
Anisotropic Local Structure of SrFe2−xNixAs2 (x = 0.00, 0.16, and 0.23) Superconductor Probed by Polarized X-ray Absorption Fine Structure Measurements

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
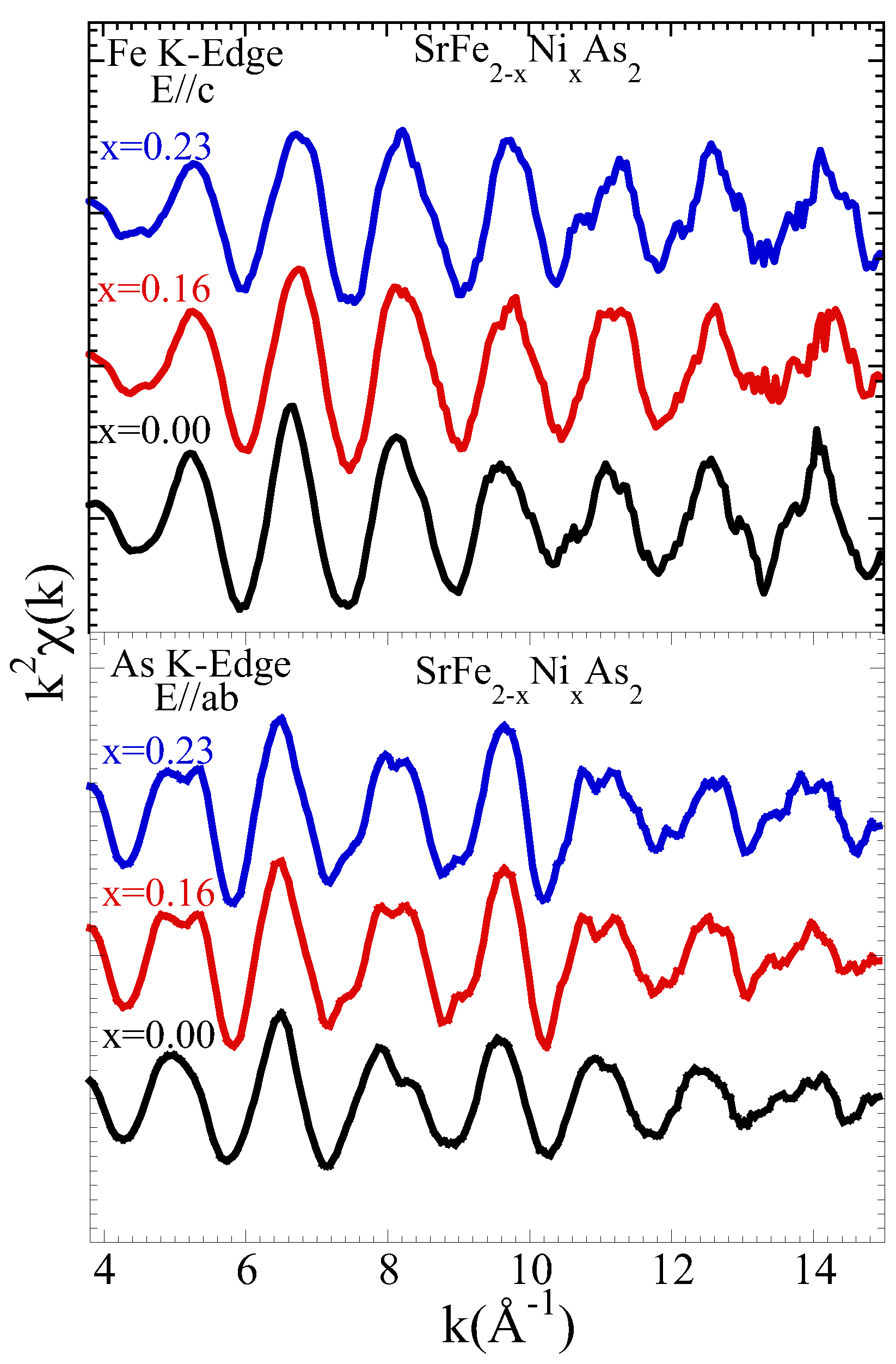
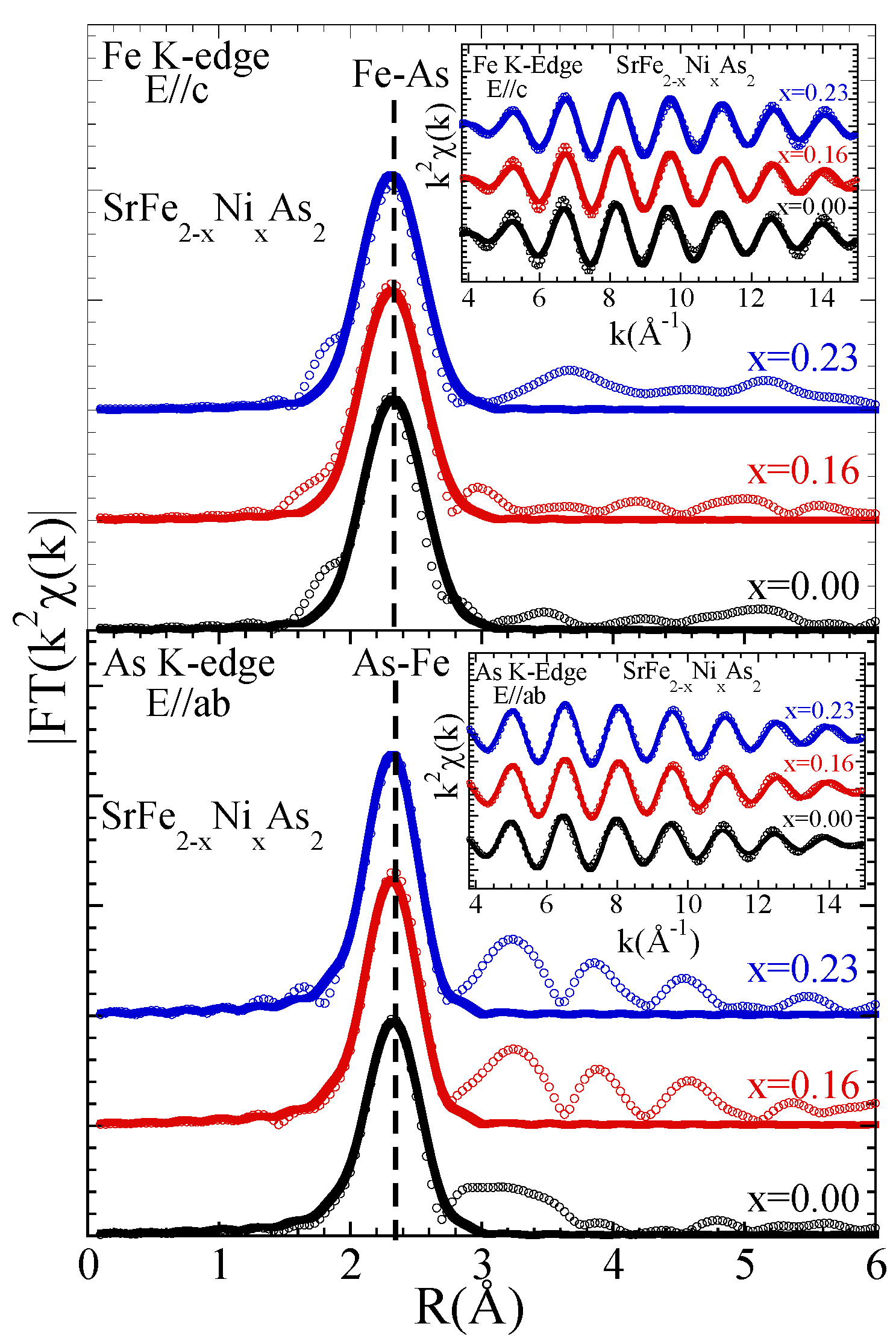
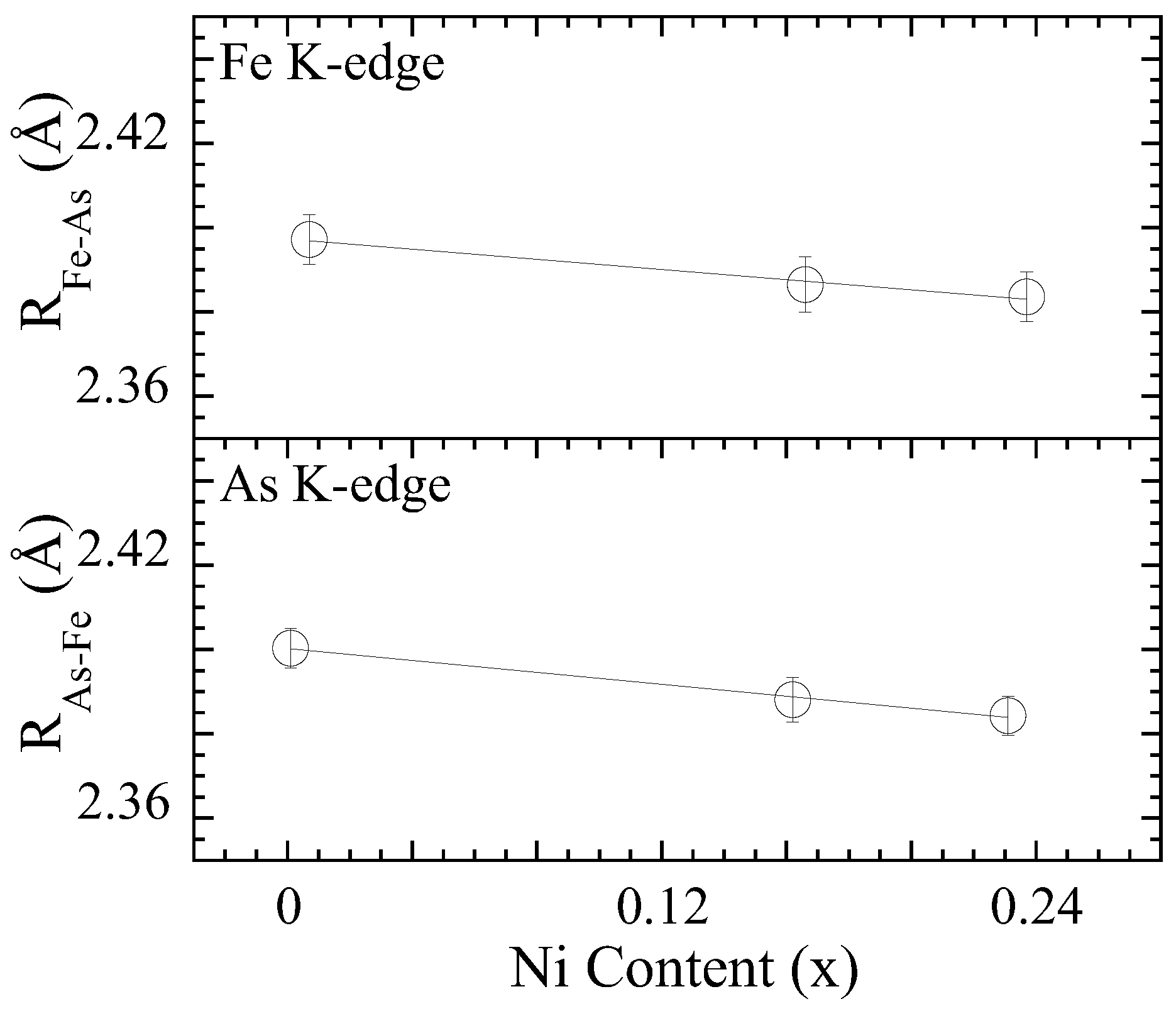
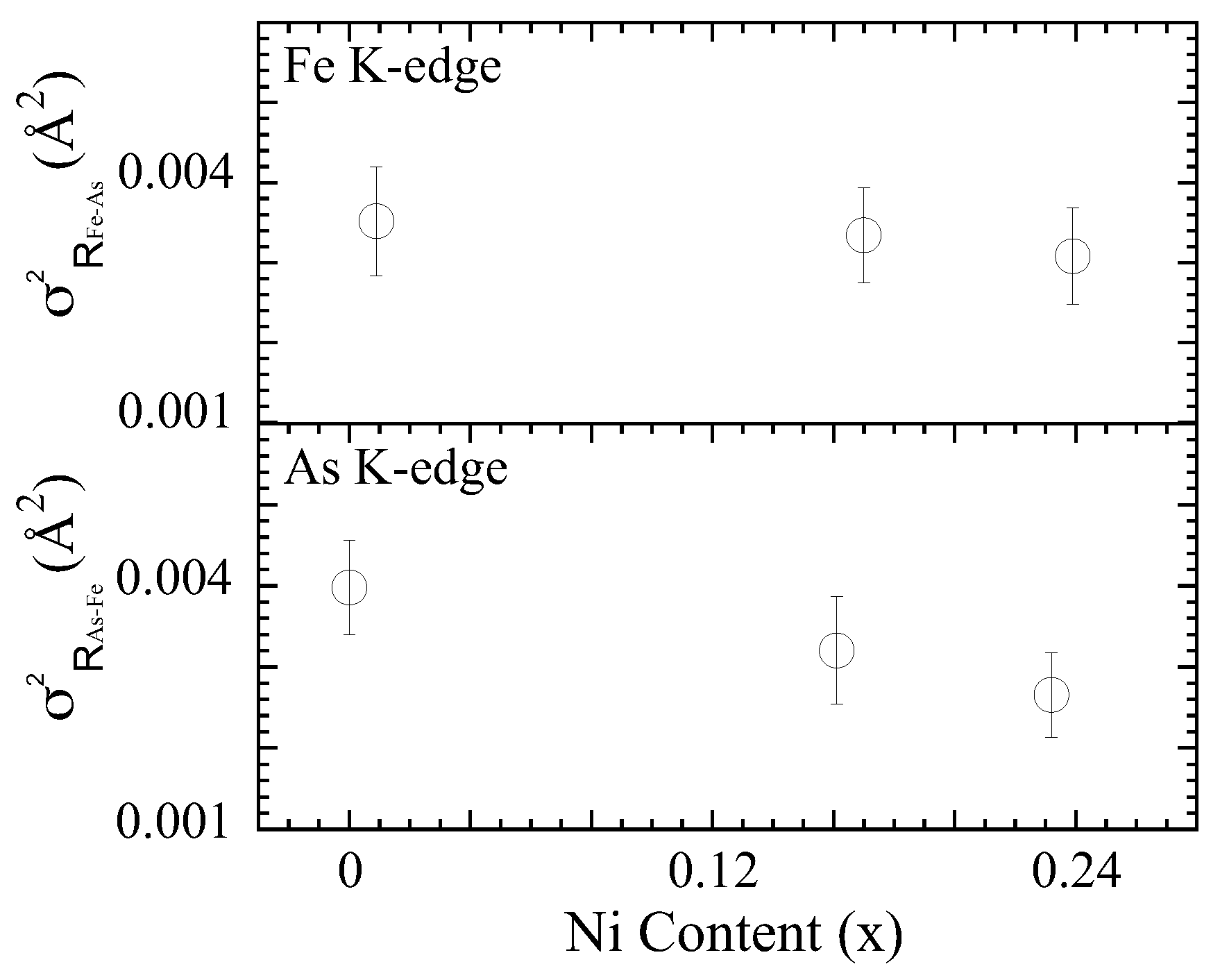
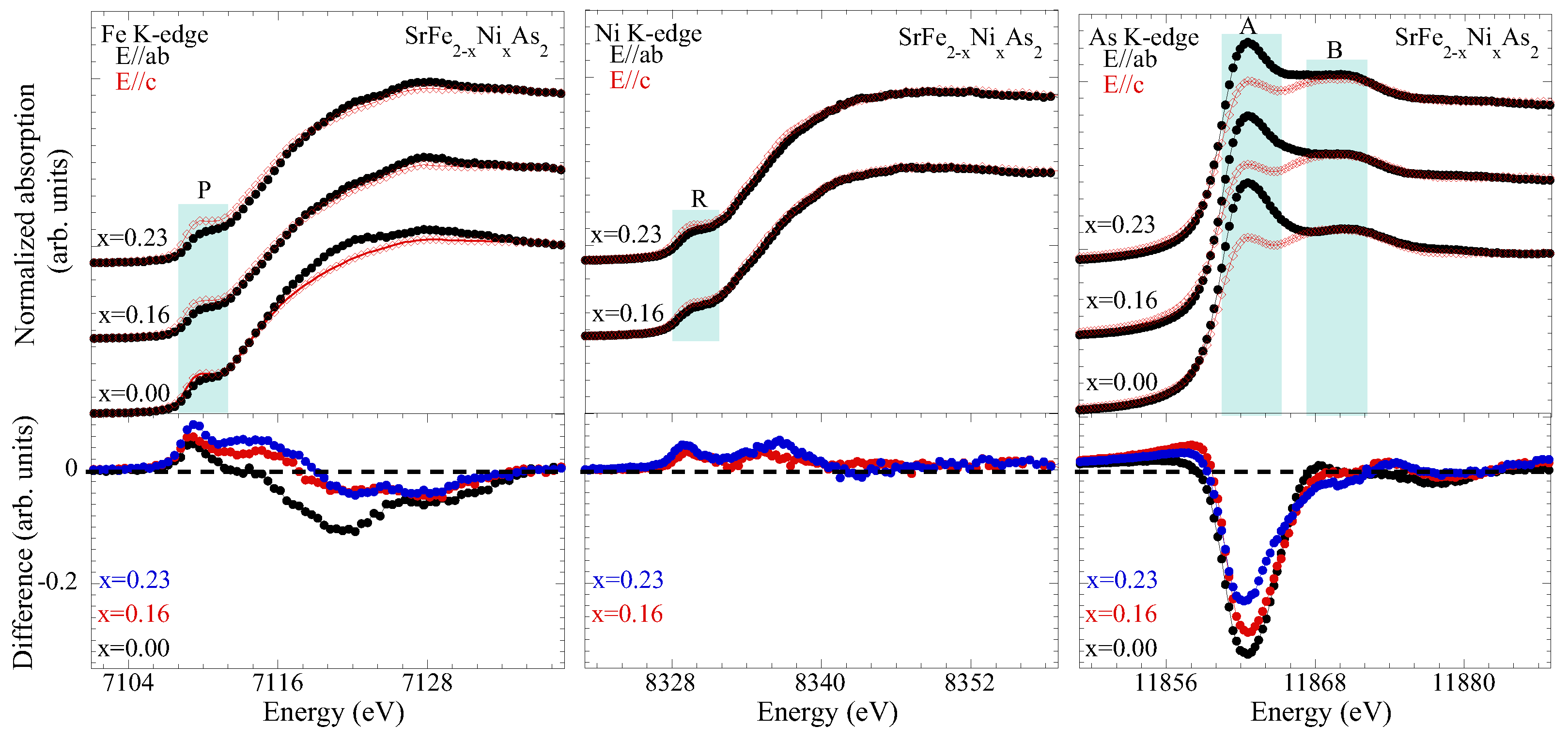
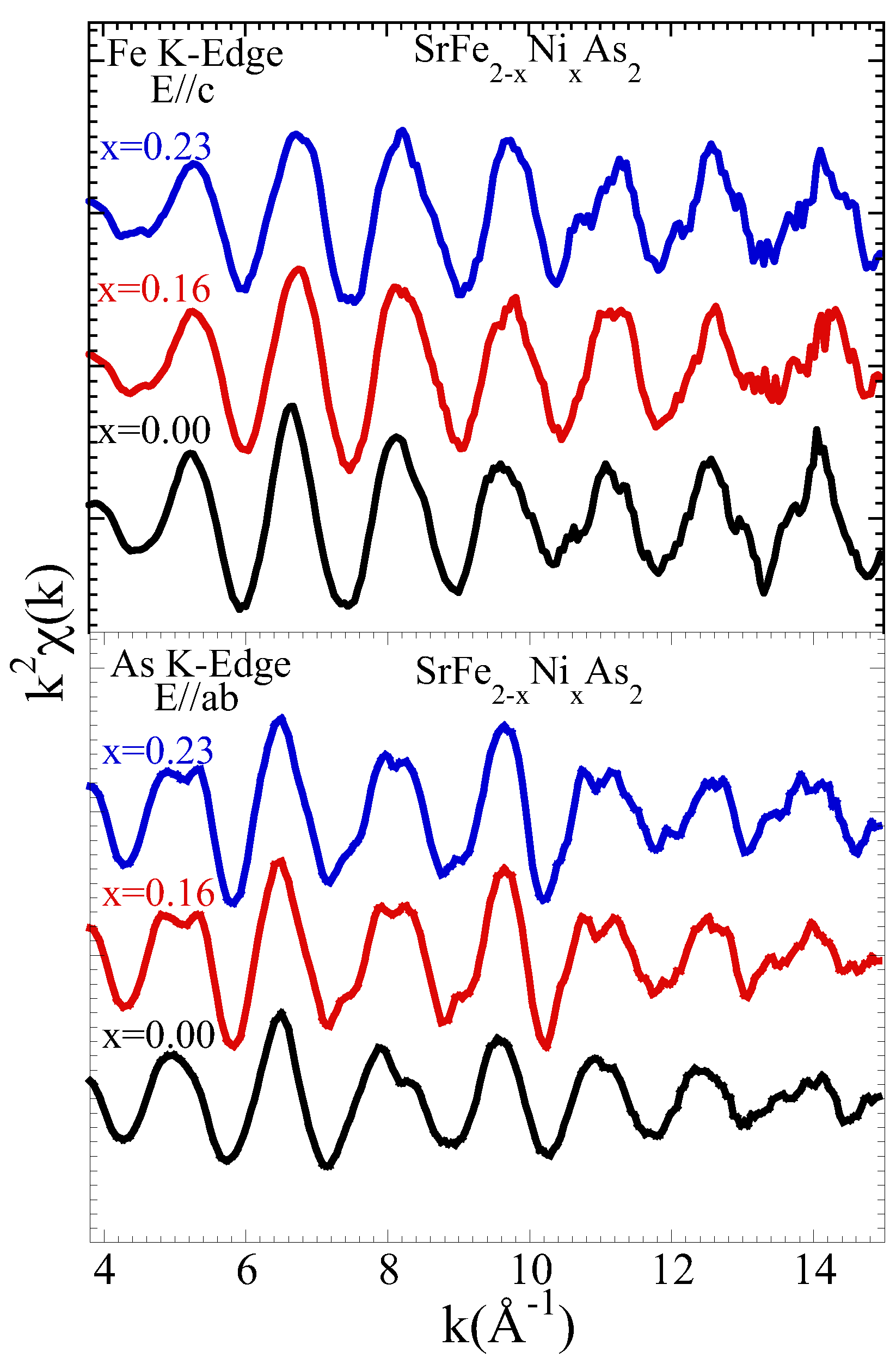
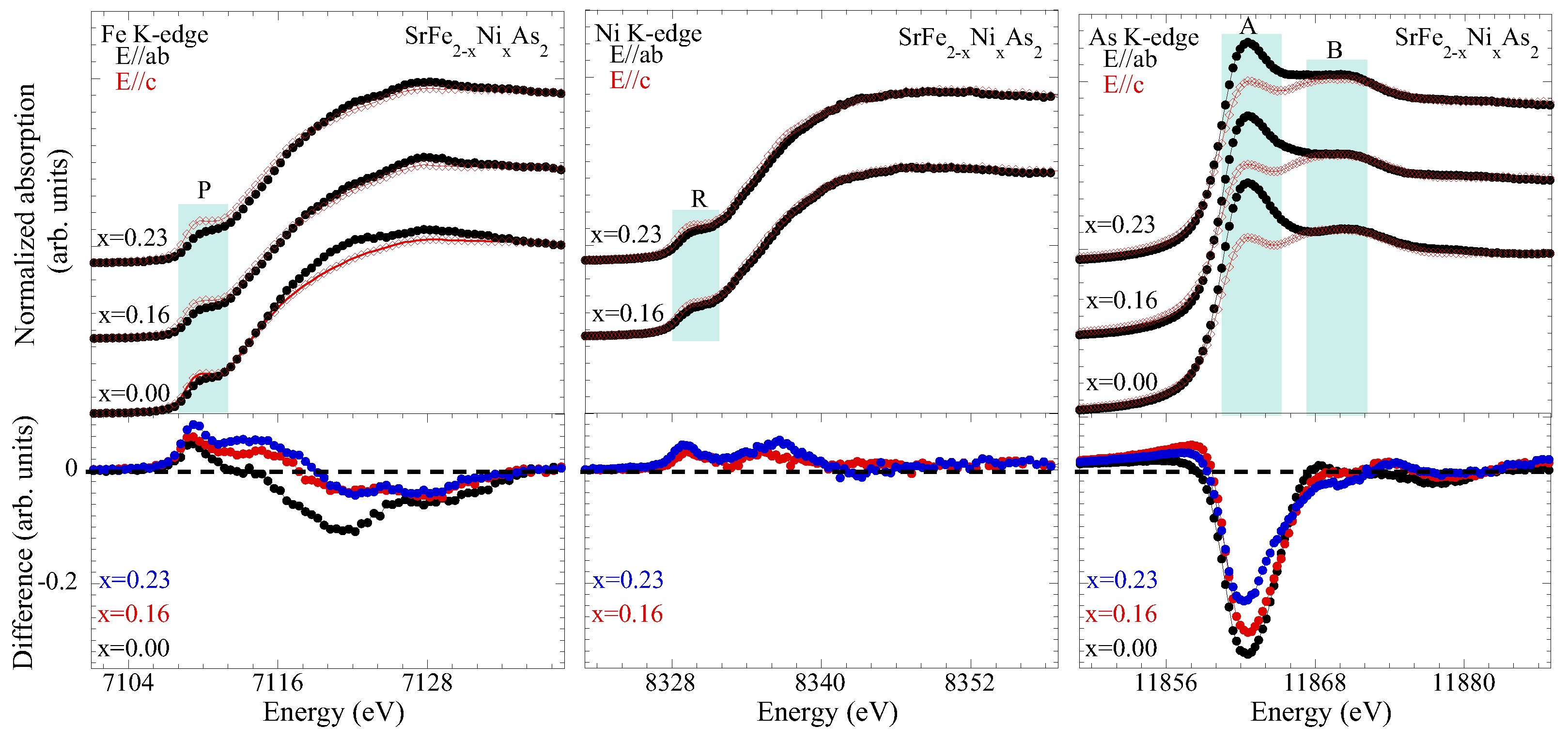
3.1. Extended X-ray Absorption Fine Structure (EXAFS) Analysis
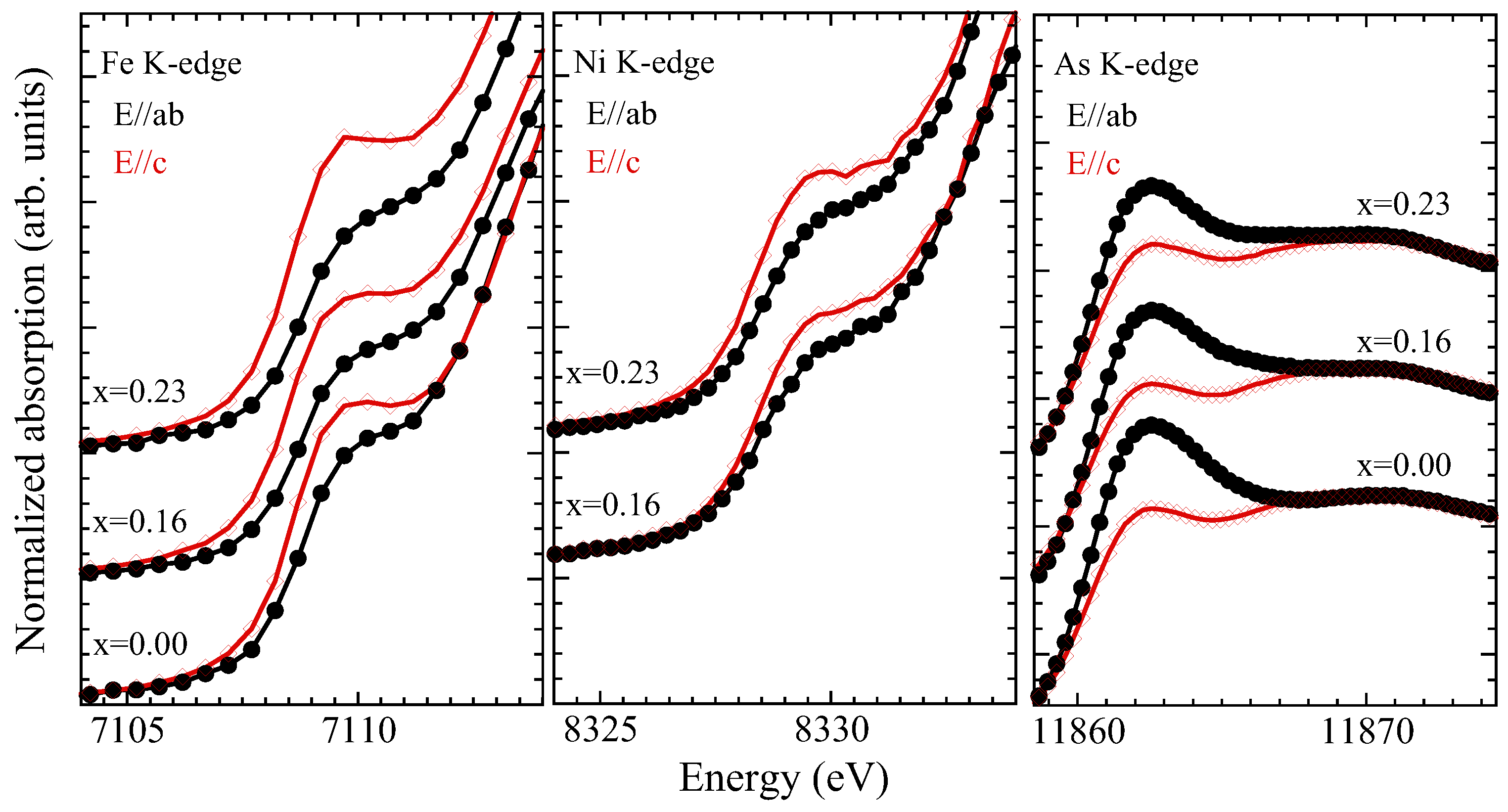
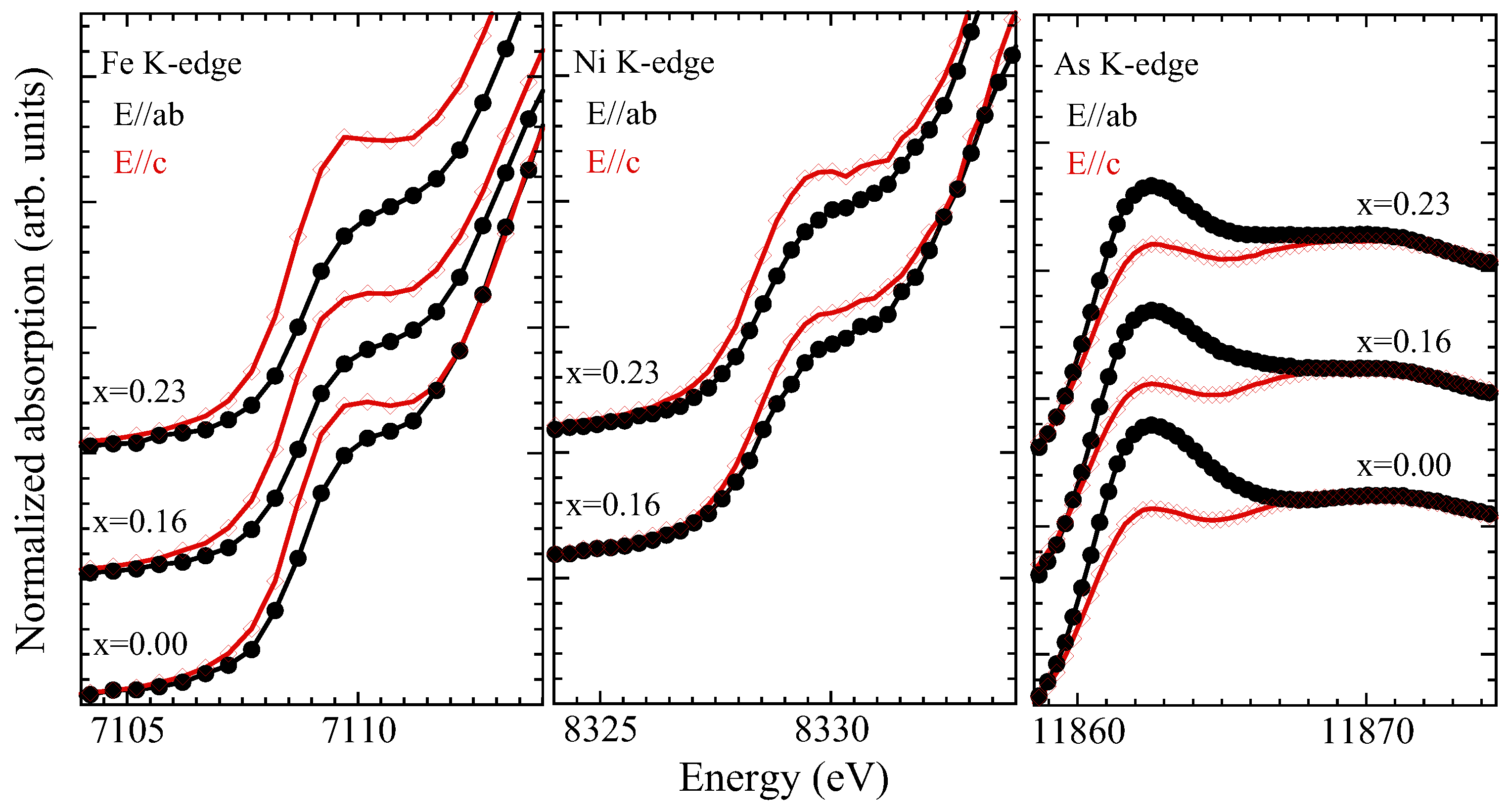
3.2. X-ray Absorption near Edge Structure (XANES) Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rotter, M.; Tegel, M.; Johrendt, D. Superconductivity at 38 K in the Iron Arsenide (Ba1-xKx)Fe2As2. Phys. Rev. Lett. 2008, 101, 107006. [Google Scholar] [CrossRef] [PubMed]
- Johnston, D.C. The Puzzle of High Temperature Superconductivity in Layered Iron Pnictides and Chalcogenides. Adv. Phys. 2010, 59, 803. [Google Scholar] [CrossRef]
- Stewart, G. Superconductivity in iron compounds. Rev. Mod. Phys. 2011, 83, 1589. [Google Scholar] [CrossRef]
- Hosono, H.; Kuroki, K. Iron-based superconductors: Current status of materials and pairing mechanism. Phys. C Supercond. Appl. 2015, 514, 399. [Google Scholar] [CrossRef]
- Sefat, A.S. Pressure effects on two superconducting iron-based families. Rep. Prog. Phys. 2011, 74, 124502. [Google Scholar] [CrossRef]
- Saha, S.; Butch, N.; Kirshenbaum, K.; Paglione, J. Evolution of bulk superconductivity in SrFe2As2 with Ni substitution. Phys. Rev. B 2009, 79, 224519. [Google Scholar] [CrossRef]
- Saparov, B.; Sefat, A.S. Annealing effects on the properties of BFe2As2 (B = Ca, Sr, Ba) superconducting parents. Dalton Trans. 2014, 43, 14971. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Fernandes, R.; Kreyssig, A.; Kim, J.; Thaler, A.; Bud’ko, S.; Canfield, P.; McQueeney, R.; Schmalian, J.; Goldman, A. Character of the structural and magnetic phase transitions in the parent and electron-doped BaFe2As2 compounds. Phys. Rev. B 2011, 83, 134522. [Google Scholar] [CrossRef]
- Jesche, A.; Caroca-Canales, N.; Rosner, H.; Borrmann, H.; Ormeci, A.; Kasinathan, D.; Klauss, H.; Luetkens, H.; Khasanov, R.; Amato, A. Strong coupling between magnetic and structural order parameters in SrFe2As2. Phys. Rev. B. 2008, 78, 180504. [Google Scholar] [CrossRef]
- Ni, N.; Nandi, S.; Kreyssig, A.; Goldman, A.; Mun, E.; Bud’ko, S.; Canfield, P. First-order structural phase transition in CaFe2As2. Phys. Rev. B 2008, 78, 014523. [Google Scholar] [CrossRef]
- Jeevan, H.; Hossain, Z.; Kasinathan, D.; Rosner, H.; Geibel, C.; Gegenwart, P. Electrical resistivity and specific heat of single-crystalline EuFe2As2: A magnetic homologue of SrFe2As2. Phys. Rev. B 2008, 78, 052502. [Google Scholar] [CrossRef]
- Canfield, P.C.; Bud’Ko, S.L. FeAs-based superconductivity: A case study of the effects of transition metal doping on BaFe2As2. Annu. Rev. Condens. Matter Phys. 2010, 1, 27. [Google Scholar] [CrossRef]
- Sefat, A.S.; Singh, D.J.; VanBebber, L.H.; Mozharivskyj, Y.; McGuire, M.A.; Jin, R.; Sales, B.C.; Keppens, V.; Mandrus, D. Absence of superconductivity in hole-doped BaFe2-xCrxAs2 single crystals. Phys. Rev. B 2009, 79, 224524. [Google Scholar] [CrossRef]
- Kim, M.; Kreyssig, A.; Thaler, A.; Pratt, D.; Tian, W.; Zarestky, J.; Green, M.; Bud’ko, S.; Canfield, P.; Mc-Queeney, R. ntiferromagnetic ordering in the absence of structural distortion in Ba(Fe1-xMnx)2As2. Phys. Rev. B 2010, 82, 220503. [Google Scholar] [CrossRef]
- Sefat, A.S.; Marty, K.; Christianson, A.D.; Saparov, B.; McGuire, M.A.; Lumsden, M.D.; Tian, W.; Sales, B.C. Effect of molybdenum 4 d hole substitution in BaFe2As2. Phys. Rev. B 2012, 85, 024503. [Google Scholar] [CrossRef]
- Kasinathan, D.; Ormeci, A.; Koch, K.; Burkhardt, U.; Schnelle, W.; Leithe-Jasper, A.; Rosner, H. AFe2As2 (A = Ca, Sr, Ba, Eu) and SrFe2-xTMxAs2 (TM = Mn, Co, Ni): Crystal structure, charge doping, magnetism and superconductivity. New J. Phys. 2009, 11, 025023. [Google Scholar] [CrossRef]
- Yan, Y.; Cheng, P.; Ying, J.; Luo, X.; Chen, F.; Zou, H.; Wang, A.; Ye, G.; Xiang, Z.; Ma, J. Structural, magnetic, and electronic transport properties of hole-doped SrFe2-xCuxAs2 single crystals. Phys. Rev. B 2013, 87, 075105. [Google Scholar] [CrossRef]
- Ni, N.; Thaler, A.; Kracher, A.; Yan, J.; Bud’ko, S.; Canfield, P. Phase diagrams of Ba (Fe1-xMx)2As2 single crystals (M = Rh and Pd). Phys. Rev. B 2009, 80, 024511. [Google Scholar] [CrossRef]
- Sefat, A.S.; Jin, R.; McGuire, M.A.; Sales, B.C.; Singh, D.J.; Mandrus, D. Superconductivity at 22 K in Co-doped BaFe2As2 crystals. Phys. Rev. Lett. 2008, 101, 117004. [Google Scholar] [CrossRef]
- Li, L.; Luo, Y.; Wang, Q.; Chen, H.; Ren, Z.; Tao, Q.; Li, Y.; Lin, X.; He, M.; Zhu, Z. Superconductivity induced by Ni doping in BaFe2As2 single crystals. New J. Phys. 2009, 11, 025008. [Google Scholar] [CrossRef]
- Leithe-Jasper, A.; Schnelle, W.; Geibel, C.; Rosner, H. Superconducting state in SrFe2-xCoxAs2 by internal doping of the iron arsenide layers. Phys. Rev. Lett. 2008, 101, 207004. [Google Scholar] [CrossRef] [PubMed]
- Han, F.; Zhu, X.; Cheng, P.; Mu, G.; Jia, Y.; Fang, L.; Wang, Y.; Luo, H.; Zeng, B.; Shen, B. Superconductivity and phase diagrams of the 4 d- and 5 d-metal-doped iron arsenides SrFe2-xMxAs2 (M = Rh, Ir, Pd). Phys. Rev. B 2009, 80, 024506. [Google Scholar] [CrossRef]
- Kumar, N.; Nagalakshmi, R.; Kulkarni, R.; Paulose, P.; Nigam, A.; Dhar, S.; Thamizhavel, A. Anisotropic magnetic and superconducting properties of CaFe2-xCoxAs2 (x = 0, 0.06) single crystals. Phys. Rev. B 2009, 79, 012504. [Google Scholar] [CrossRef]
- Kumar, N.; Chi, S.; Chen, Y.; Rana, K.G.; Nigam, A.; Thamizhavel, A.; William Ratcliff, I.; Dhar, S.; Lynn, J.W. Evolution of the bulk properties, structure, magnetic order, and superconductivity with Ni doping in CaFe2-xNixAs2. Phys. Rev. B 2009, 80, 144524. [Google Scholar] [CrossRef]
- Jiang, S.; Xing, H.; Xuan, G.; Ren, Z.; Wang, C.; Xu, Z.-A.; Cao, G. Superconductivity and local-moment magnetism in Eu(Fe0.89Co0.11)2As2. Phys. Rev. B 2009, 80, 184514. [Google Scholar] [CrossRef]
- Li, L.; Cao, H.; McGuire, M.A.; Kim, J.S.; Stewart, G.R.; Sefat, A.S. Role of magnetism in superconductivity of BaFe2As2: Study of 5 d Au-doped crystals. Phys. Rev. B 2015, 92, 094504. [Google Scholar] [CrossRef]
- Chu, J.H.; Analytis, J.G.; Kucharczyk, C.; Fisher, I.R. Determination of the phase diagram of the electron-doped superconductor Ba(Fe1-xCox)2As2. Phys. Rev. B 2009, 79, 014506. [Google Scholar] [CrossRef]
- Harnagea, L.; Singh, S.; Friemel, G.; Leps, N.; Bombor, D.; Abdel-Hafiez, M.; Wolter, A.; Hess, C.; Klingeler, R.; Behr, G. Phase diagram of the iron arsenide superconductors Ca (Fe1-xCox)2As2 (0≤ x ≤ 0.2). Phys. Rev. B 2011, 83, 094523. [Google Scholar] [CrossRef]
- Butch, N.; Saha, S.; Zhang, X.; Kirshenbaum, K.; Greene, R.; Paglione, J. Effective carrier type and field dependence of the reduced-Tc superconducting state in SrFe2-xNixAs2. Phys. Rev. B 2010, 81, 024518. [Google Scholar] [CrossRef]
- Krellner, C.; Caroca-Canales, N.; Jesche, A.; Rosner, H.; Ormeci, A.; Geibel, C. Magnetic and structural transitions in layered iron arsenide systems: AFe2As2 versus RFeAsO. Phys. Rev. B 2008, 78, 100504. [Google Scholar] [CrossRef]
- Tegel, M.; Rotter, M.; Weiss, V.; Schappacher, F.M.; Pöttgen, R.; Johrendt, D. Structural and magnetic phase transitions in the ternary iron arsenides SrFe2As2 and EuFe2As2. J. Condens. Matter Phys. 2008, 20, 452201. [Google Scholar] [CrossRef]
- Kissikov, T.; Sarkar, R.; Lawson, M.; Bush, B.; Timmons, E.; Tanatar, M.; Prozorov, R.; Bud’ko, S.; Canfield, P.; Fernandes, R. Uniaxial strain control of spin-polarization in multicomponent nematic order of BaFe2As2. arXiv 2017, arXiv:1704.03566. [Google Scholar] [CrossRef]
- Mizukami, Y.; Konczykowski, M.; Kawamoto, Y.; Kurata, S.; Kasahara, S.; Hashimoto, K.; Mishra, V.; Kreisel, A.; Wang, Y.; Hirschfeld, P.J.; et al. Disorder-induced topological change of the superconducting gap structure in iron pnictides. Nat. Commun. 2014, 5, 5657. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Butch, N.; Kirshenbaum, K.; Paglione, J.; Zavalij, P. Superconducting and ferromagnetic phases induced by lattice distortions in stoichiometric SrFe2As2 single crystals. Phys. Rev. Lett. 2009, 103, 037005. [Google Scholar] [CrossRef] [PubMed]
- Koningsberger, D.C.; Prins, R. X-ray Absorption: Principles, Applications, Techniques of EXAFS, SEXAFS, XANES. In Chemical Analysis: A Series of Monographs on Analytical Chemistry and Its Applications; Wiley: Hoboken, NJ, USA, 1988. [Google Scholar]
- Tröger, L.; Arvanitis, D.; Baberschke, K.; Michaelis, H.; Grimm, U.; Zschech, E. Full correction of the self-absorption in soft-fluorescence extended X-ray-absorption fine structure. Phys. Rev. B 1992, 46, 3283. [Google Scholar] [CrossRef] [PubMed]
- Pugliese, G.M.; Simonelli, L.; Tortora, L.; Tomassucci, G.; Iyo, A.; Eisaki, H.; Mizokawa, T.; Saini, N.L. Determination of the local structure of the La0.5−x Na0.5+xFe2As2 superconductor. Phys. Rev. B 2022, 105, 024519. [Google Scholar] [CrossRef]
- Gurman, S.J. Interpretation of EXAFS data. J. Synchrotron Rad. 1995, 2, 56. [Google Scholar] [CrossRef]
- Garcia, J.; Bianconi, A.; Benfatto, M.; Natoli, C.R. Coordination geometry of transitionmetal ions in dilute solutions by XANES. J. Phys. Colloq. 1986, 47, C8-49–C8-54. [Google Scholar] [CrossRef]
- De Groot, F.; Vanko, G.; Glatzel, P. The 1s X-ray absorption pre-edge structures in transition metal oxides. J. Condens. Matter Phys. 2009, 21, 104207. [Google Scholar] [CrossRef]
- Joseph, B.; Iadecola, A.; Simonelli, L.; Mizuguchi, Y.; Takano, Y.; Mizokawa, T.; Saini, N. A study of the electronic structure of FeSe1−xTex chalcogenides by Fe and Se K-edge X-ray absorption near edge structure measurements. J. Condens. Matter Phys. 2010, 22, 485702. [Google Scholar] [CrossRef]
- Modrow, H.; Bucher, S.; Rehr, J.; Ankudinov, A. Calculation and interpretation of K-shell X-ray absorption near-edge structure of transition metal oxides. Phys. Rev. B 2003, 67, 035123. [Google Scholar] [CrossRef]
- De Montellano, A.G.S.O.; de Leon, J.M.; Saini, N.L.; Bianconi, A. Effect of rare earth substitution in the density of electronic states of LnOFeAs. J. Appl. Phys. 2012, 111, 112631. [Google Scholar] [CrossRef]
- Xu, W.; Joseph, B.; Iadecola, A.; Marcelli, A.; Chu, W.S.; Gioacchino, D.D.; Bianconi, A.; Wu, Z.Y.; Saini, N.L. Arsenic K-edge XANES study of REFeAsO oxypnictides. Europhys. Lett. 2010, 90, 57001. [Google Scholar] [CrossRef]
- Kuroki, K.; Usui, H.; Onari, S.; Arita, R.; Aoki, H. Pnictogen height as a possible switch between high-Tc nodeless and low-Tc nodal pairings in the iron-based superconductors. Phys. Rev. B 2009, 79, 224511. [Google Scholar] [CrossRef]
- Kontani, H.; Onari, S. Orbital-fluctuation-mediated superconductivity in iron pnictides: Analysis of the five-orbital Hubbard-Holstein model. Phys. Rev. Lett. 2010, 104, 157001. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hacisalihoglu, M.Y.; Tortora, L.; Tomassucci, G.; Simonelli, L.; Saini, N.L. Anisotropic Local Structure of SrFe2−xNixAs2 (x = 0.00, 0.16, and 0.23) Superconductor Probed by Polarized X-ray Absorption Fine Structure Measurements. Materials 2024, 17, 1301. https://doi.org/10.3390/ma17061301
Hacisalihoglu MY, Tortora L, Tomassucci G, Simonelli L, Saini NL. Anisotropic Local Structure of SrFe2−xNixAs2 (x = 0.00, 0.16, and 0.23) Superconductor Probed by Polarized X-ray Absorption Fine Structure Measurements. Materials. 2024; 17(6):1301. https://doi.org/10.3390/ma17061301
Chicago/Turabian StyleHacisalihoglu, M. Y., L. Tortora, G. Tomassucci, L. Simonelli, and N. L. Saini. 2024. "Anisotropic Local Structure of SrFe2−xNixAs2 (x = 0.00, 0.16, and 0.23) Superconductor Probed by Polarized X-ray Absorption Fine Structure Measurements" Materials 17, no. 6: 1301. https://doi.org/10.3390/ma17061301