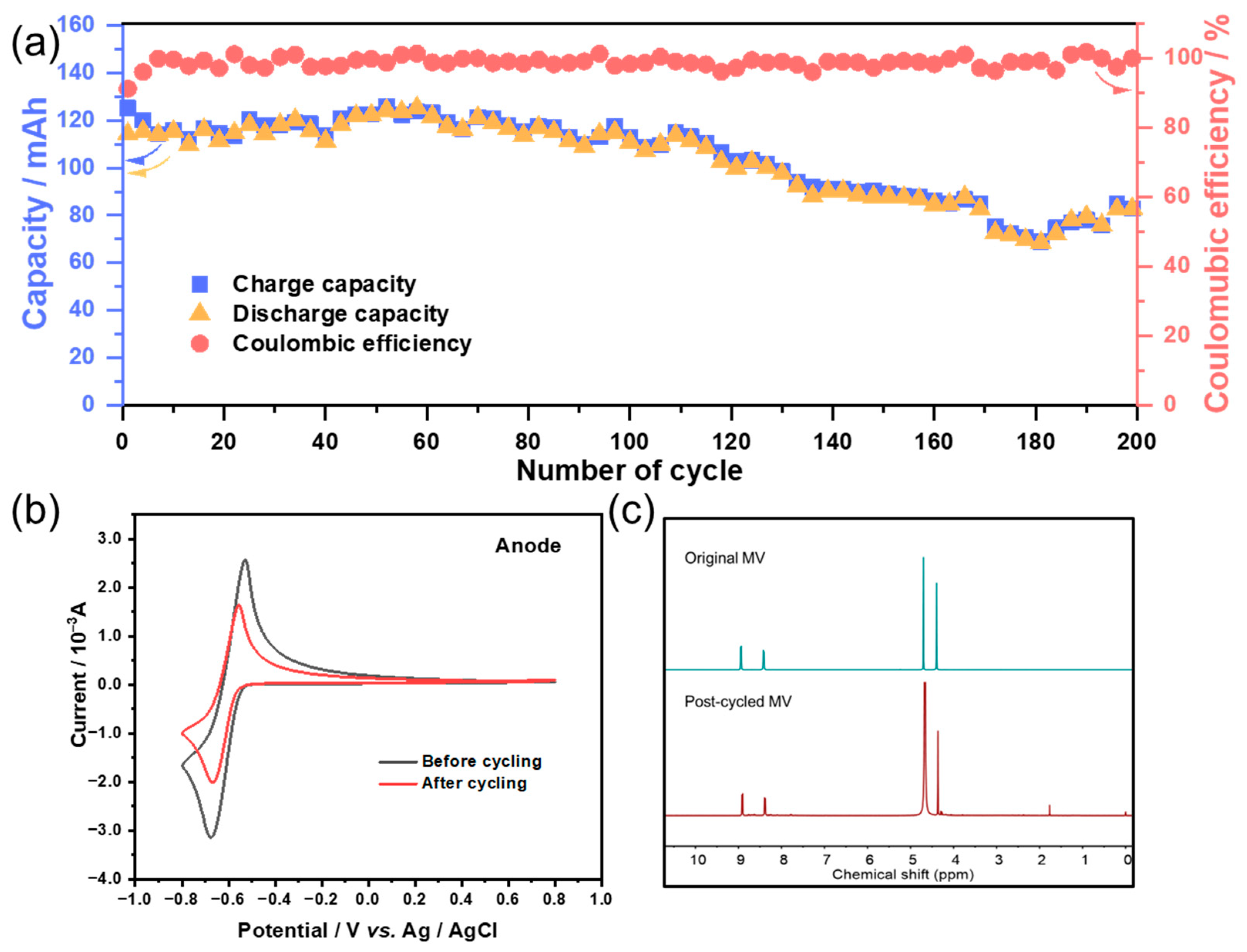
3.3. Flow Battery Performance
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AORFB | Aqueous organic redox flow battery |
| CV | Cyclic voltammetry |
| LSV | Linear sweep voltammetry |
| RDE | Rotating disk electrode measurements |
| TEMPO | 2,2,6,6-tetramethylpiperidin-1-oxyl |
| MV | Methyl viologen |
| CE | Coulombic efficiency |
| D | Diffusion coefficient |
| K0 | Electron-transfer rate constant |
| DCM | Dichloromethane |
| ACN | Acetonitrile |
| TEMP-BPy | (2,2,6,6-tetramethyl-1-piperidinyloxy)carbonyl-ethyl-(4-(pyridin-4-yl)benzyl) ammonium bromide |
References
- Luo, J.; Wang, A.P.; Hu, M.; Liu, T.L. Materials challenges of aqueous redox flow batteries. MRS Energy Sustain. 2022, 9, 1–12. [Google Scholar] [CrossRef]
- Ben Cheikh, N.; Ben Zaied, Y. Understanding the drivers of the renewable energy transition. Econ. Anal. Policy 2024, 82, 604–612. [Google Scholar] [CrossRef]
- Pedraza, E.; de la Cruz, C.; Mavrandonakis, A.; Ventosa, E.; Rubio-Presa, R.; Sanz, R.; Senthilkumar, S.T.; Navalpotro, P.; Marcilla, R. Unprecedented Aqueous Solubility of TEMPO and its Application as High Capacity Catholyte for Aqueous Organic Redox Flow Batteries. Adv. Energy Mater. 2023, 13, 2301929. [Google Scholar] [CrossRef]
- Hu, B.; Tang, Y.; Luo, J.; Grove, G.; Guo, Y.; Liu, T.L. Improved radical stability of viologen anolytes in aqueous organic redox flow batteries. Chem. Commun. 2018, 54, 6871–6874. [Google Scholar] [CrossRef]
- Chang, Z.; Henkensmeier, D.; Chen, R. One-step cationic grafting of 4-hydroxy-TEMPO and its application in a hybrid redox flow battery with a crosslinked PBI membrane. ChemSusChem 2017, 10, 3193–3197. [Google Scholar] [CrossRef]
- Hu, B.; Luo, J.; Hu, M.; Yuan, B.; Liu, T.L. A pH-Neutral, Metal-Free Aqueous Organic Redox Flow Battery Employing an Ammonium Anthraquinone Anolyte. Angew. Chem. Int. Ed. 2019, 58, 16629–16636. [Google Scholar] [CrossRef]
- Choi, C.; Kim, S.; Kim, R.; Choi, Y.; Kim, S.; Jung, H.-y.; Yang, J.H.; Kim, H.-T. A review of vanadium electrolytes for vanadium redox flow batteries. Renew. Sustain. Energy Rev. 2017, 69, 263–274. [Google Scholar] [CrossRef]
- Li, L.; Kim, S.; Wang, W.; Vijayakumar, M.; Nie, Z.; Chen, B.; Zhang, J.; Xia, G.; Hu, J.; Graff, G.; et al. A stable vanadium redox-flow battery with high energy density for large-scale energy storage. Adv. Energy Mater. 2011, 1, 394–400. [Google Scholar] [CrossRef]
- Winsberg, J.; Hagemann, T.; Janoschka, T.; Hager, M.D.; Schubert, U.S. Redox-flow batteries: From metals to organic redox-active materials. Angew. Chem. Int. Ed. 2017, 56, 686–711. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, Y.; Cai, H.; Yang, J. TEMPO and its derivatives in organic redox-flow batteries. Univ. Chem. 2017, 32, 32–44. [Google Scholar] [CrossRef]
- Liu, T.; Wei, X.; Nie, Z.; Sprenkle, V.; Wang, W. A Total Organic Aqueous Redox Flow Battery Employing a Low Cost and Sustainable Methyl Viologen Anolyte and 4-HO-TEMPO Catholyte. Adv. Energy Mater. 2015, 6, 1501449. [Google Scholar] [CrossRef]
- Zhou, W.; Liu, W.; Qin, M.; Chen, Z.; Xu, J.; Cao, J.; Li, J. Fundamental properties of TEMPO-based catholytes for aqueous redox flow batteries: Effects of substituent groups and electrolytes on electrochemical properties, solubilities and battery performance. RSC Adv. 2020, 10, 21839–21844. [Google Scholar] [CrossRef] [PubMed]
- Winsberg, J.; Stolze, C.; Schwenke, A.; Muench, S.; Hager, M.D.; Schubert, U.S. Aqueous 2,2,6,6-tetramethylpiperidine-N-oxyl catholytes for a high-capacity and high current density oxygen-insensitive hybrid-flow battery. ACS Energy Lett. 2017, 2, 411–416. [Google Scholar] [CrossRef]
- Liu, B.; Tang, C.W.; Jiang, H.; Jia, G.; Zhao, T. Carboxyl-Functionalized TEMPO Catholyte Enabling High-Cycling-Stability and High-Energy-Density Aqueous Organic Redox Flow Batteries. ACS Sustain. Chem. Eng. 2021, 9, 6258–6265. [Google Scholar] [CrossRef]
- Liu, Y.; Goulet, M.-A.; Tong, L.; Liu, Y.; Ji, Y.; Wu, L.; Gordon, R.G.; Aziz, M.J.; Yang, Z.; Xu, T. A Long-Lifetime All-Organic Aqueous Flow Battery Utilizing TMAP-TEMPO Radical. Chem 2019, 5, 1861–1870. [Google Scholar] [CrossRef]
- Hu, B.; Hu, M.; Luo, J.; Liu, T.L. A Stable, Low Permeable TEMPO Catholyte for Aqueous Total Organic Redox Flow Batteries. Adv. Energy Mater. 2022, 12, 2102577. [Google Scholar] [CrossRef]
- Jin, S.; Fell, E.M.; Vina-Lopez, L.; Jing, Y.; Michalak, P.W.; Gordon, R.G.; Aziz, M.J. Near Neutral pH Redox Flow Battery with Low Permeability and Long-Lifetime Phosphonated Viologen Active Species. Adv. Energy Mater. 2020, 10, 2000100. [Google Scholar] [CrossRef]
- Chang, Z.; Henkensmeier, D.; Chen, R. Shifting redox potential of nitroxyl radical by introducing an imidazolium substituent and its use in aqueous flow batteries. J. Power Sources 2019, 418, 11–16. [Google Scholar] [CrossRef]
- Fan, H.; Hu, B.; Li, H.; Ravivarma, M.; Feng, Y.; Song, J. Conjugate-driven electron density delocalization of piperidine nitroxyl radical for stable aqueous Zinc hybrid flow batteries. Angew. Chem. Int. Ed. 2022, 61, e202115908. [Google Scholar] [CrossRef]
- Hu, S.; Wang, L.; Yuan, X.; Xiang, Z.; Huang, M.; Luo, P.; Liu, Y.; Fu, Z.; Liang, Z. Viologen-Decorated TEMPO for Neutral Aqueous Organic Redox Flow Batteries. Energy Mater. Adv. 2021, 2021, 9795237. [Google Scholar] [CrossRef]
- Soares, B.G.; Ferreira, S.C.; Livi, S. Modification of anionic and cationic clays by zwitterionic imidazolium ionic liquid and their effect on the epoxy-based nanocomposites. Appl. Clay Sci. 2017, 135, 347–354. [Google Scholar] [CrossRef]
- Janoschka, T.; Friebe, C.; Hager, M.D.; Martin, N.; Schubert, U.S. An approach toward replacing vanadium: A single organic molecule for the anode and cathode of an aqueous redox-flow battery. ChemistryOpen 2017, 6, 216–220. [Google Scholar] [CrossRef]
- Winsberg, J.; Stolze, C.; Muench, S.; Liedl, F.; Hager, M.D.; Schubert, U.S. TEMPO/Phenazine combi-molecule: A redox-active material for symmetric aqueous redox-flow batteries. ACS Energy Lett. 2016, 1, 976–980. [Google Scholar] [CrossRef]
- Hagemann, T.; Strumpf, M.; Schröter, E.; Stolze, C.; Grube, M.; Nischang, I.; Hager, M.D.; Schubert, U.S. (2,2,6,6-tetramethylpiperidin-1-yl)oxyl-containing zwitterionic polymer as catholyte species for high-capacity aqueous polymer redox flow batteries. Chem. Mater. 2019, 31, 7987–7999. [Google Scholar] [CrossRef]
- Luo, J.; Hu, B.; Debruler, C.; Liu, T.L. A π-conjugation extended Viologen as a two-electron storage anolyte for total organic aqueous redox flow batteries. Angew. Chem. Int. Ed. 2018, 57, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods: Fundamental and Applications; John Wiley & Sons, Inc.: New York, NY, USA, 2001. [Google Scholar]
- González-Meza, O.A.; Larios-Durán, E.R.; Gutiérrez-Becerra, A.; Casillas, N.; Escalante, J.I.; Bárcena-Soto, M. Development of a Randles-Ševčík-like equation to predict the peak current of cyclic voltammetry for solid metal hexacyanoferrates. J. Solid State Electrochem. 2019, 23, 3123–3133. [Google Scholar] [CrossRef]
- Rubio-Presa, R.; Lubián, L.; Borlaf, M.; Ventosa, E.; Sanz, R. Addressing practical use of viologen-derivatives in redox flow batteries through molecular engineering. ACS Mater. Lett. 2023, 5, 798–802. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, T.; Burnett, M.; Green, K. Electronic influence of substitution on the pyridine ring within NNN pincer-type molecules. Dalton Trans. 2020, 49, 2356–2363. [Google Scholar] [CrossRef] [PubMed]
- Burešová, Z.; Klikar, M.; Mazúr, P.; Mikešová, M.; Kvíčala, J.; Bystron, T.; Bureš, F. Redox Property Tuning in Bipyridinium Salts. Front. Chem. 2021, 8, 2020. [Google Scholar] [CrossRef]
- Janoschka, T.; Martin, N.; Hager, M.D.; Schubert, U.S. An Aqueous Redox-Flow Battery with High Capacity and Power: The TEMPTMA/MV System. Angew. Chem. Int. Ed. 2016, 55, 14427–14430. [Google Scholar] [CrossRef]
- Fu, H.; Zhang, C.; Wang, H.; Du, B.; Nie, J.; Xu, J.; Chen, L. Stable aqueous redox flow battery assembled in air atmosphere employing an anionic terpolymer as active cathode material. J. Power Sources 2022, 545, 231905. [Google Scholar] [CrossRef]
- Wang, H.; Sayed, S.Y.; Luber, E.J.; Olsen, B.C.; Shirurkar, S.M.; Venkatakrishnan, S.; Tefashe, U.M.; Farquhar, A.K.; Smotkin, E.S.; McCreery, R.L.; et al. Redox flow batteries: How to determine electrochemical kinetic parameters. ACS Nano 2020, 14, 2575–2584. [Google Scholar] [CrossRef] [PubMed]
- Janoschka, T.; Martin, N.; Martin, U.; Friebe, C.; Morgenstern, S.; Hiller, H.; Hager, M.D.; Schubert, U.S. An aqueous, polymer-based redox-flow battery using non-corrosive, safe, and low-cost materials. Nature 2015, 527, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Yang, F.; Niu, Z.; Wu, H.; He, Y.; Zhu, H.; Ye, J.; Zhao, Y.; Zhang, X. Enhanced cyclability of organic redox flow batteries enabled by an artificial bipolar molecule in neutral aqueous electrolyte. J. Power Sources 2019, 417, 83–89. [Google Scholar] [CrossRef]
- Pan, M.; Gao, L.; Liang, J.; Zhang, P.; Lu, S.; Lu, Y.; Ma, J.; Jin, Z. Reversible Redox Chemistry in Pyrrolidinium-Based TEMPO Radical and Extended Viologen for High-Voltage and Long-Life Aqueous Redox Flow Batteries. Adv. Energy Mater. 2022, 12, 2103478. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, Q.; Zhang, X.; Ran, J.; Han, X.; Yang, Z.; Xu, T. Degradation of electrochemical active compounds in aqueous organic redox flow batteries. Curr. Opin. Electrochem. 2022, 32, 100895. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, Q.; Ren, Y.; He, C.; Nie, J.; Du, B. A Bipyridine-Ester Dual-Modified 2,2,6,6-Tetramethylpiperidin-1-oxyl Derivative for Aqueous Organic Redox Flow Batteries. Materials 2025, 18, 2770. https://doi.org/10.3390/ma18122770
Zheng Q, Ren Y, He C, Nie J, Du B. A Bipyridine-Ester Dual-Modified 2,2,6,6-Tetramethylpiperidin-1-oxyl Derivative for Aqueous Organic Redox Flow Batteries. Materials. 2025; 18(12):2770. https://doi.org/10.3390/ma18122770
Chicago/Turabian StyleZheng, Qianqian, Yanwen Ren, Cuicui He, Jingjing Nie, and Binyang Du. 2025. "A Bipyridine-Ester Dual-Modified 2,2,6,6-Tetramethylpiperidin-1-oxyl Derivative for Aqueous Organic Redox Flow Batteries" Materials 18, no. 12: 2770. https://doi.org/10.3390/ma18122770
APA StyleZheng, Q., Ren, Y., He, C., Nie, J., & Du, B. (2025). A Bipyridine-Ester Dual-Modified 2,2,6,6-Tetramethylpiperidin-1-oxyl Derivative for Aqueous Organic Redox Flow Batteries. Materials, 18(12), 2770. https://doi.org/10.3390/ma18122770