
Local Structure Displacements and Electronic Structure of Sb-Substituted Rock-Salt Type AgBi1−xSbxSe0.8S0.6Te0.6 System

 , , ,
, , ,  , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
3. Results and Discussion
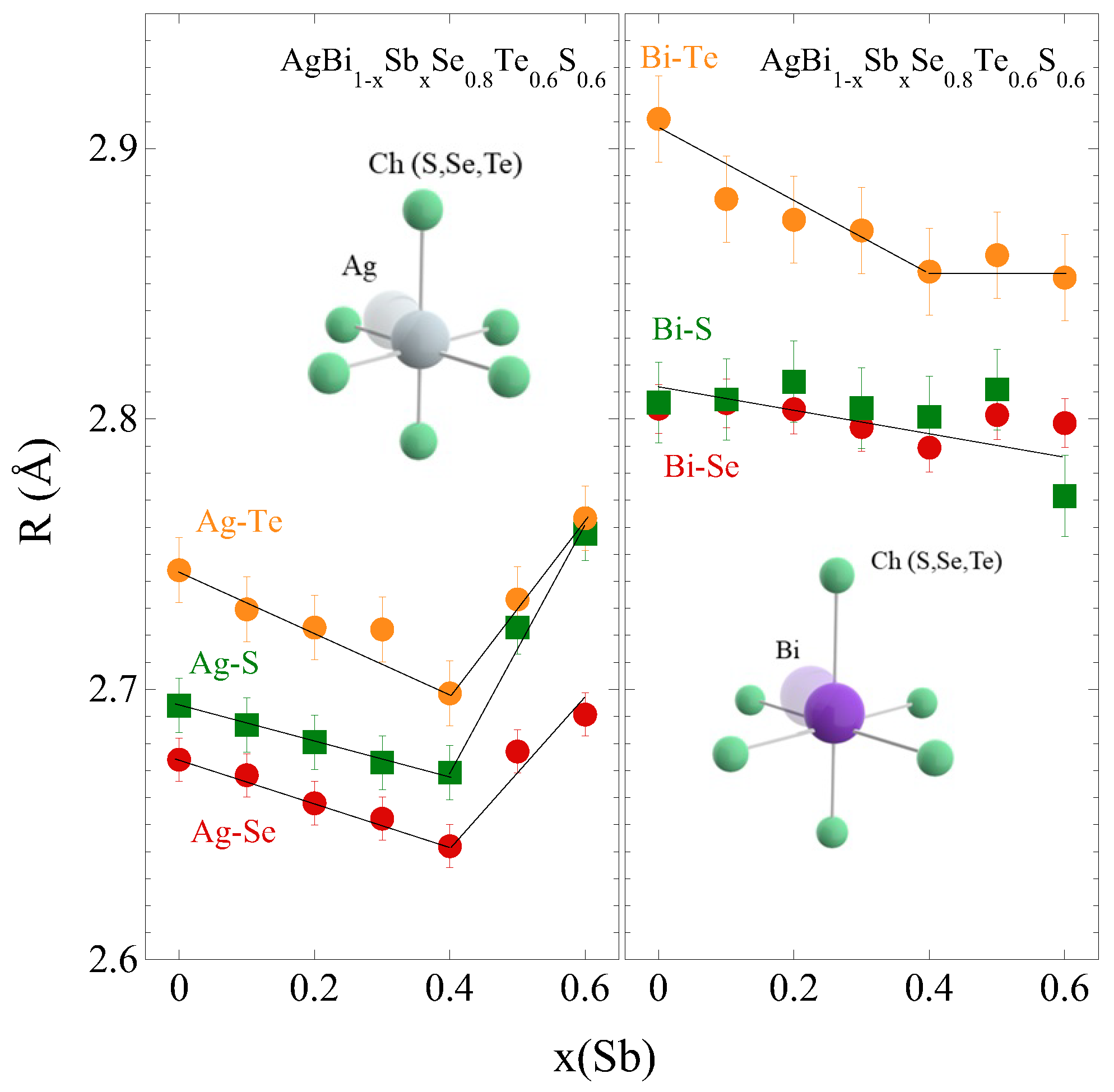
3.1. The Local Structure Investigations by EXAFS Spectroscopy
3.2. The Local Geometry by XANES Spectroscopy
3.3. The Electronic Structure Studies by XPS
4. Summary and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Snyder, G.J.; Toberer, E.S. Complex thermoelectric materials. Nat. Mater. 2008, 7, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Goldsmid, H.J. Introduction to thermoelectricity. In Springer Series in Materials Science; Springer: Berlin, Germany, 2010; Volume 121. [Google Scholar]
- Zhang, X.; Zhao, L.D. Thermoelectric materials: Energy conversion between heat and electricity. J. Mater. 2015, 1, 92–105. [Google Scholar] [CrossRef]
- Yan, Q.; Kanatzidis, M.G. High-performance thermoelectrics and challenges for practical devices. Nat. Mater. 2022, 21, 503–513. [Google Scholar] [CrossRef]
- Champier, D. Thermoelectric generators: A review of applications. Energy Convers. Manag. 2017, 140, 167–181. [Google Scholar] [CrossRef]
- Mukherjee, M.; Srivastava, A.; Singh, A.K. Recent advances in designing thermoelectric materials. J. Mater. Chem. C 2022, 10, 12524–12555. [Google Scholar] [CrossRef]
- Wernick, J.H.; Geller, S.; Benson, K.E. Constitution of the AgSbSe2-AgSbTe2-AgBiSe2-AgBiTe2 system. J. Phys. Chem. Solids 1958, 7, 240–248. [Google Scholar] [CrossRef]
- Morelli, D.T.; Jovovic, V.; Heremans, J.P. Intrinsically Minimal Thermal Conductivity in Cubic I-V-VI2 Semiconductors. Phys. Rev. Lett. 2008, 101, 035901. [Google Scholar] [CrossRef]
- Biswas, K.; He, J.; Blum, I.D.; Wu, C.I.; Hogan, T.P.; Seidman, D.N.; Dravid, V.P.; Kanatzidis, M.G. High-performance bulk thermoelectrics with all-scale hierarchical architectures. Nature 2012, 489, 414–418. [Google Scholar] [CrossRef]
- Pan, L.; Berardan, D.; Dragoe, N. High thermoelectric properties of n-type AgBiSe2. J. Am. Chem. Soc. 2013, 135, 4914–4917. [Google Scholar] [CrossRef]
- Jang, H.; Toriyama, M.Y.; Abbey, S.; Frimpong, B.; Male, J.P.; Snyder, J.F.; Jung, Y.S.; Oh, M.W. Suppressing Charged Cation Antisites via Se Vapor Annealing Enables p-Type Dopability in AgBiSe2–SnSe Thermoelectrics. Adv. Mater. 2022, 34, 2204132. [Google Scholar] [CrossRef]
- Boecher, F.; Culver, S.P.; Peilstoecker, J.; Weldert, K.S.; Zeier, W.G. Vacancy and anti-site disorder scattering in AgBiSe2 thermoelectrics. Dalton Trans. 2017, 46, 3906–3914. [Google Scholar] [CrossRef]
- Bernges, T.; Peilstöcker, J.; Dutta, M.; Ohno, S.; Culver, S.P.; Biswas, K.; Zeier, W.G. Local Structure and Influence of Sb Substitution on the Structure-Transport Properties in AgBiSe2. Inorg. Chem. 2019, 58, 9236–9245. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Hou, S.; Xue, W.; Yin, L.; Liu, Y.; Wang, X.; Chen, C.; Mao, J.; Zhang, Q. Manipulation of phase structure and Se vacancy to enhance the average thermoelectric performance of AgBiSe2. Mater. Today Phys. 2022, 27, 100837. [Google Scholar] [CrossRef]
- Feng, Z.; Zhang, X.; Wang, Y.; Zhang, J.; Jia, T.; Cao, B.; Zhang, Y. Thermoelectric optimization of AgBiSe2 by defect engineering for room-temperature applications. Phys. Rev. B 2019, 99, 155203. [Google Scholar] [CrossRef]
- Seshita, A.; Yamashita, A.; Fujita, T.; Katase, T.; Miura, A.; Nakahira, Y.; Moriyoshi, C.; Kuroiwa, Y.; Mizuguchi, Y. Stabilization and high thermoelectric performance of high-entropy-type cubic AgBi(S, Se, Te)2. J. Alloys Compd. 2024, 1004, 175679. [Google Scholar] [CrossRef]
- Seshita, A.; Yamashita, A.; Katase, T.; Mizuguchi, Y. High entropy effect on thermoelectric properties of nonequilibrium cubic phase of AgBiSe2−2xSxTex with x = 0–0.6. Dalton Trans. 2024, 53, 14830–14838. [Google Scholar] [CrossRef]
- Xia, Q.; Ying, P.; Han, Z.; Li, X.; Xu, L.; Cui, J. Chemical Composition Engineering Leading to the Significant Improvement in the Thermoelectric Performance of AgBiSe2-Based n-Type Solid Solutions. ACS Appl. Energy Mater. 2021, 4, 2899–2907. [Google Scholar] [CrossRef]
- Yamashita, A.; Goto, Y.; Miura, A.; Moriyoshi, C.; Kuroiwa, Y.; Mizuguchi, Y. n-Type thermoelectric metal chalcogenide (Ag,Pb,Bi)(S,Se,Te) designed by multi-site-type high-entropy alloying. Mater. Res. Lett. 2021, 9, 366–372. [Google Scholar] [CrossRef]
- Zhao, T.; Zhu, H.; Zhang, B.; Zheng, S.; Li, N.; Wang, G.; Wang, G.; Lu, X.; Zhou, X. High thermoelectric performance of tellurium-free n-type AgBi1−xSbxSe2 with stable cubic structure enabled by entropy engineering. Acta Mater. 2021, 220, 117291. [Google Scholar] [CrossRef]
- Armstrong, R.; Faust, J., Jr.; Tiller, W. A Structural Study of the Compound AgSbTe2. J. Appl. Phys. 1960, 31, 1954–1959. [Google Scholar] [CrossRef]
- Hockings, E.F. The thermal conductivity of silver antimony telluride. J. Phys. Chem. Solids 1959, 10, 341–342. [Google Scholar] [CrossRef]
- Wolfe, R.; Wernick, J.H.; Haszko, S.E. Anomalous Hall Effect in AgSbTe2. J. Appl. Phys. 1960, 31, 1959–1964. [Google Scholar] [CrossRef]
- Nielsen, M.D.; Ozolins, V.; Heremans, J.-P. Lone pair electrons minimize lattice thermal conductivity. Energy Environ. Sci. 2013, 6, 570–578. [Google Scholar] [CrossRef]
- Zhang, S.; Zhu, T.; Yang, S.; Yu, C.; Zhao, X. Phase compositions, nanoscale microstructures and thermoelectric properties in Ag2−ySbyTe1+y alloys with precipitated Sb2Te3 plates. Acta Mater. 2010, 58, 4160–4169. [Google Scholar] [CrossRef]
- Ko, Y.-H.; Oh, M.-W.; Lee, J.K.; Park, S.-D.; Kim, K.-J.; Choi, Y.S. Structural studies of AgSbTe2 under pressure: Experimental and theoretical analyses. Curr. Appl. Phys. 2014, 14, 1538–1542. [Google Scholar] [CrossRef]
- Ma, J.; Delaire, O.; May, A.F.; Carlton, C.E.; McGuire, M.A.; VanBebber, L.H.; Abernathy, D.L.; Ehlers, G.; Hong, T.; Huq, A.; et al. Glass-like phonon scattering from a spontaneous nanostructure in AgSbTe2. Nat. Nanotechnol. 2013, 8, 445–451. [Google Scholar] [CrossRef]
- Shinya, H.; Masago, A.; Fukushima, T.; Katayama-Yoshida, H. Inherent instability by antibonding coupling in AgSbTe2. Jpn. J. Appl. Phys. 2016, 55, 041801. [Google Scholar] [CrossRef]
- Du, B.; Yan, Y.; Tang, X. Variable-Temperature In Situ X-ray Diffraction Study of the Thermodynamic Evolution of AgSbTe2 Thermoelectric Compound. J. Electron. Mater. 2015, 44, 2118–2123. [Google Scholar] [CrossRef]
- Cao, J.; Dong, J.; Saglik, S.; Zhang, D.; Faye Duran Solco, S.; Joel Wen Jie You, I.; Liu, H.; Zhu, Q.; Xu, J.; Wu, J.; et al. Non-equilibrium strategy for enhancing thermoelectric properties and improving stability of AgSbTe2. Nano Energy 2023, 107, 108118. [Google Scholar] [CrossRef]
- Seshita, A.; Yamashita, A.; Mizuguchi, Y. Point defect scattering engineering for effective lattice thermal conductivity reduction in high-entropy-type thermoelectric material with rock salt structure. Appl. Phys. Express 2025, APEX-108511. [Google Scholar]
- Koningsberger, D.C.; Prins, R. X-ray Absorption: Principles, Applications, Techniques of EXAFS, SEXAFS, XANES. In Chemical Analysis: A Series of Monographs on Analytical Chemistry and Its Applications; Wiley: Hoboken, NJ, USA, 1988. [Google Scholar]
- Bunker, G. Introduction to XAFS; Cambridge University Press: Cambridge, UK, 2010. [Google Scholar]
- Huang, W.; Zhu, Y.; Liu, Y.; Tao, S.; Yang, C.; Diao, Q.; Hong, Z.; Han, H.; Liu, L.; Xu, W. Long-range ordering and local structural disordering of BiAgSe2 and BiAgSeTe thermoelectrics. Phys. Chem. Chem. Phys. 2021, 23, 24328–24335. [Google Scholar] [CrossRef]
- Tortora, L.; Seshita, A.; Hacisalihoglu, M.Y.; Yamashita, A.; Tomassucci, G.; Minati, F.; Skorynina, A.; Simonelli, L.; Mizokawa, T.; Mizuguchi, Y.; et al. Local structure and anomalous chemical potential shift in the AgBiSe2−2xTexSx system. Phys. Rev. B 2025, 111, 094203. [Google Scholar] [CrossRef]
- Simonelli, L.; Marini, C.; Olszewski, W.; Ávila Pérez, M.; Ramanan, N.; Guilera, G.; Cuartero, V.; Klementiev, K. CLAESS: The hard X-ray absorption beamline of the ALBA CELLS synchrotron. Cogent Phys. 2016, 3, 1231987. [Google Scholar] [CrossRef]
- Ravel, B.; Newville, M. ARTEMIS: Interactive graphical data analysis using IFEFFIT. Phys. Scr. 2006, 115, 1007. [Google Scholar]
- Igor Pro 9, Igor Pro; WaveMetrics Inc.: Lake Oswego, OR, USA 2018.
- Gurman, S.J. Interpretation of EXAFS data. J. Synchrotron Radiat. 1995, 2, 56–63. [Google Scholar] [CrossRef]
- Pugliese, G.M.; Tortora, L.; Paris, E.; Wakita, T.; Terashima, K.; Puri, A.; Nagao, M.; Higashinaka, R.; Matsuda, T.D.; Aoki, Y.; et al. The Local Structure of the BiS2 Layer in RE(O,F)BiS2 Determined by In-Plane Polarized X-ray Absorption Measurements. Physchem 2021, 1, 250–258. [Google Scholar] [CrossRef]
- Wang, S.; Sun, Y.; Yang, J.; Duan, B.; Wu, L.; Zhang, W.; Yang, J. High thermoelectric performance in Te-free (Bi,Sb)2Se3 via structural transition induced band convergence and chemical bond softening. Energy Environ. Sci. 2016, 9, 3436–3447. [Google Scholar] [CrossRef]
- Losťák, P.; Drasǎr, Č.; Süssmann, H.; Reinshaus, P.; Novotný, R.; Benes, L. Preparation and some physical properties of (Bi1−xSbx)2Se3 single crystals. J. Cryst. Growth 1997, 179, 144–152. [Google Scholar]
- Rezaei, N.; Hashemifar, S.J.; Akbarzadeh, H. Thermoelectric properties of AgSbTe2 from first-principles calculations. J. Appl. Phys. 2014, 116, 103705. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tortora, L.; Seshita, A.; Tomassucci, G.; Minati, F.; Skorynina, A.; Simonelli, L.; Yamashita, A.; Mizuguchi, Y.; Saini, N.L. Local Structure Displacements and Electronic Structure of Sb-Substituted Rock-Salt Type AgBi1−xSbxSe0.8S0.6Te0.6 System. Materials 2025, 18, 2578. https://doi.org/10.3390/ma18112578
Tortora L, Seshita A, Tomassucci G, Minati F, Skorynina A, Simonelli L, Yamashita A, Mizuguchi Y, Saini NL. Local Structure Displacements and Electronic Structure of Sb-Substituted Rock-Salt Type AgBi1−xSbxSe0.8S0.6Te0.6 System. Materials. 2025; 18(11):2578. https://doi.org/10.3390/ma18112578
Chicago/Turabian StyleTortora, Lorenzo, Asato Seshita, Giovanni Tomassucci, Francesco Minati, Alina Skorynina, Laura Simonelli, Aichi Yamashita, Yoshikazu Mizuguchi, and Naurang L. Saini. 2025. "Local Structure Displacements and Electronic Structure of Sb-Substituted Rock-Salt Type AgBi1−xSbxSe0.8S0.6Te0.6 System" Materials 18, no. 11: 2578. https://doi.org/10.3390/ma18112578
APA StyleTortora, L., Seshita, A., Tomassucci, G., Minati, F., Skorynina, A., Simonelli, L., Yamashita, A., Mizuguchi, Y., & Saini, N. L. (2025). Local Structure Displacements and Electronic Structure of Sb-Substituted Rock-Salt Type AgBi1−xSbxSe0.8S0.6Te0.6 System. Materials, 18(11), 2578. https://doi.org/10.3390/ma18112578